1. Introduction
Genomic selection (GS) has changed plant breeding over the past decade, fundamentally transforming genetic evaluation and selection. By integrating genomic data into predictive models, GS has accelerated breeding cycles, improved selection precision, and enhanced genetic gains [
1,
2]. Unlike traditional methods reliant on extensive phenotypic evaluations, GS leverages genome-wide markers to predict genotype performance, reducing the costs and time associated with field trials [
3]. This innovation has been pivotal in addressing global challenges such as food security and climate change by enabling the rapid development of high-yielding, resilient crop varieties [
4]. Today, GS is a cornerstone of modern plant breeding, integrating cutting-edge technologies and big data analytics to drive sustainability and innovation.
GS has been successfully applied across diverse crops, enhancing yield potential and disease resistance in maize and wheat [
2], accelerating the development of stress-tolerant rice varieties [
5], and shortening breeding cycles in perennials like sugarcane and oil palm [
6]. Its ability to predict genetic potential using genome-wide markers has significantly reduced the need for extensive phenotypic evaluations. Additionally, GS has improved genetic gains for complex traits such as drought tolerance and nutrient use efficiency, underscoring its transformative impact on modern agriculture [
7].
The GBLUP (genomic best linear unbiased prediction) statistical model remains one of the most popular and widely used approaches in genomic prediction due to its simplicity, robustness, and interpretability. Despite the emergence of modern machine learning methods, GBLUP is preferred in many cases because it is computationally efficient and provides reliable predictions, especially for traits controlled by many small-effect loci [
8]. Its linear mixed-model framework accounts for genetic relationships using genomic relationship matrices, making it particularly suitable for plant and animal breeding programs [
1]. While machine learning methods like random forests and deep learning can capture complex non-linear interactions, they often require large datasets, extensive hyperparameter tuning, and are prone to overfitting when data are limited [
2]. In contrast, GBLUP provides a balance between accuracy and simplicity, ensuring stable performance across a variety of traits and environments [
9,
10]. Its widespread adoption by GS underscores its reliability and practical advantages, particularly in agricultural contexts where interpretability and computational feasibility are critical.
Given the computational efficiency and widespread use of GBLUP in genomic prediction, there is significant interest in exploring strategies to enhance its predictive power. Combining GBLUP with quantile mapping (QM) and outlier detection techniques offers a promising avenue for improvement. Quantile mapping can address biases in the distribution of predicted values by aligning them more closely with the observed data, thereby increasing prediction accuracy and ensuring a better calibration [
11]. Outlier detection, on the other hand, enhances the robustness of the model by identifying and removing data points that disproportionately influence predictions, which is especially crucial in genomic datasets prone to noise and inconsistencies [
12]. Together, these methods can, in theory, synergistically improve GBLUP by refining its inputs and outputs, ultimately leading to more reliable predictions. This combined approach not only leverages the interpretability and computational advantages of GBLUP but also integrates advanced techniques to address limitations inherent to genomic datasets, making it a powerful tool for plant and animal breeding.
QM is widely utilized across disciplines for bias correction and improving data alignment. In climate science, QM adjusts biases in model outputs, enhancing the accuracy of temperature and precipitation projections for reliable climate assessments [
13]. In hydrology, it refines streamflow and rainfall-runoff predictions, crucial for flood and drought evaluations [
14]. In remote sensing, QM harmonizes satellite-derived data with ground-based observations, improving environmental dataset utility [
15]. Beyond environmental sciences, QM is applied in genomics for aligning predicted values with observed data, enhancing prediction accuracy, and in economics for bias correction in income and risk assessments. Its versatility makes QM a critical tool across multiple fields.
Outlier detection plays a critical role in improving predictions in machine learning by identifying and mitigating the impact of anomalous data points that can distort model performance. By detecting and removing outliers, models achieve a better generalization, reduced bias, and enhanced accuracy, especially in regression and classification tasks. Methods such as statistical thresholds, clustering, and advanced algorithms like isolation forests are commonly applied to detect outliers in diverse datasets. Outlier detection has shown effectiveness in applications such as genomic prediction, fraud detection, and environmental modeling, where precise predictions are essential for decision-making [
16]. These approaches refine training data quality and ultimately lead to more robust and reliable machine learning models [
17,
18]. These studies underscore the importance of addressing outliers to enhance the reliability of genomic prediction models.
As already mentioned, previous studies have shown that quantile mapping (QM) and outlier detection can enhance GBLUP for genomic predictions, which motivated this study. QM improves calibration by aligning predicted values with observed distributions, addressing biases from GBLUP’s normality assumptions. Outlier detection enhances robustness by mitigating the impact of extreme values that could distort variance estimates and bias predictions. Given these prior findings, this study aimed to further evaluate their effectiveness. To strengthen the rationale, it is important to explicitly reference previous studies, clarify how these methods theoretically improve predictions, and demonstrate their impact through comparative analyses.
By leveraging QM for bias correction and four outlier detection methods (Invchi, Logit, Meanp, and SumZ) to refine the training set, this study aims to maximize the predictive potential of GBLUP across diverse datasets. The benchmark analysis, conducted on 14 real datasets, evaluates predictive accuracy using Pearson’s correlation (COR) and normalized mean square error (NRMSE), showcasing the synergistic effects of combining these complementary methods. However, for simplicity, we present full results below for three datasets, Disease, EYT_1, and Wheat_1, as well as results across datasets. We studied GBLUP alone and GBLUP in combination with quantile mapping (QM) and four outlier detection models (Invchi, Logit, Meanp, and SumZ) making a total of 10 genomic prediction models. Several results for datasets are shown in
Appendix A,
Appendix B and
Appendix C.
2. Results
The results are presented in four sections.
Section 1,
Section 2 and
Section 3 present the results for the datasets Disease, EYT_1, and Wheat_1.
Section 4 provides the results across datasets.
Appendix A provides the tables of results corresponding to datasets Disease, EYT_1, Wheat_1, and across datasets.
Appendix B and
Appendix Cprovide the figures and tables of results for the other datasets included in the study: Maize, Japonica, Indica, Groundnut, EYT_2, EYT_3, Wheat_2, Wheat_3, Wheat_4, Wheat_5, and Wheat_6. The results are provided in terms of the metrics of Pearson’s correlation (COR) and normalized mean square error (NRMSE). The assignment of datasets to the appendices was random, that is, not based on any specific criteria.
As described in the
Section 4 below, we compared the genomic prediction accuracy of 10 different model options: GBLUP alone; GBLUP combined only with quantile mapping (QM); GBLUP combined with the four outlier detection methods (Invchi, Logit, Meanp, and SumZ); and GBLUP combined with the four combinations of quantile mapping (QM) with the four outlier detection methods (QM_Invchi, QM_Logit, QM_Meanp, and QM_SumZ).
