

Drug Resistance: The Role of Sphingolipid Metabolism
 , and
, and
Abstract
1. Introduction
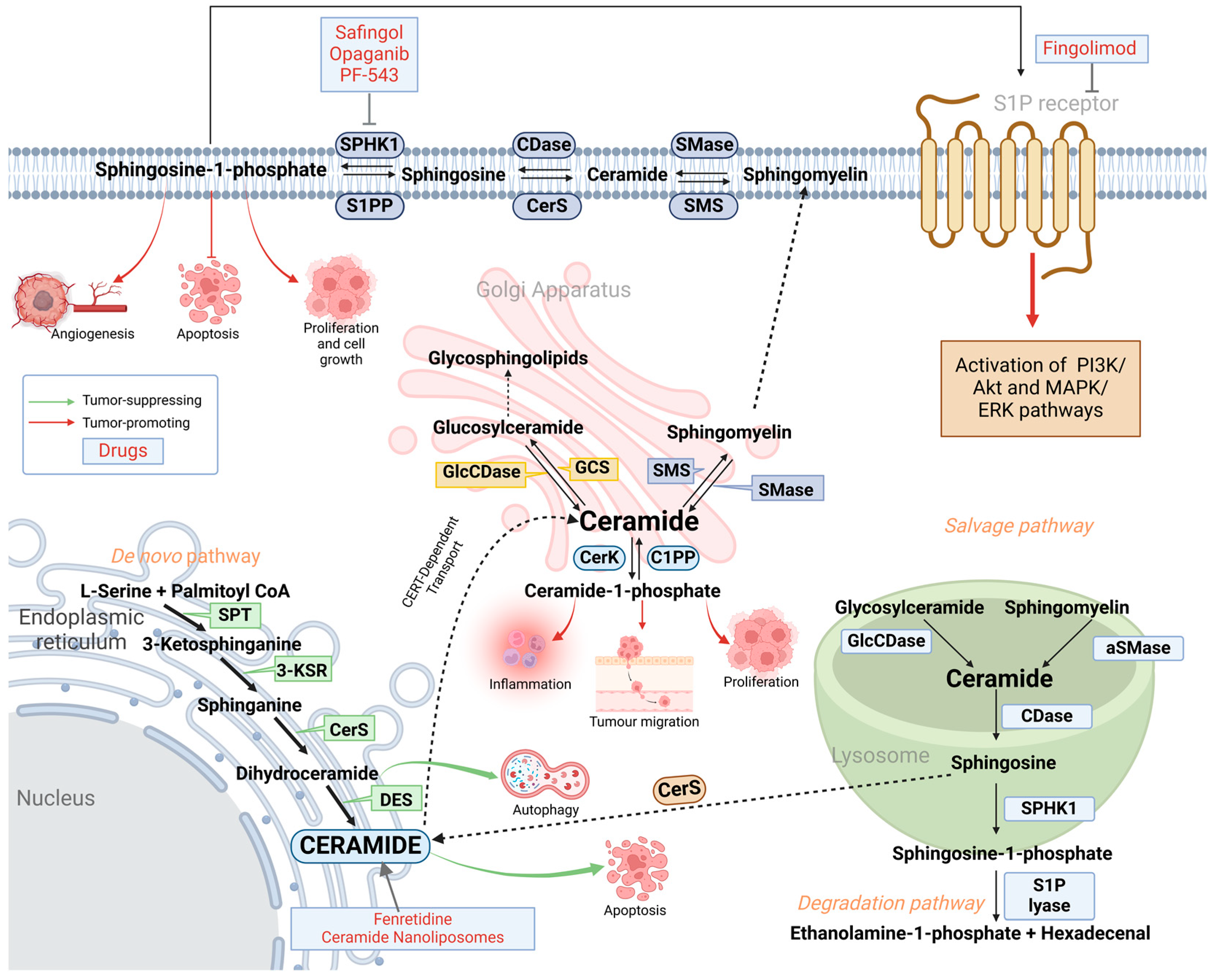
2. Sphingolipid Metabolism: De Novo, Salvage and Degradation Pathways
3. Enzymes of Sphingolipid Metabolism Involved in Cancer-Related Drug Resistance
3.1. Glucosylceramide Synthase (GCS) Causes Drug Resistance Through Upregulation of Multidrug Resistance Proteins
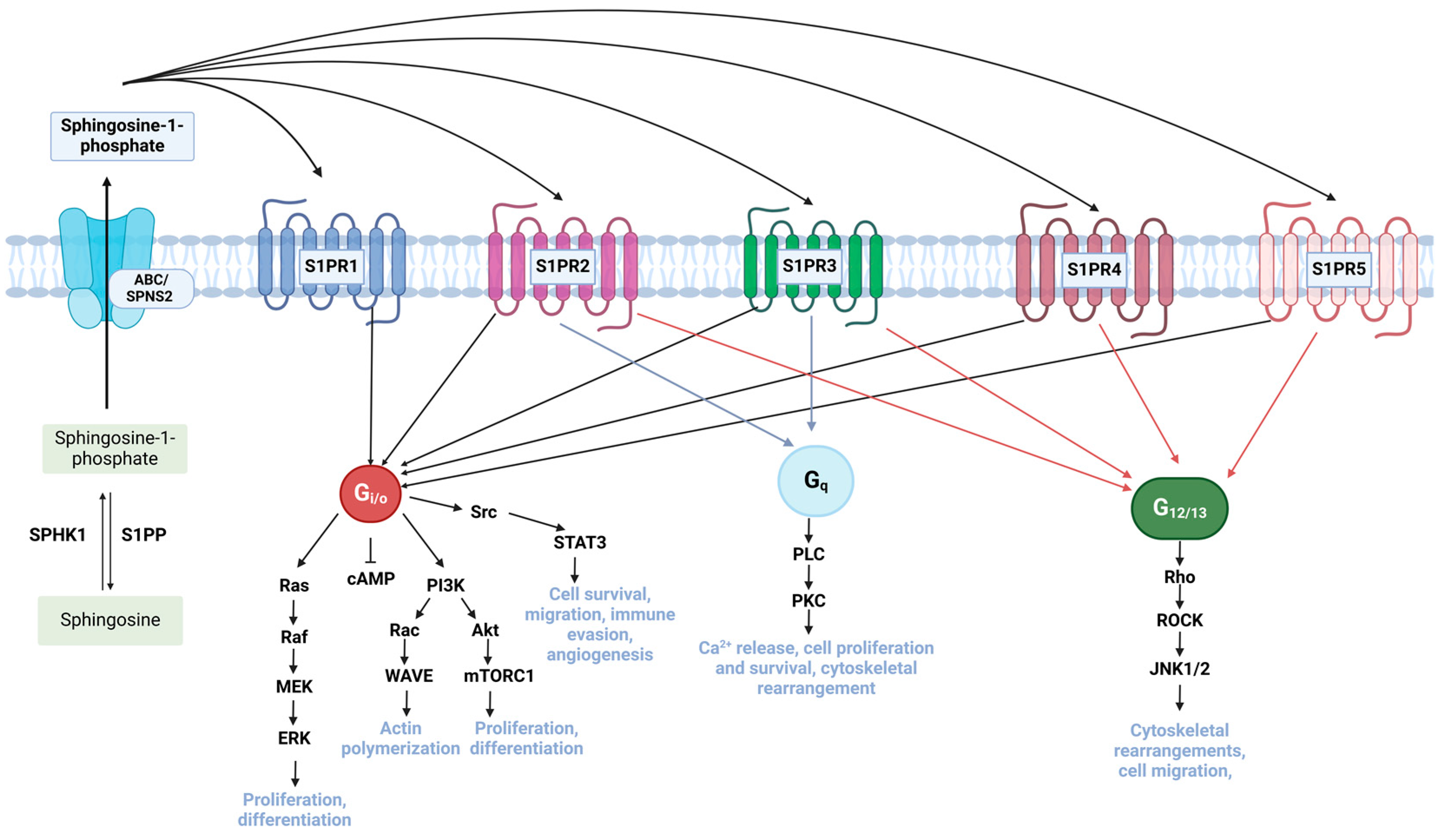
3.2. Dysregulation of Sphingosine Kinase (SPHK) and Sphingosine-1-Phosphate (S1P) Promotes Drug Resistance Through Activation of Pro-Tumorigenic Pathways
3.3. Abnormal Acid Ceramidase (AC) Levels Promote Drug Efflux in Cancer Cells
3.4. Downregulation of Sphingomyelinases (SMase) Mediates Apoptosis Resistance
4. Clinical Implications
4.1. Targeting S1P Signalling Pathway
4.2. Induction of Apoptosis in Cancer Cells by Ceramide Nanoliposomes
4.3. Natural Compounds Targeting Abnormal Sphingolipid Expression in Cancer Therapy
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, K.; Kumar, K.; Singh, A.; Tripathi, A.; Tiwari, L. Drug Resistance in Cancer Therapy: Mechanisms, Challenges and Strategies. Asian J. Nurs. Educ. Res. 2024, 14, 95–100. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; LLeonart, M.E. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Körner, C.; Fröhlich, F. Compartmentation and functions of sphingolipids. Curr. Opin. Cell Biol. 2022, 74, 104–111. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Salas, A.; Ponnusamy, S.; Senkal, C.E.; Meyers-Needham, M.; Selvam, S.P.; Saddoughi, S.A.; Apohan, E.; Sentelle, R.D.; Smith, C.; Gault, C.R.; et al. Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood 2011, 117, 5941–5952. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036.e9. [Google Scholar] [CrossRef]
- La Monica, S.; Vacondio, F.; Eltayeb, K.; Lodola, A.; Volta, F.; Viglioli, M.; Ferlenghi, F.; Galvani, F.; Galetti, M.; Bonelli, M.; et al. Targeting glucosylceramide synthase induces antiproliferative and proapoptotic effects in osimertinib-resistant NSCLC cell models. Sci. Rep. 2024, 14, 6491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; DiVittore, N.A.; Young, M.M.; Jia, Z.; Xie, K.; Ritty, T.M.; Kester, M.; Fox, T.E. Altered sphingolipid metabolism in patients with metastatic pancreatic cancer. Biomolecules 2013, 3, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key regulators of apoptosis and pivotal players in cancer drug resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef]
- Bataller, M.; Sánchez-García, A.; Garcia-Mayea, Y.; Mir, C.; Rodriguez, I.; LLeonart, M.E. The Role of Sphingolipids Metabolism in Cancer Drug Resistance. Front. Oncol. 2021, 11, 807636. [Google Scholar] [CrossRef] [PubMed]
- Sasset, L.; Zhang, Y.; Dunn, T.M.; Di Lorenzo, A. Sphingolipid De Novo Biosynthesis: A Rheostat of Cardiovascular Homeostasis. Trends Endocrinol. Metab. 2016, 27, 807–819. [Google Scholar] [CrossRef]
- Tidhar, R.; Futerman, A.H. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2511–2518. [Google Scholar] [CrossRef]