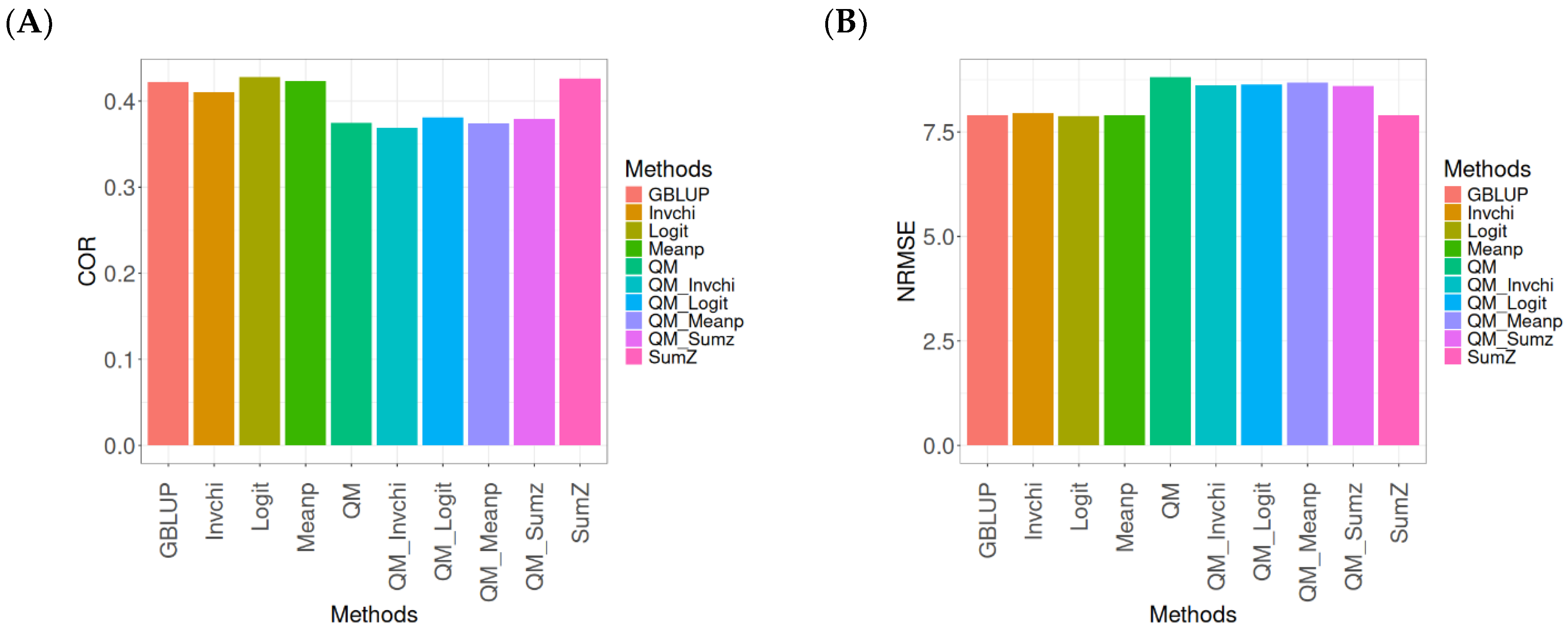
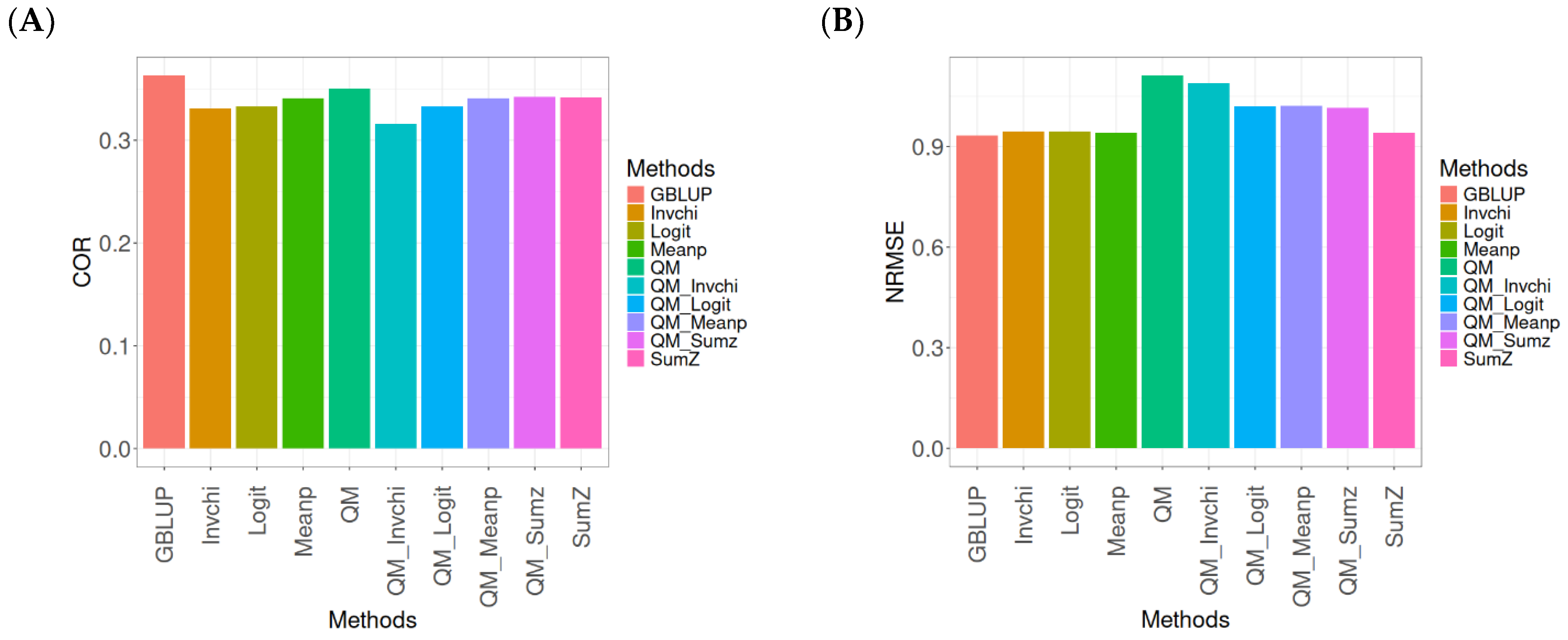
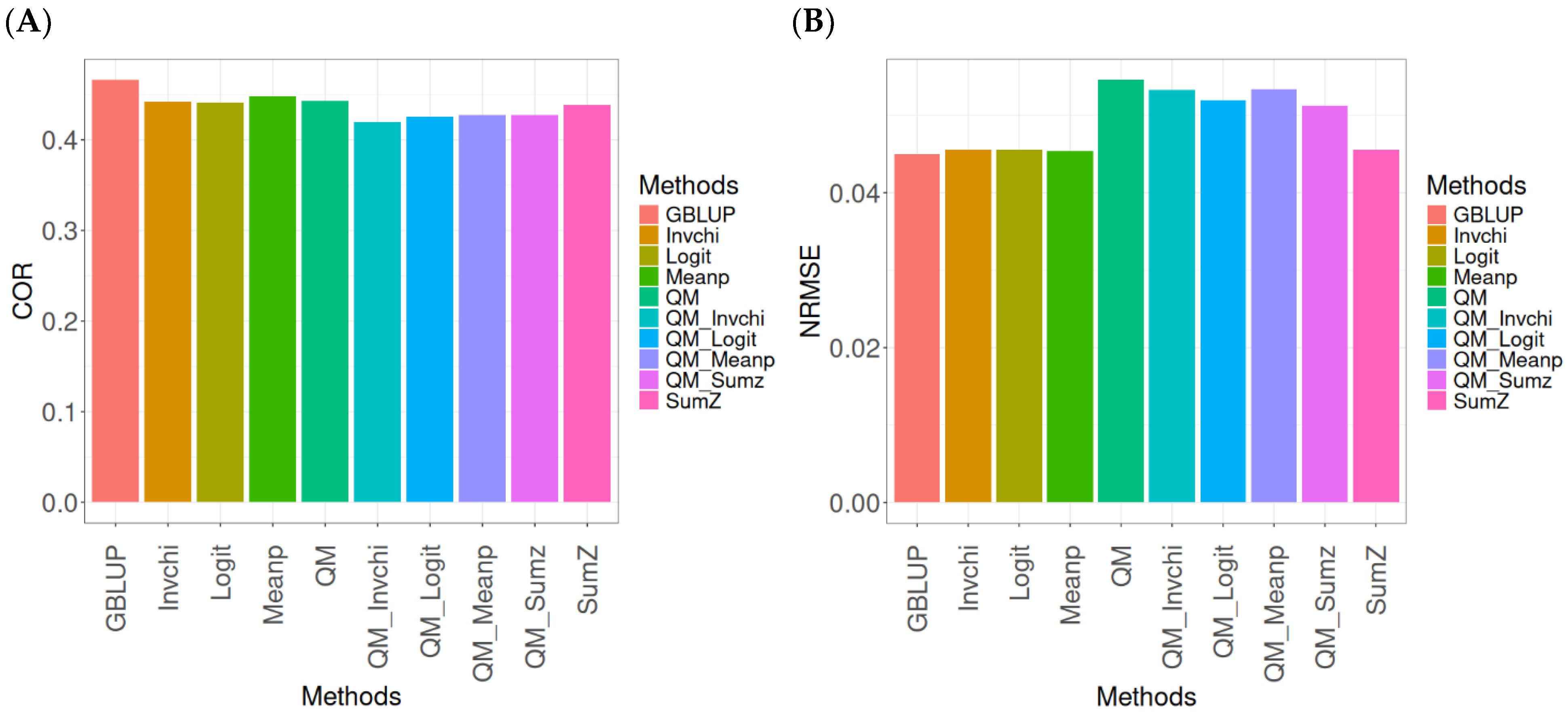
2.1. Disease
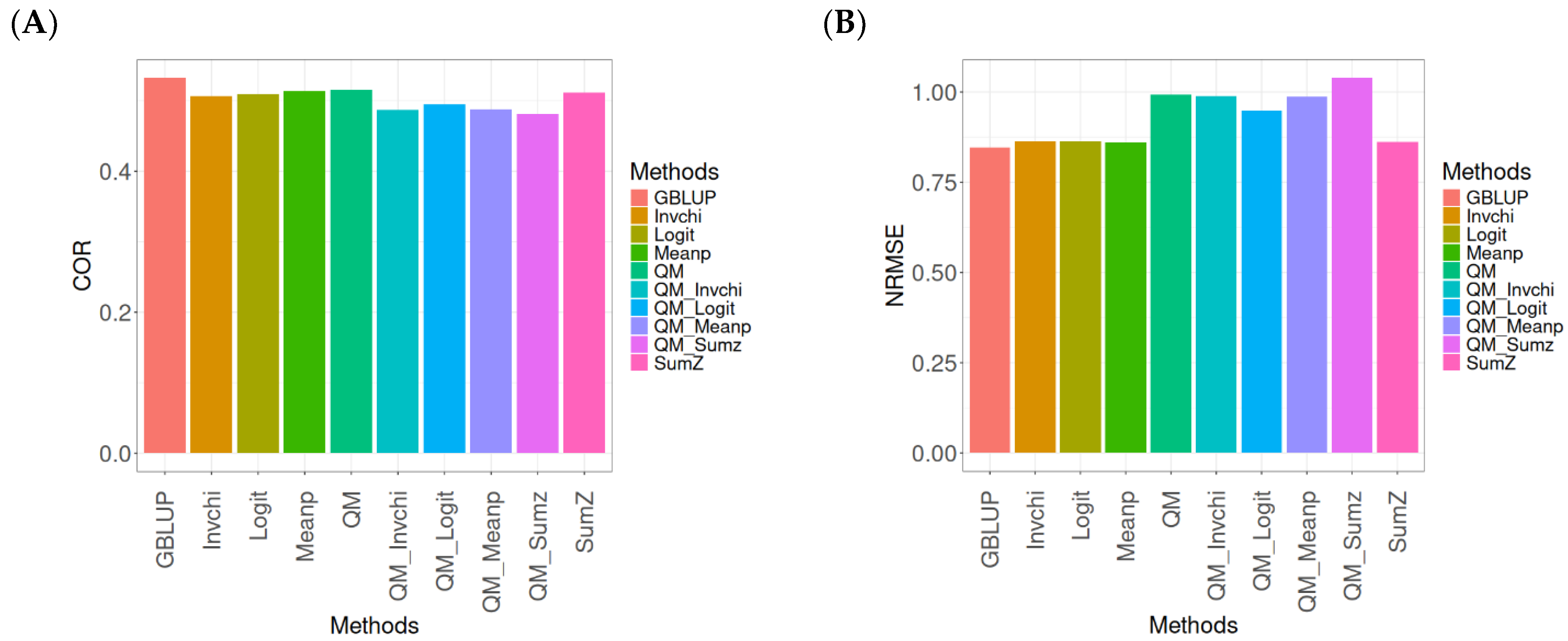
Figure 1 presents the results for the Disease dataset under a comparative analysis of the GBLUP, Invchi, Logit, Meanp, Sumz, QM, QM_Invchi, QM_Logit, QM_Meanp, QM_Sumz, and Sumz models in terms of their predictive efficiency measured by COR and NRMSE. For more details, see
Table A1 (in
Appendix A).
The analysis of Pearson’s correlation between observed and predicted values (
Figure 1A) for the Disease dataset reveals that the GBLUP method stands out as the most effective approach, achieving a correlation of 0.1766, which is 0.8567% greater than QM’s correlation of 0.1751. In comparison to other methods, GBLUP significantly outperforms Meanp (0.1728, 2.1991% less effective), QM_Meanp (0.1661, 6.3215% less effective), SumZ (0.1630, 8.3436% less effective), QM_Sumz (0.1586, 11.3493% less effective), Logit (0.1559, 13.2777% less effective), Invchi (0.1552, 13.7887% less effective), QM_Invchi (0.1530, 15.4248% less effective), and QM_Logit (0.1528, 15.5759% less effective).
Regarding the NRMSE metric between observed and predicted values (
Figure 1B) for the Disease dataset, the results indicate that the GBLUP method achieves the lowest average NRMSE, making it the most effective option. GBLUP yields a value of 0.4313, which is 0.1159% better than Meanp (0.4318) and 0.5565% better than SumZ (0.4337). Additionally, GBLUP outperforms Logit (0.4345) by 0.7419% and Invchi (0.4346) by 0.7651%. Notably, GBLUP also shows significant advantages over QM_Logit (0.4984) by 15.5576%, QM_Invchi (0.4986) by 15.604%, QM_Sumz (0.4987) by 15.6272%, QM_Meanp (0.5072) by 17.598%, and QM (0.5234) by 21.354%.
Overall, the analysis of the Disease dataset indicates that the GBLUP method is the most effective approach, demonstrating a higher Pearson’s correlation compared to other methods, including QM and Meanp. This trend is also reflected in the NRMSE metric, where GBLUP achieves the lowest average NRMSE, confirming its superior performance. Its advantages over a range of alternative methods, including various quantile mapping strategies, further solidify the reliability and effectiveness of GBLUP for predictive tasks in this context.
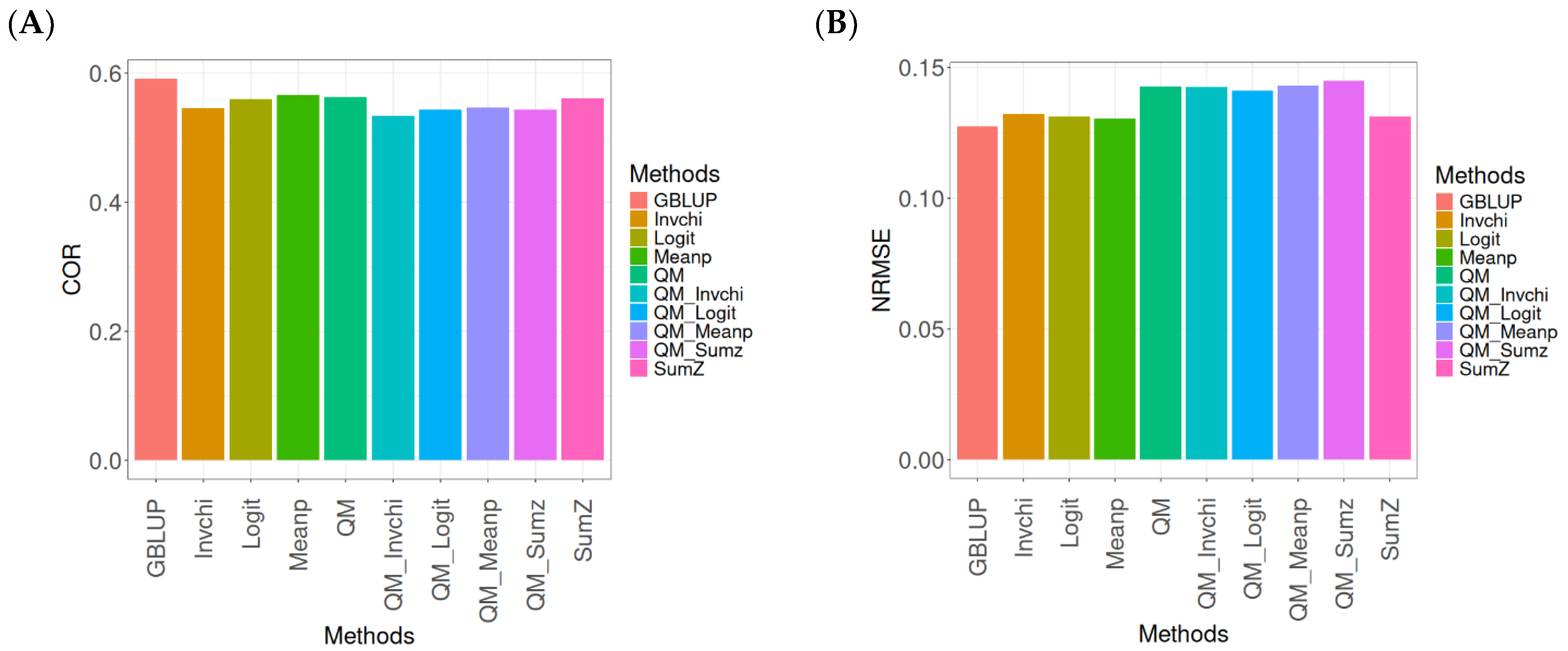
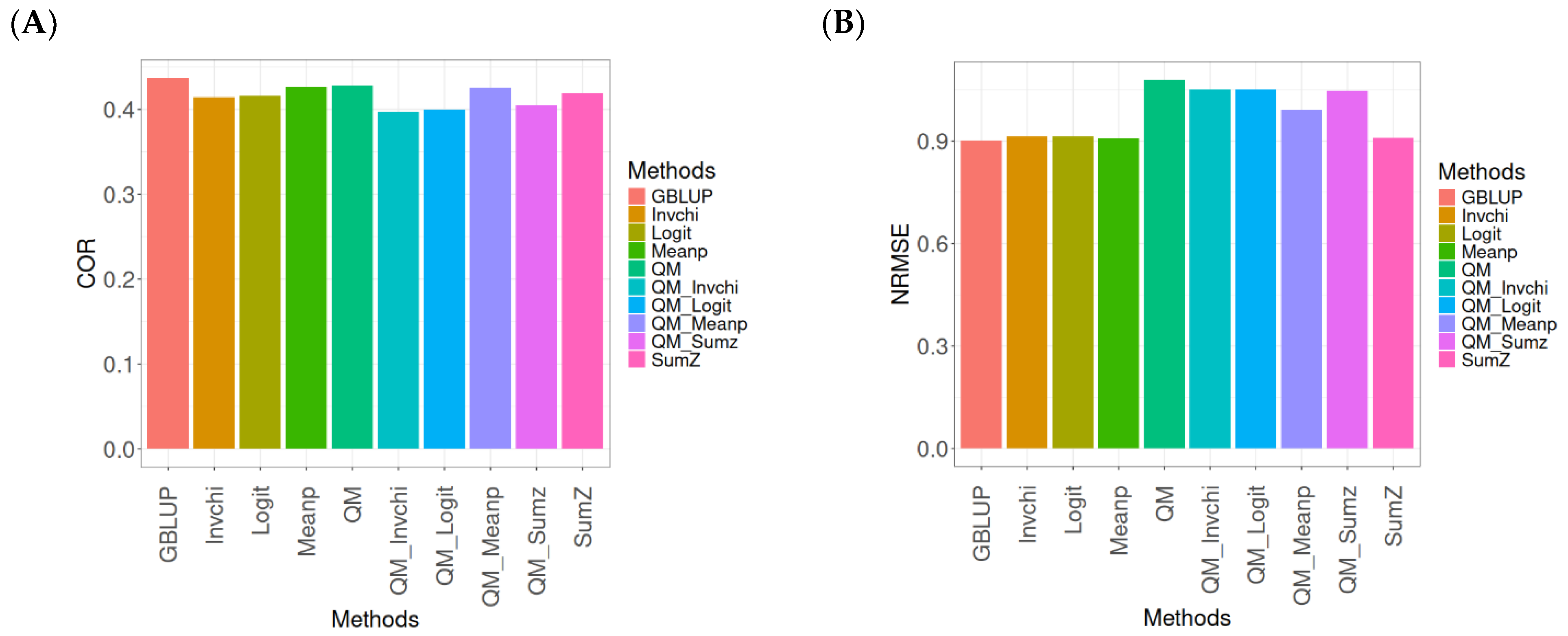
2.2. EYT_1
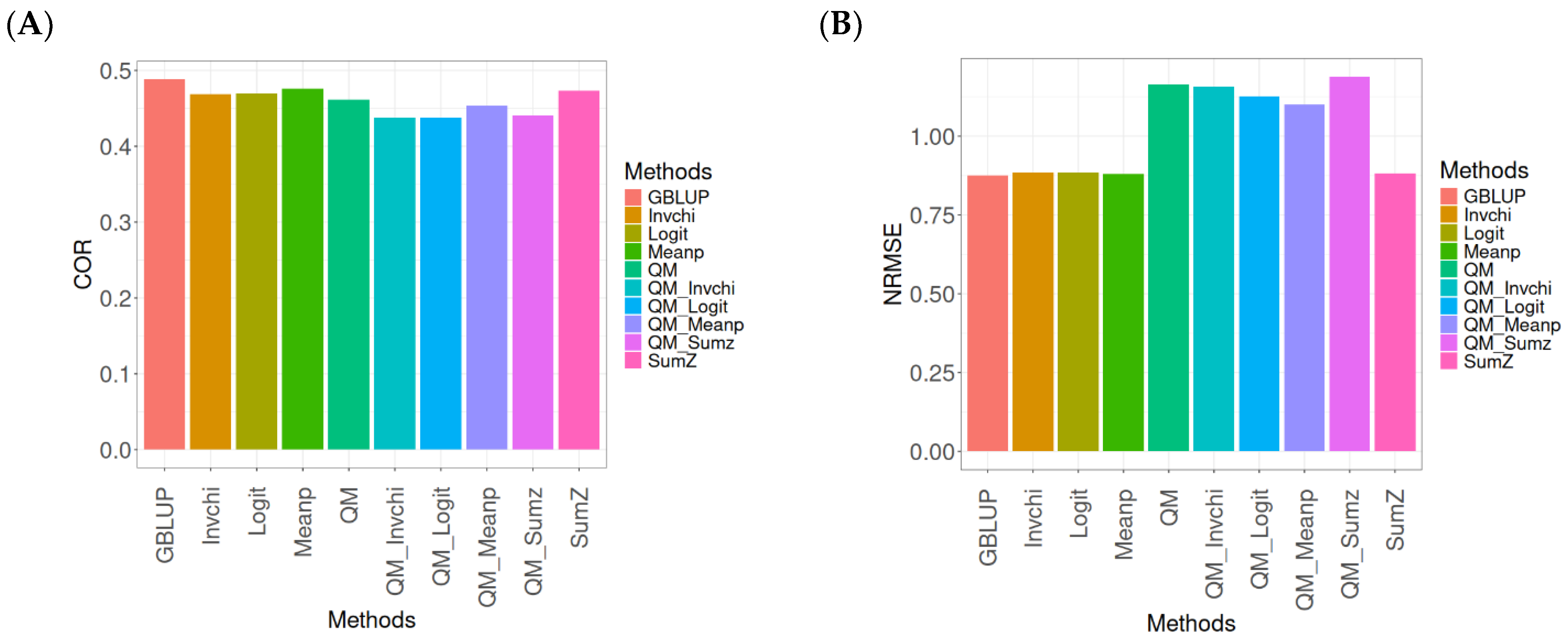
The results for the models evaluated on the EYT_1 dataset (
Figure 2) were assessed using the same metrics, COR and NRMSE. For more details, see
Table A2 (in
Appendix A).
The evaluation of Pearson’s correlation between observed and predicted values (
Figure 2A) for the EYT_1 dataset indicates that the GBLUP method emerges as the most effective strategy, attaining a correlation of 0.4659, which is 3.9955% greater than Meanp’s correlation of 0.4480. In relation to other