- Pralhada Rao, R.; Vaidyanathan, N.; Rengasamy, M.; Mammen Oommen, A.; Somaiya, N.; Jagannath, M.R. Sphingolipid metabolic pathway: An overview of major roles played in human diseases. J. Lipids 2013, 2013, 178910. [Google Scholar] [CrossRef]
- Xiao, S.; Peng, K.; Li, C.; Long, Y.; Yu, Q. The role of sphingosine-1-phosphate in autophagy and related disorders. Cell Death Discov. 2023, 9, 380. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Yao, L.; Xu, J.; Zhang, L.; Zheng, T.; Liu, L.; Zhang, L. Physicochemical stability-increasing effects of anthocyanin via a co-assembly approach with an amphiphilic peptide. Food Chem. 2021, 362, 130101. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, N.; Hanafusa, K.; Hotta, T.; Oshima, E.; Iwabuchi, K.; Nakayama, H. Multiplicity of Glycosphingolipid-Enriched Microdomain-Driven Immune Signaling. Int. J. Mol. Sci. 2021, 22, 9565. [Google Scholar] [CrossRef] [PubMed]
- Gouazé, V.; Yu, J.Y.; Bleicher, R.J.; Han, T.-Y.; Liu, Y.-Y.; Wang, H.; Gottesman, M.M.; Bitterman, A.; Giuliano, A.E.; Cabot, M.C. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol. Cancer Ther. 2004, 3, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Kreitzburg, K.M.; van Waardenburg, R.C.A.M.; Yoon, K.J. Sphingolipid metabolism and drug resistance in ovarian cancer. Cancer Drug Resist. 2018, 1, 181–197. [Google Scholar] [CrossRef]
- Gouazé, V.; Liu, Y.-Y.; Prickett, C.S.; Yu, J.Y.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res. 2005, 65, 3861–3867. [Google Scholar] [CrossRef]
- Wegner, M.-S.; Gruber, L.; Mattjus, P.; Geisslinger, G.; Grösch, S. The UDP-glucose ceramide glycosyltransferase (UGCG) and the link to multidrug resistance protein 1 (MDR1). BMC Cancer 2018, 18, 153. [Google Scholar] [CrossRef]
- Wojtal, K.A.; de Vries, E.; Hoekstra, D.; van Ijzendoorn, S.C.D. Efficient trafficking of MDR1/P-glycoprotein to apical canalicular plasma membranes in HepG2 cells requires PKA-RIIalpha anchoring and glucosylceramide. Mol. Biol. Cell 2006, 17, 3638–3650. [Google Scholar] [CrossRef]
- Roh, J.-L.; Kim, E.H.; Park, J.Y.; Kim, J.W. Inhibition of Glucosylceramide Synthase Sensitizes Head and Neck Cancer to Cisplatin. Mol. Cancer Ther. 2015, 14, 1907–1915. [Google Scholar] [CrossRef]
- Selvam, S.P.; Ogretmen, B. Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–27. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Alkafaas, S.S.; Elsalahaty, M.I.; Ismail, D.F.; Radwan, M.A.; Elkafas, S.S.; Loutfy, S.A.; Elshazli, R.M.; Baazaoui, N.; Ahmed, A.E.; Hafez, W.; et al. The emerging roles of sphingosine 1-phosphate and SphK1 in cancer resistance: A promising therapeutic target. Cancer Cell Int. 2024, 24, 89. [Google Scholar] [CrossRef]
- Ihlefeld, K.; Vienken, H.; Claas, R.F.; Blankenbach, K.; Rudowski, A.; ter Braak, M.; Koch, A.; Van Veldhoven, P.P.; Pfeilschifter, J.; Meyer zu Heringdorf, D. Upregulation of ABC transporters contributes to chemoresistance of sphingosine 1-phosphate lyase-deficient fibroblasts. J. Lipid Res. 2015, 56, 60–69. [Google Scholar] [CrossRef]
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell. Signal. 2017, 34, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xie, R.J.; Geng, X.X.; Luo, X.H.; Han, B.; Cheng, M.L. Effect of Danshao Huaxian capsule on expression of matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 in fibrotic liver of rats. World J. Gastroenterol. 2005, 11, 4953–4956. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Yao, Z.; Sui, Y.; Fang, S. SphK1 Promotes Cancer Progression through Activating JAK/STAT Pathway and Up-Regulating S1PR1 Expression in Colon Cancer Cells. Anticancer. Agents Med. Chem. 2022, 22, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissié, J.; Malavaud, B.; et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar] [CrossRef]
- Katsuta, E.; Yan, L.; Nagahashi, M.; Raza, A.; Sturgill, J.L.; Lyon, D.E.; Rashid, O.M.; Hait, N.C.; Takabe, K. Doxorubicin effect is enhanced by sphingosine-1-phosphate signaling antagonist in breast cancer. J. Surg. Res. 2017, 219, 202–213. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Marfe, G.; Di Stefano, C.; Gambacurta, A.; Ottone, T.; Martini, V.; Abruzzese, E.; Mologni, L.; Sinibaldi-Salimei, P.; de Fabritis, P.; Gambacorti-Passerini, C.; et al. Sphingosine kinase 1 overexpression is regulated by signaling through PI3K, AKT2, and mTOR in imatinib-resistant chronic myeloid leukemia cells. Exp. Hematol. 2011, 39, 653–665.e6. [Google Scholar] [CrossRef]
- Parveen, F.; Bender, D.; Law, S.-H.; Mishra, V.K.; Chen, C.-C.; Ke, L.-Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef]
- Cho, S.M.; Kwon, H.J. Acid ceramidase, an emerging target for anti-cancer and anti-angiogenesis. Arch. Pharm. Res. 2019, 42, 232–243. [Google Scholar] [CrossRef]
- Turner, L.S.; Cheng, J.C.; Beckham, T.H.; Keane, T.E.; Norris, J.S.; Liu, X. Autophagy is increased in prostate cancer cells overexpressing acid ceramidase and enhances resistance to C6 ceramide. Prostate Cancer Prostatic Dis. 2011, 14, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Clifford, R.E.; Govindarajah, N.; Bowden, D.; Sutton, P.; Glenn, M.; Darvish-Damavandi, M.; Buczacki, S.; McDermott, U.; Szulc, Z.; Ogretmen, B.; et al. Targeting Acid Ceramidase to Improve the Radiosensitivity of Rectal Cancer. Cells 2020, 9, 2693. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Bai, A.; Beckham, T.H.; Marrison, S.T.; Yount, C.L.; Young, K.; Lu, P.; Bartlett, A.M.; Wu, B.X.; Keane, B.J.; et al. Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse. J. Clin. Investig. 2013, 123, 4344–4358. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Song, P.; Ogretmen, B. Roles of bioactive sphingolipids in cancer biology and therapeutics. In Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 49, pp. 413–440. [Google Scholar] [CrossRef]
- Goñi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef]
- Grammatikos, G.; Teichgräber, V.; Carpinteiro, A.; Trarbach, T.; Weller, M.; Hengge, U.R.; Gulbins, E. Overexpression of Acid Sphingomyelinase Sensitizes Glioma Cells to Chemotherapy. Antioxid. Redox Signal. 2007, 9, 1449–1456. [Google Scholar] [CrossRef]
- Gramatzki, D.; Herrmann, C.; Happold, C.; Becker, K.A.; Gulbins, E.; Weller, M.; Tabatabai, G. Glioma cell death induced by irradiation or alkylating agent chemotherapy is independent of the intrinsic ceramide pathway. PLoS ONE 2013, 8, e63527. [Google Scholar] [CrossRef]