approaches, GBLUP significantly surpasses QM (0.4429, 5.193% less effective), Invchi (0.4417, 5.4788% less effective), Logit (0.4414, 5.5505% less effective), SumZ (0.4389, 6.1517% less effective), QM_Meanp (0.4273, 9.0335% less effective), QM_Sumz (0.4270, 9.1101% less effective), QM_Logit (0.4257, 9.4433% less effective), and QM_Invchi (0.4193, 11.1138% less effective).
Regarding the NRMSE metric between observed and predicted values (
Figure 2B) for the EYT_1 dataset, the findings reveal that the GBLUP method achieves the lowest average NRMSE, establishing it as the most effective choice. GBLUP has a value of 0.0450, which is 0.8889% greater than Meanp (0.0454) and 1.1111% better than Invchi (0.0455). Additionally, GBLUP outperforms Logit (0.0456) and SumZ (0.0456) by 1.3333%. Notably, GBLUP also exhibits significant advantages over QM_Sumz (0.0512) by 13.7778%, QM_Logit (0.0519) by 15.3333%, QM_Invchi (0.0533) by 18.4444%, QM_Meanp (0.0534) by 18.6667%, and QM (0.0545) by 21.1111%.
Overall, the analysis of the EYT_1 dataset indicates that the GBLUP method consistently outperforms other strategies, displaying both the highest Pearson’s correlation and the lowest NRMSE. This establishes GBLUP as the most effective choice compared to Meanp, Invchi, and the various quantile mapping methods. Its superior performance across both metrics underscores its reliability and potential for the enhancement of predictive accuracy in related applications.