- Allam, R.M.; Al-Abd, A.M.; Khedr, A.; Sharaf, O.A.; Nofal, S.M.; Khalifa, A.E.; Mosli, H.A.; Abdel-Naim, A.B. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol. Lett. 2018, 291, 77–85. [Google Scholar] [CrossRef]
- Hait, N.C.; Avni, D.; Yamada, A.; Nagahashi, M.; Aoyagi, T.; Aoki, H.; Dumur, C.I.; Zelenko, Z.; Gallagher, E.J.; Leroith, D.; et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERα expression and enhances hormonal therapy for breast cancer. Oncogenesis 2015, 4, e156. [Google Scholar] [CrossRef]
- Rupp, T.; Pelouin, O.; Genest, L.; Legrand, C.; Froget, G.; Castagné, V. Therapeutic potential of Fingolimod in triple negative breast cancer preclinical models. Transl. Oncol. 2021, 14, 100926. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Alling, N.; Jackson, E.A.; Yagoub, D.; Haass, N.K.; Allen, J.D.; Martinello-Wilks, R. Evaluation of cell cycle arrest in estrogen responsive MCF-7 breast cancer cells: Pitfalls of the MTS assay. PLoS ONE 2011, 6, e20623. [Google Scholar] [CrossRef]
- Shen, Y.; Cai, M.; Xia, W.; Liu, J.; Zhang, Q.; Xie, H.; Wang, C.; Wang, X.; Zheng, S. FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Lett. 2007, 254, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kalhori, V.; Magnusson, M.; Asghar, M.Y.; Pulli, I.; Törnquist, K. FTY720 (Fingolimod) attenuates basal and sphingosine-1-phosphate-evoked thyroid cancer cell invasion. Endocr. Relat. Cancer 2016, 23, 457–468. [Google Scholar] [CrossRef]
- Yin, P.; Xue, Y.; Wang, T.; Zhong, D.; Li, G. The Therapeutic Targets of Fingolimod (FTY720) Are Involved in Pathological Processes in the Frontal Cortex of Alzheimer’s Disease Patients: A Network Pharmacology Study. Front. Aging Neurosci. 2021, 13, 609679. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Li, H.; Hong, X.; Ying, G.; Lu, X.; Zhuang, L.; Wu, S. TRIM14 promotes colorectal cancer cell migration and invasion through the SPHK1/STAT3 pathway. Cancer Cell Int. 2018, 18, 202. [Google Scholar] [CrossRef]
- Lin, S.; Pandruvada, S.; Yu, H. Inhibition of Sphingosine-1-Phosphate Receptor 2 by JTE013 Promoted Osteogenesis by Increasing Vesicle Trafficking, Wnt/Ca(2+), and BMP/Smad Signaling. Int. J. Mol. Sci. 2021, 22, 12060. [Google Scholar] [CrossRef]
- Li, P.-H.; Wu, J.-X.; Zheng, J.-N.; Pei, D.-S. A sphingosine kinase-1 inhibitor, SKI-II, induces growth inhibition and apoptosis in human gastric cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 10381–10385. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting NFĸB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 2011, 11, 678–689. [Google Scholar] [CrossRef]
- Ling, L.-U.; Tan, K.-B.; Lin, H.; Chiu, G.N.C. The role of reactive oxygen species and autophagy in safingol-induced cell death. Cell Death Dis. 2011, 2, e129. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Huang, Y.; Kumazoe, M.; Lesnick, C.; Yamada, S.; Ueda, N.; Suzuki, T.; Yamashita, S.; Kim, Y.H.; Fujimura, Y.; et al. Sphingosine Kinase-1 Protects Multiple Myeloma from Apoptosis Driven by Cancer-Specific Inhibition of RTKs. Mol. Cancer Ther. 2015, 14, 2303–2312. [Google Scholar] [CrossRef]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; White, M.D.; Meacham, W.D.; Slaughter, E.M.; Muir, S.E.; Elliott, S.; Rhodes, L.V.; Ashe, H.B.; Wiese, T.E.; Smith, C.D.; et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology 2010, 151, 5124–5135. [Google Scholar] [CrossRef]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, F.R.; Pearson, J.M.; Tan, S.-F.; Cheon, H.; Xing, J.C.; Dunton, W.; Feith, D.J.; Loughran, T.P.J. Sphingosine kinase-2 is overexpressed in large granular lymphocyte leukaemia and promotes survival through Mcl-1. Br. J. Haematol. 2020, 190, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef]
- Ciner, A.; Gourdin, T.; Davidson, J.; Parette, M.; Walker, S.J.; Fox, T.E.; Jiang, Y. A phase I study of the ceramide nanoliposome in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2024, 93, 23–29. [Google Scholar] [CrossRef]
- Orienti, I.; Francescangeli, F.; De Angelis, M.L.; Fecchi, K.; Bongiorno-Borbone, L.; Signore, M.; Peschiaroli, A.; Boe, A.; Bruselles, A.; Costantino, A.; et al. A new bioavailable fenretinide formulation with antiproliferative, antimetabolic, and cytotoxic effects on solid tumors. Cell Death Dis. 2019, 10, 529. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Xu, Y.; Wang, C.; Ding, Y.; Gao, M.; Ma, C.; Ma, X.; Li, L. Mixed micelles of TPGS and Soluplus® for co-delivery of paclitaxel and fenretinide: In vitro and in vivo anticancer study. Pharm. Dev. Technol. 2020, 25, 865–873. [Google Scholar] [CrossRef]
- Zhang, H.; Mi, J.-Q.; Fang, H.; Wang, Z.; Wang, C.; Wu, L.; Zhang, B.; Minden, M.; Yang, W.-T.; Wang, H.-W.; et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc. Natl. Acad. Sci. USA 2013, 110, 5606–5611. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Davis, T.S.; Kester, M.; Loughran, T.P.J.; Cabot, M.C. Dynamics of ceramide generation and metabolism in response to fenretinide--Diversity within and among leukemia. Leuk. Res. 2015, 39, 1071–1078. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.E.; Maurer, B.J.; Reynolds, C.P.; Kang, M.; Singh, H.; Crandon, S.; Murgo, A.J.; Doroshow, J.H. Phase I trial of fenretinide lym-x-sorb oral powder in adults with solid tumors and lymphomas. Anticancer. Res. 2011, 31, 961–966. [Google Scholar]
- Colombo, N.; Formelli, F.; Cantù, M.G.; Parma, G.; Gasco, M.; Argusti, A.; Santinelli, A.; Montironi, R.; Cavadini, E.; Baglietto, L.; et al. A phase I-II preoperative biomarker trial of fenretinide in ascitic ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1914–1919. [Google Scholar] [CrossRef]
- Aristarco, V.; Serrano, D.; Maisonneuve, P.; Guerrieri-Gonzaga, A.; Lazzeroni, M.; Feroce, I.; Macis, D.; Cavadini, E.; Albertazzi, E.; Jemos, C.; et al. Fenretinide in Young Women at Genetic or Familial Risk of Breast Cancer: A Placebo-Controlled Biomarker Trial. Cancer Prev. Res. 2024, 17, 255–263. [Google Scholar] [CrossRef]
- Puntoni, M.; Petrera, M.; Campora, S.; Garrone, E.; Defferrari, C.; Torrisi, R.; Johansson, H.; Bruno, S.; Curotto, A.; DeCensi, A. Prognostic Significance of VEGF after Twenty-Year Follow-up in a Randomized Trial of Fenretinide in Non-Muscle-Invasive Bladder Cancer. Cancer Prev. Res. 2016, 9, 437–444. [Google Scholar] [CrossRef]