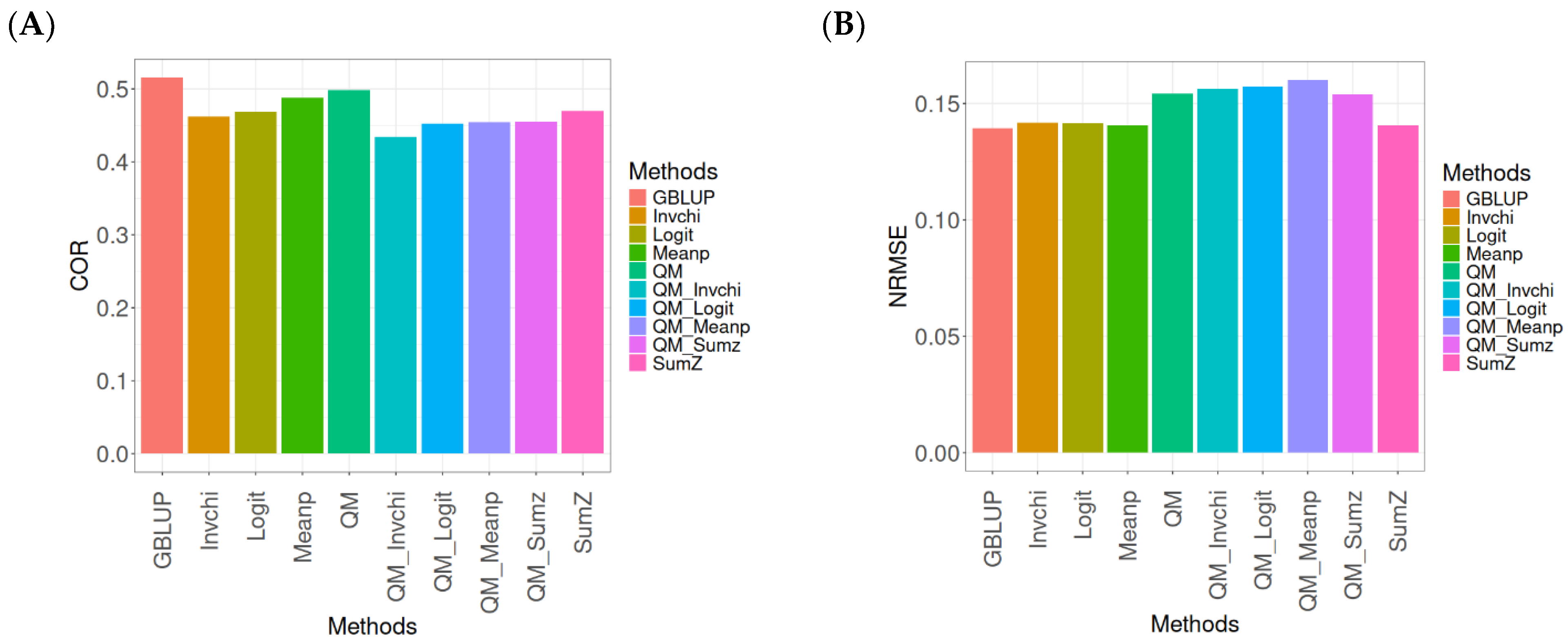
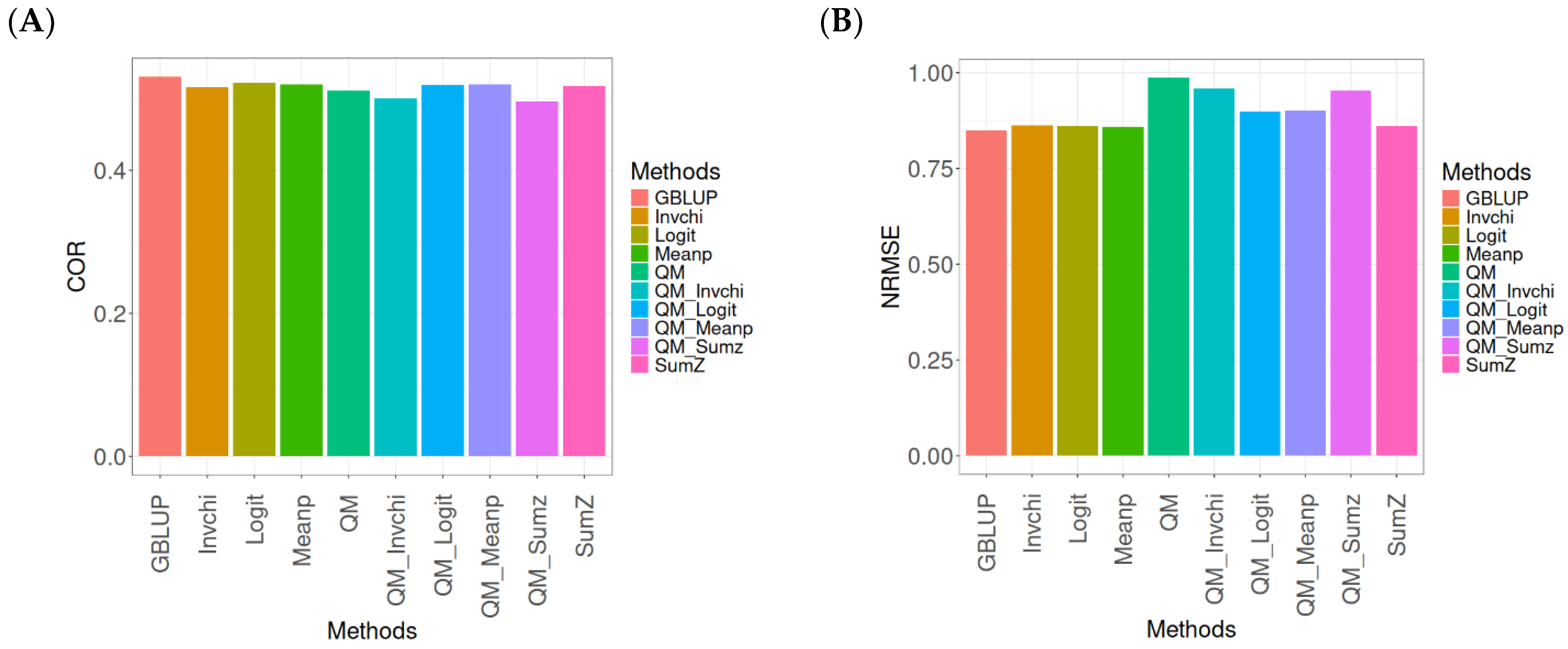
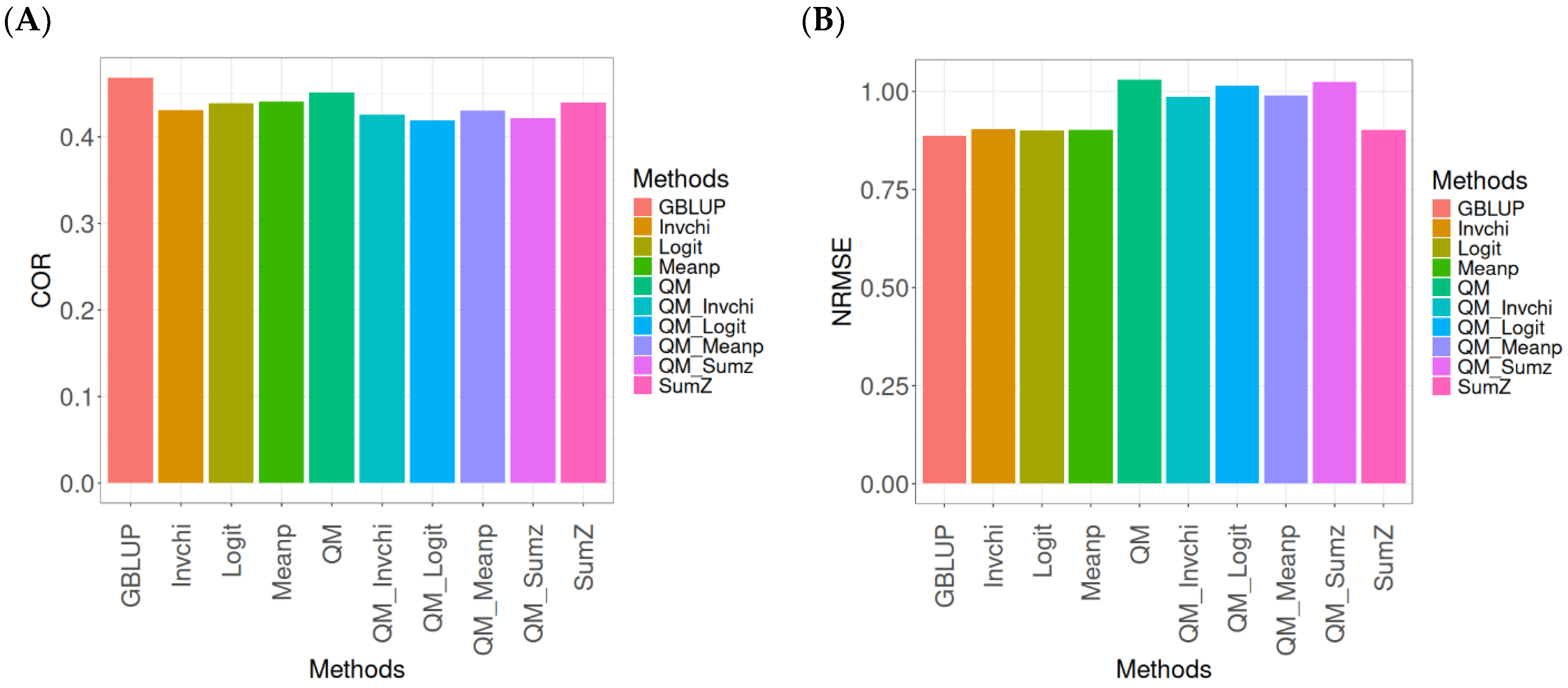
2.3. Wheat_1
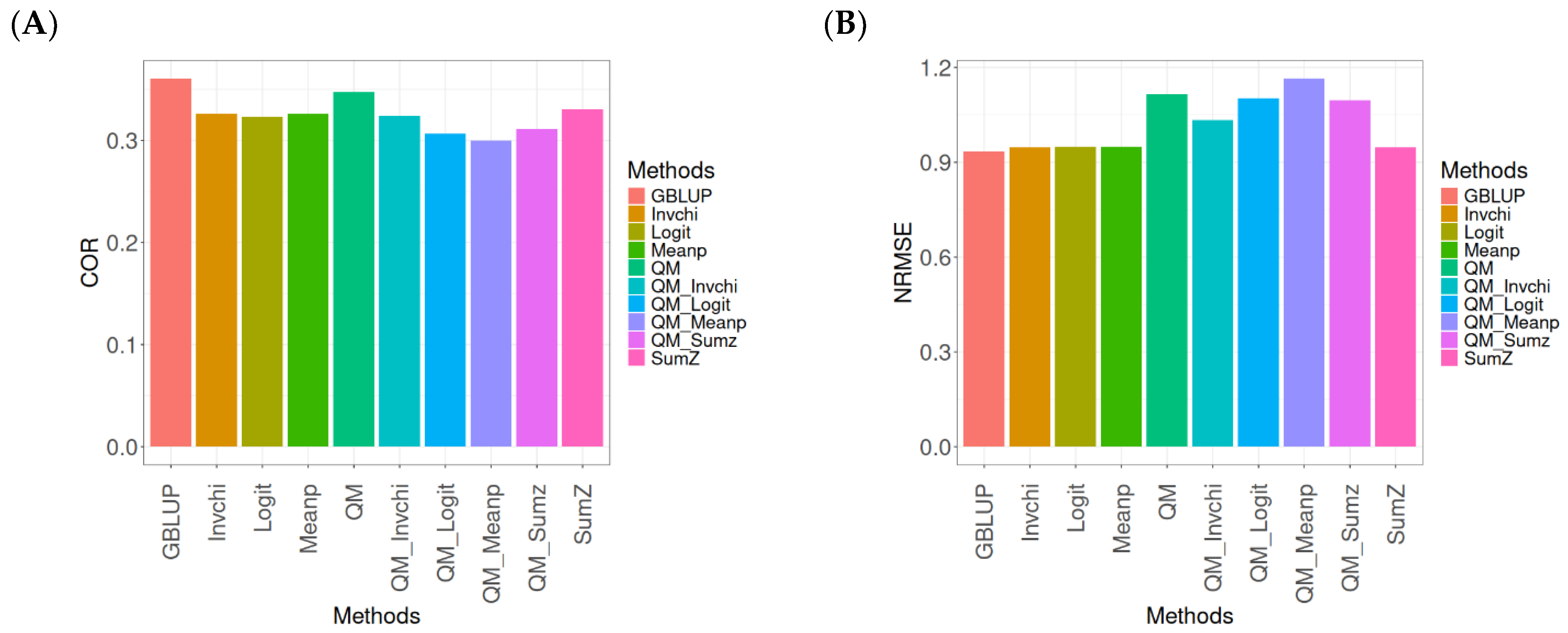
This section presents the results of the genomic prediction models evaluated on the Wheat_1 data, considering the same metrics as before. For more details, see
Table A3 (in
Appendix A).
The assessment of Pearson’s correlation between observed and predicted values (
Figure 3A) for the Wheat_1 dataset shows that the GBLUP method emerges as the most effective strategy, achieving a correlation of 0.4682, which is 3.8598% greater than Meanp’s correlation of 0.4406. In comparison to other methods, GBLUP significantly outperforms QM (0.4508, 6.2642% less effective), SumZ (0.4400, 6.4091% less effective), Logit (0.4387, 6.7244% less effective), Invchi (0.4314, 8.5304% less effective), QM_Meanp (0.4299, 8.909% less effective), QM_Invchi (0.4256, 10.0094% less effective), QM_Sumz (0.4214, 11.1058% less effective), and QM_Logit (0.4187, 11.8223% less effective).
Regarding the NRMSE metric between observed and predicted values (
Figure 3B) for the Wheat_1 dataset, the findings indicate that the GBLUP method achieves the lowest average NRMSE, establishing it as the most effective option. GBLUP has a value of 0.887, which is 1.5671% better than Logit (0.9009) and 1.6347% greater than Meanp (0.9015). Additionally, GBLUP outperforms SumZ (0.9016) by 1.646% and Invchi (0.9047) by 1.9955%. Notably, GBLUP also presents significant advantages over QM_Invchi (0.9866) by 11.2289%, QM_Meanp (0.9895) by 11.5558%, QM_Logit (1.0148) by 14.4081%, QM_Sumz (1.0238) by 15.4228%, and QM (1.0293) by 16.0428%.
The assessment of the Wheat_1 dataset reveals that the GBLUP method is the most effective strategy, achieving a higher Pearson’s correlation compared to other approaches, including Meanp and remaining methods. The performance of GBLUP is not only superior in correlation but also presents the lowest average NRMSE, further establishing its effectiveness. It significantly outperforms other methods, such as Logit and SumZ, as well as a range of quantile mapping strategies, indicating its reliability for predictive tasks. Overall, the consistent advantages of GBLUP reinforce its position as the preferred method in this context.