- Govindarajah, N.; Clifford, R.; Bowden, D.; Sutton, P.A.; Parsons, J.L.; Vimalachandran, D. Sphingolipids and acid ceramidase as therapeutic targets in cancer therapy. Crit. Rev. Oncol. Hematol. 2019, 138, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Lepannetier, S.; Zanou, N.; Yerna, X.; Emeriau, N.; Dufour, I.; Masquelier, J.; Muccioli, G.; Tajeddine, N.; Gailly, P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016, 60, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Maines, L.W.; Keller, S.N.; Smith, C.D. Opaganib (ABC294640) Induces Immunogenic Tumor Cell Death and Enhances Checkpoint Antibody Therapy. Int. J. Mol. Sci. 2023, 24, 16901. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Hoepner, R.; Kleiter, I. Fingolimod for multiple sclerosis and emerging indications: Appropriate patient selection, safety precautions, and special considerations. Ther. Clin. Risk Manag. 2016, 12, 261–272. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef]
- Thomas, K.; Proschmann, U.; Ziemssen, T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin. Pharmacother. 2017, 18, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Nganga, R.; Oleinik, N.; Kim, J.; Selvam, S.P.; De Palma, R.; Johnson, K.A.; Parikh, R.Y.; Gangaraju, V.; Peterson, Y.; Dany, M.; et al. Receptor-interacting Ser/Thr kinase 1 (RIPK1) and myosin IIA-dependent ceramidosomes form membrane pores that mediate blebbing and necroptosis. J. Biol. Chem. 2019, 294, 502–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, C.; Hu, Y.; Dong, C.; Song, Y.; Song, E. C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCζ tumor-suppressive activities and regulating integrin affinity modulation. Sci. Rep. 2015, 5, 9275. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Kumar, G.S. Sanguinarine and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 155–172. [Google Scholar] [CrossRef]
- Rahman, A.; Pallichankandy, S.; Thayyullathil, F.; Galadari, S. Critical role of H2O2 in mediating sanguinarine-induced apoptosis in prostate cancer cells via facilitating ceramide generation, ERK1/2 phosphorylation, and Par-4 cleavage. Free Radic. Biol. Med. 2019, 134, 527–544. [Google Scholar] [CrossRef]
- Kono, K.; Tanaka, M.; Ogita, T.; Hosoya, T.; Kohama, T. F-12509A, a new sphingosine kinase inhibitor, produced by a discomycete. J. Antibiot. 2000, 53, 459–466. [Google Scholar] [CrossRef]
- Bu, Y.; Wu, H.; Deng, R.; Wang, Y. Therapeutic Potential of SphK1 Inhibitors Based on Abnormal Expression of SphK1 in Inflammatory Immune Related-Diseases. Front. Pharmacol. 2021, 12, 733387. [Google Scholar] [CrossRef]
- Jiang, B.; Song, J.; Jin, Y. A flavonoid monomer tricin in Gramineous plants: Metabolism, bio/chemosynthesis, biological properties, and toxicology. Food Chem. 2020, 320, 126617. [Google Scholar] [CrossRef]
- Muraki, K.; Ohnishi, K.; Takezawa, A.; Suzuki, H.; Hatano, N.; Muraki, Y.; Hamzah, N.; Foster, R.; Waldmann, H.; Nussbaumer, P.; et al. Na+ entry through heteromeric TRPC4/C1 channels mediates (−)Englerin A-induced cytotoxicity in synovial sarcoma cells. Sci. Rep. 2017, 7, 16988. [Google Scholar] [CrossRef] [PubMed]