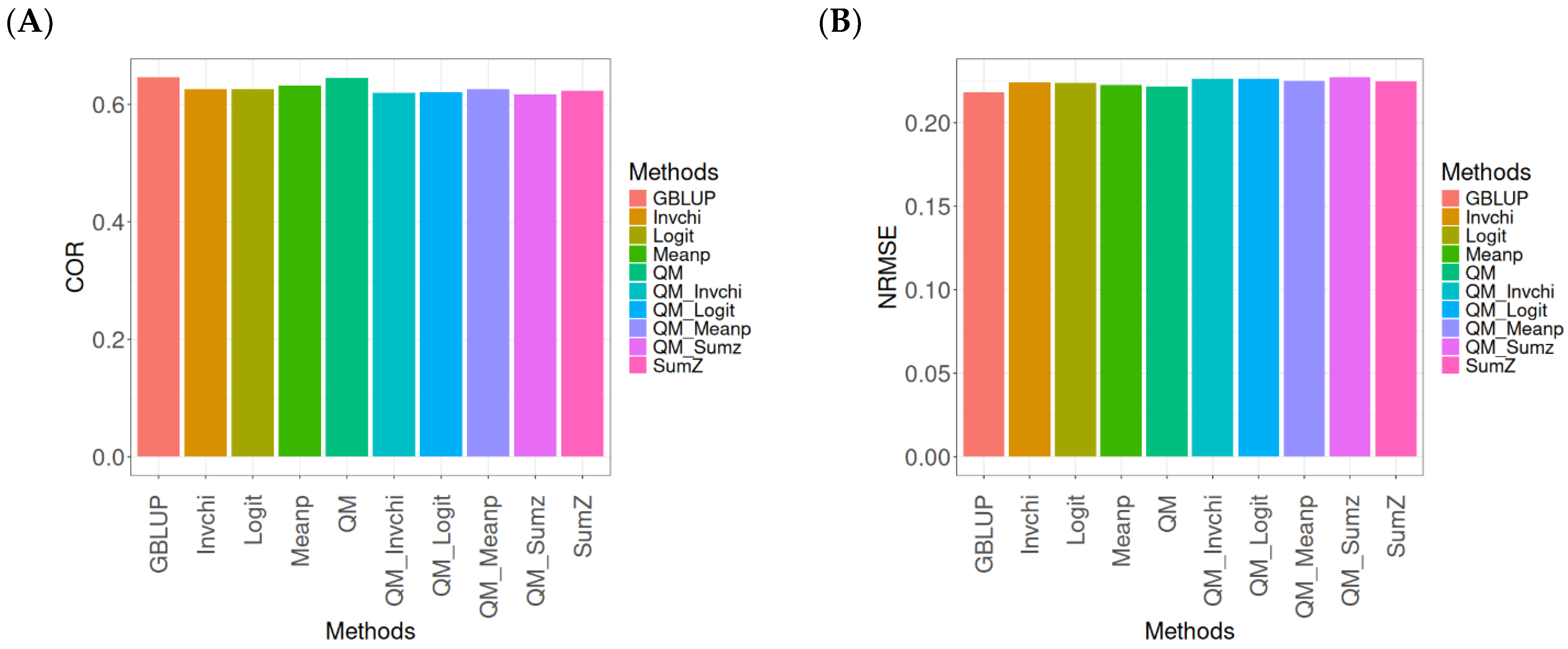
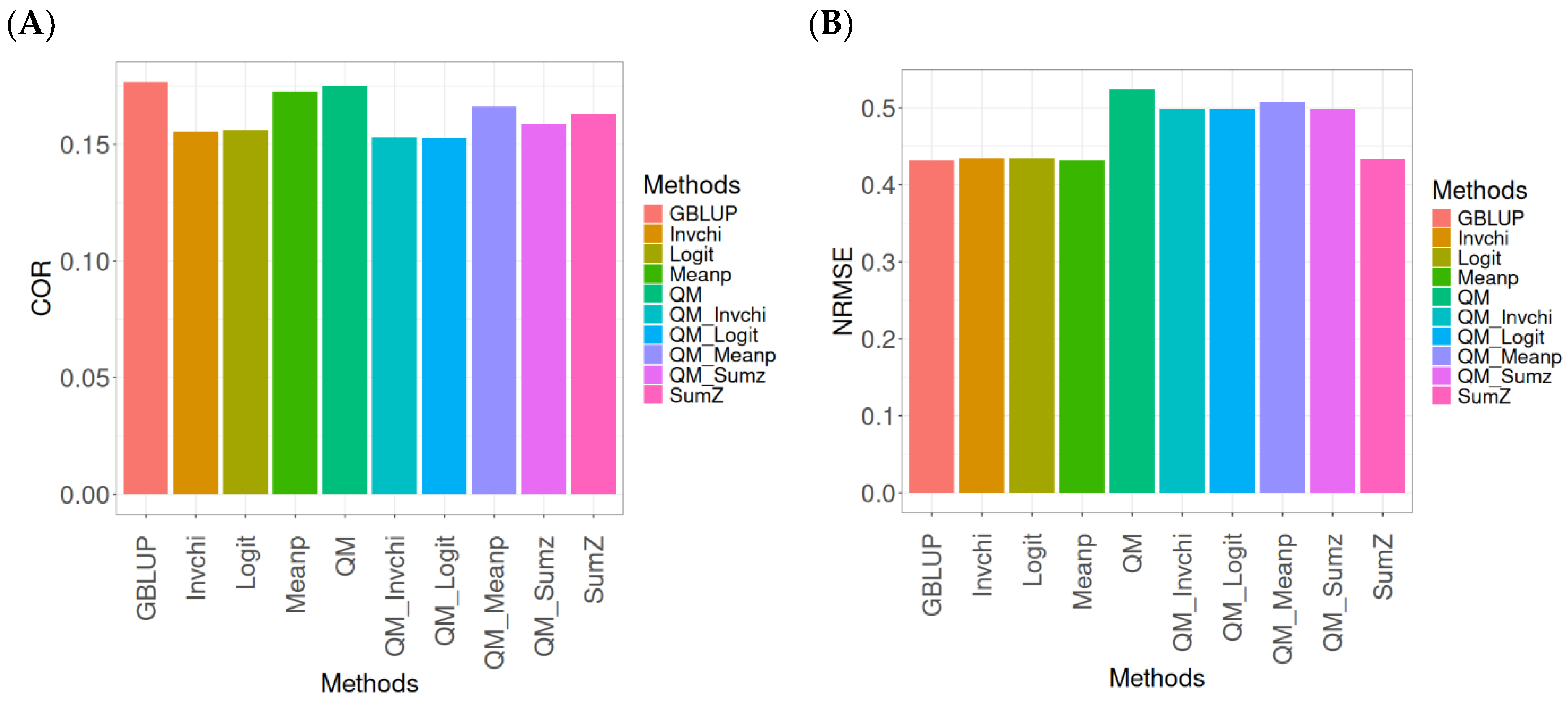
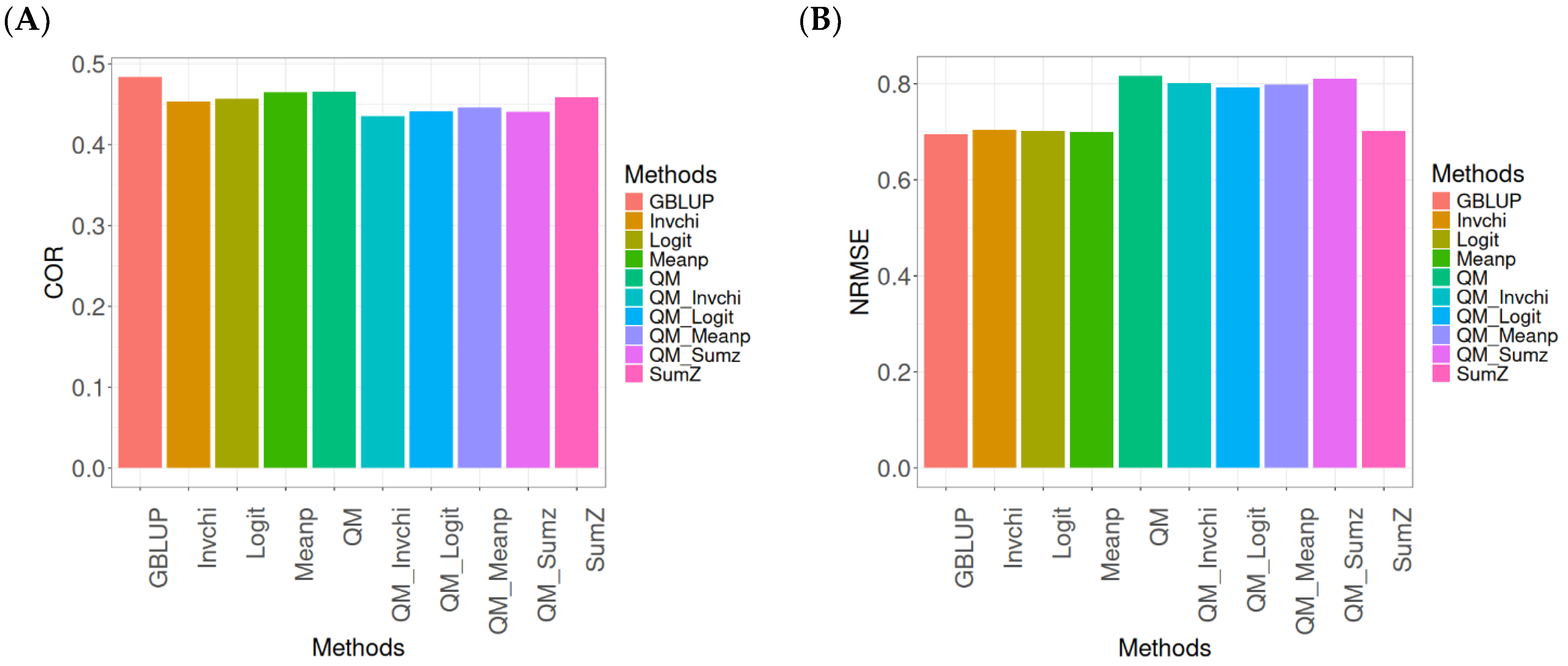
2.4. Across Data
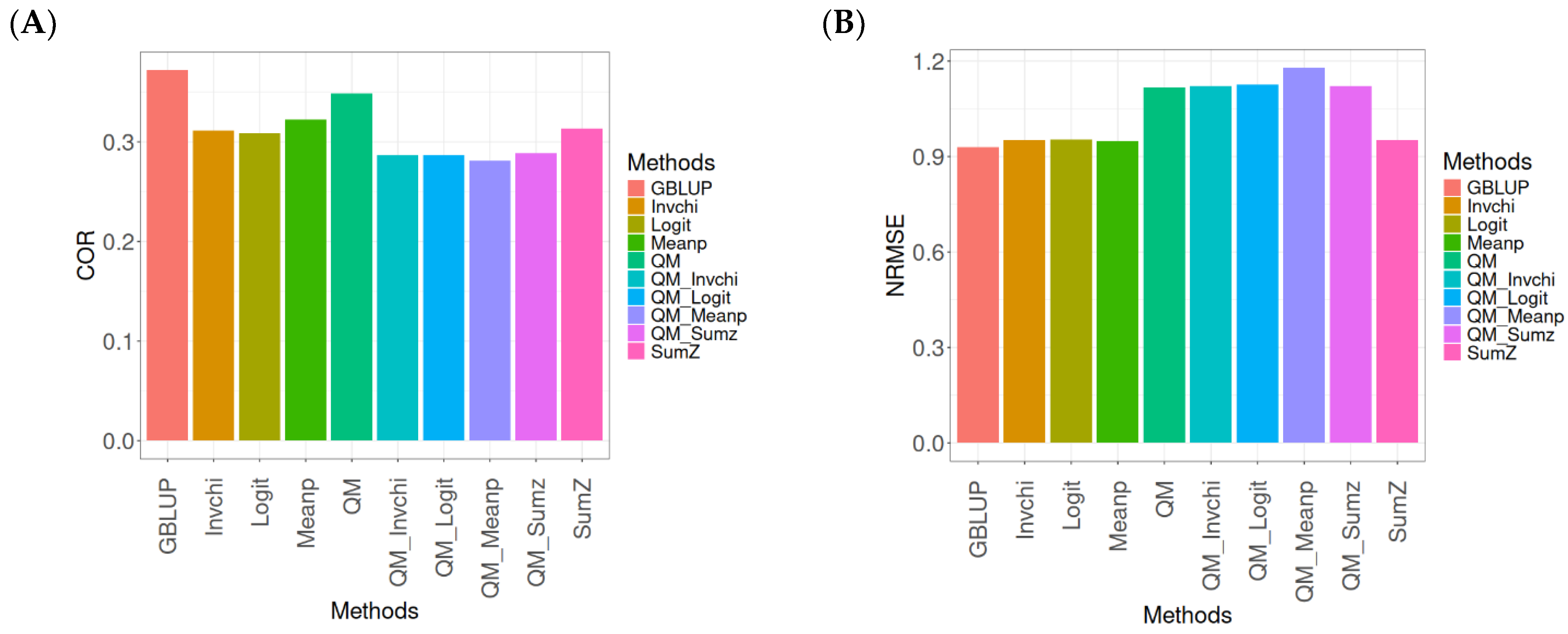
In this section, the analysis of the results presented across datasets is given under the same model and metrics as before. For more details, see
Table A4 (in
Appendix A).
The assessment of Pearson’s correlation between observed and predicted values (
Figure 4A) across datasets highlights the GBLUP method as the most effective strategy, achieving a correlation of 0.4834, which is 3.9794% greater than Meanp’s correlation of 0.4649. In comparison to other methods, GBLUP significantly outperforms QM (0.4659, 3.7562% less effective), SumZ (0.4584, 5.4538% less effective), Logit (0.4569, 5.8% less effective), and Invchi (0.4533, 6.6402% less effective). Notably, GBLUP also shows advantages over various quantile mapping methods, including QM_Meanp (0.4458, 8.4343% less effective), QM_Logit (0.4412, 9.5648% less effective), QM_Sumz (0.4405, 9.7389% less effective), and QM_Invchi (0.4355, 10.9989% less effective).
The assessment of the NRMSE metric between observed and predicted values (
Figure 4B) across datasets indicates that the GBLUP method achieves the lowest average NRMSE, establishing it as the most effective option. GBLUP has a value of 0.6954, which is 0.7046% better than Meanp (0.7003) and 0.9347% greater than SumZ (0.7019). Additionally, GBLUP outperforms Logit (0.7019) and Invchi (0.7043) by 0.9347% and 1.2798%, respectively. Notably, GBLUP also presents significant advantages over various quantile mapping methods, including QM_Logit (0.7928) by 14.0063%, QM_Meanp (0.7976) by 14.6966%, QM_Invchi (0.8018) by 15.3005%, QM_Sumz (0.8110) by 16.6235%, and QM (0.8160) by 17.3425%.
The assessment of Pearson’s correlation across datasets reveals that the GBLUP method is the most effective approach, achieving a higher correlation compared to other methods, including Meanp and various quantile mapping strategies. GBLUP not only excels in correlation but also records the lowest average NRMSE, solidifying its status as the most reliable option. Its performance surpasses that of Logit and SumZ, as well as several quantile mapping methods, indicating a clear advantage. Overall, GBLUP’s consistent effectiveness across both metrics reinforces its preference for predictive tasks in this context.
3. Discussion
The successful implementation of GS in plant breeding faces several challenges, including the need for high-quality genomic and phenotypic data, appropriate statistical models, and robust validation strategies. One key hurdle is the limited availability of large, diverse datasets required to capture the genetic architecture of complex traits and account for genotype-by-environment interactions, which are critical in breeding programs targeting multiple environments [
2,
19]. Additionally, computational demands increase significantly with the inclusion of high-dimensional genomic data, requiring advancements in algorithms and computational resources. Another challenge lies in translating GP predictions into actionable breeding decisions, demanding integration with traditional breeding practices and decision-support tools [