- Batova, A.; Altomare, D.; Creek, K.E.; Naviaux, R.K.; Wang, L.; Li, K.; Green, E.; Williams, R.; Naviaux, J.C.; Diccianni, M.; et al. Englerin A induces an acute inflammatory response and reveals lipid metabolism and ER stress as targetable vulnerabilities in renal cell carcinoma. PLoS ONE 2017, 12, e0172632. [Google Scholar] [CrossRef]
- Fujiwara, T.; Liu, B.; Niu, W.; Hashimoto, K.; Nambu, H.; Yakura, T. Practical Synthesis of Pachastrissamine (Jaspine B), 2-epi-Pachastrissamine, and the 2-epi-Pyrrolidine Analogue. Chem. Pharm. Bull. 2016, 64, 179–188. [Google Scholar] [CrossRef]
- Bogdanova, A.; Kello, M.; Macejova, A.; Nosalova, N.; Petik, P.; Takac, P.; Martinkova, M.; Mezeiova, E.; Mirossay, L.; Gal, P.; et al. Jaspine B Hydrochloride-induced Apoptosis in HeLa Cells Is Associated With Disrupted Sphingolipid Metabolism and Ceramide Overload. Anticancer Res. 2021, 41, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- García-Seisdedos, D.; Babiy, B.; Lerma, M.; Casado, M.E.; Martínez-Botas, J.; Lasunción, M.A.; Pastor, Ó.; Busto, R. Curcumin stimulates exosome/microvesicle release in an in vitro model of intracellular lipid accumulation by increasing ceramide synthesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2020, 1865, 158638. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Koneru, B.; Wei, S.-J.; Chen, W.H.; Makena, M.R.; Urias, E.; Kang, M.H.; Reynolds, C.P. Fenretinide via NOXA Induction, Enhanced Activity of the BCL-2 Inhibitor Venetoclax in High BCL-2-Expressing Neuroblastoma Preclinical Models. Mol. Cancer Ther. 2019, 18, 2270–2282. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Q.; Zhou, X.; Huang, J. SphK1 functions downstream of IGF-1 to modulate IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells: A preliminary in vitro study. J. Cancer 2019, 10, 4264–4269. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H.J.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef]
- Lorvik, K.B.; Bogen, B.; Corthay, A. Fingolimod blocks immunosurveillance of myeloma and B-cell lymphoma resulting in cancer development in mice. Blood 2012, 119, 2176–2177. [Google Scholar] [CrossRef]
- Ntranos, A.; Hall, O.; Robinson, D.P.; Grishkan, I.V.; Schott, J.T.; Tosi, D.M.; Klein, S.L.; Calabresi, P.A.; Gocke, A.R. FTY720 impairs CD8 T-cell function independently of the sphingosine-1-phosphate pathway. J. Neuroimmunol. 2014, 270, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Yuan, J.; Zhang, J.; Li, S.; Lin, W.; Cheng, B. Bioactive sphingolipids as emerging targets for signal transduction in cancer development. Biochim. Biophys. Acta (BBA) Rev. Cancer 2024, 1879, 189176. [Google Scholar] [CrossRef] [PubMed]
- Canals, D.; Perry, D.M.; Jenkins, R.W.; Hannun, Y.A. Drug targeting of sphingolipid metabolism: Sphingomyelinases and ceramidases. Br. J. Pharmacol. 2011, 163, 694–712. [Google Scholar] [CrossRef] [PubMed]
- Diaz Escarcega, R.; McCullough, L.D.; Tsvetkov, A.S. The Functional Role of Sphingosine Kinase 2. Front. Mol. Biosci. 2021, 8, 683767. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Name | Mechanism of Action | Cancer Type | Status | References |
|---|---|---|---|---|
| Fingolimod (FTY720) | Inhibits S1P signaling by acting as a functional antagonist of S1PR1 | Prostate cancer | Preclinical studies | [51] |
| Breast cancer | Preclinical studies | [52,53,54] | ||
| Pancreatic cancer | Preclinical studies | [55] | ||
| Thyroid cancer | Preclinical studies | [56] | ||
| Colorectal cancer | Preclinical studies | [57,58] | ||
| JTE013 | Selectively inhibits S1PR2 | Preclinical studies | [59] | |
| SK1-I/II (BML-258) | Competitive SPHK1/2 inhibitor | Gastric cancer | Preclinical studies | [60] |
| Glioblastoma | Preclinical studies | [58] | ||
| Breast cancer | Preclinical studies | [61] | ||
| Colorectal cancer | Preclinical studies | [58] | ||
| Safingol | SPHK1 inhibitor | Breast and Colon cancer | Preclinical studies | [62] |
| Multiple Myeloma | Preclinical studies | [63] | ||
| Sphingomab (sonepcizumab) | Monoclonal antibody neutralizing S1P | Metastatic renal cell carcinoma | Phase II clinical trial | [64] |
| Opaganib (ABC294640) | Selectively inhibits SPHK2 | Breast cancer | Phase I clinical trial for advanced solid tumors. Phase II clinical trial for hepatocellular carcinoma. Preclinical studies for triple negative breast cancer and granular lymphocyte leukemia. | [61,65,66] |
| Granular lymphocyte leukemia | Preclinical studies | [67] | ||
| Advanced solid tumors | Phase I clinical trial | [68] | ||
| Ceramide nanoliposomes | Ceramide inducer | Advanced solid tumors. | Phase I clinical trials | [69] |
| Fenretinide | DES inhibitor | Lung and colorectal cancer | Preclinical studies | [70] |
| Ovarian and breast cancer | Preclinical studies | [71] | ||
| AML | Preclinical studies | [72,73] | ||
| Solid tumours and lymphoma | Phase I clinical trial | [74] | ||
| Ascitic Ovarian Cancer | Phase I-II clinical trial | [75] | ||
| Breast cancer | Phase II clinical trial | [76] | ||
| Bladder cancer | Phase II clinical trial | [77] | ||
| LCL521 | Acid ceramidase inhibitor | Head and neck cancer | Preclinical studies | [78] |
| Carmoflur | Acid ceramidase inhibitor | Glioblastoma | Preclinical studies | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhakupova, A.; Zeinolla, A.; Kokabi, K.; Sergazy, S.; Aljofan, M. Drug Resistance: The Role of Sphingolipid Metabolism. Int. J. Mol. Sci. 2025, 26, 3716. https://doi.org/10.3390/ijms26083716
Zhakupova A, Zeinolla A, Kokabi K, Sergazy S, Aljofan M. Drug Resistance: The Role of Sphingolipid Metabolism. International Journal of Molecular Sciences. 2025; 26(8):3716. https://doi.org/10.3390/ijms26083716
Chicago/Turabian StyleZhakupova, Assem, Adelina Zeinolla, Kamilya Kokabi, Shynggys Sergazy, and Mohamad Aljofan. 2025. "Drug Resistance: The Role of Sphingolipid Metabolism" International Journal of Molecular Sciences 26, no. 8: 3716. https://doi.org/10.3390/ijms26083716
APA StyleZhakupova, A., Zeinolla, A., Kokabi, K., Sergazy, S., & Aljofan, M. (2025). Drug Resistance: The Role of Sphingolipid Metabolism. International Journal of Molecular Sciences, 26(8), 3716. https://doi.org/10.3390/ijms26083716