20]. Addressing these issues involves interdisciplinary collaboration and significant investment in training, data curation, and infrastructure to fully leverage the potential of GP in enhancing genetic gains and breeding efficiency.
Improving the efficiency of GS in plant breeding relies on strategies that enhance prediction accuracy, optimize resource allocation, and integrate GS into breeding pipelines. One successful approach is the use of multi-environment trials (MET) to capture genotype-by-environment interactions, enabling better predictions across diverse target environments [
2]. Sparse testing schemes, which involve phenotyping only a subset of genotypes in certain environments, are also effective in reducing costs while maintaining prediction accuracy when paired with robust statistical models [
21,
22]. Additionally, leveraging complementary data sources such as high-throughput phenotyping and environmental covariates can further enhance GS accuracy by providing insights into complex trait architectures [
23]. Implementing these strategies requires investment in advanced data management systems and interdisciplinary collaboration to fully integrate GS into breeding programs and maximize genetic gains.
Despite its potential, the practical application of GS in plant breeding remains highly challenging due to complexities such as the need for high-quality genomic and phenotypic data, the variability in genotype-by-environment interactions, and the computational burden of analyzing large datasets. The effectiveness of GS often depends on the accuracy of prediction models, which can be hindered by limited training data, especially for less-studied traits or environments [
24]. Furthermore, the integration of GS into breeding programs requires adapting existing workflows and overcoming economic and logistical barriers, such as the cost of genotyping and the need for skilled personnel [
2]. To address these limitations, researchers are actively exploring novel approaches, including integrating environmental data, leveraging machine learning techniques, and developing strategies like sparse testing to improve the efficiency and scalability of GS [
25]. These efforts aim to refine GS methodologies and make them more applicable to real-world breeding scenarios.
For this reason, this study explored the use of quantile mapping and the removal of outlier observations within a GBLUP framework to improve the predictive accuracy of the conventional GBLUP model. In theory, these combinations have the potential to enhance the prediction accuracy of GBLUP by addressing critical issues such as the influence of extreme values and non-normality in the data. Quantile mapping, by transforming the distribution of predictions to better align with observed values, can correct systematic biases that often undermine model performance. Simultaneously, outlier removal helps reduce noise and ensures that the model focuses on patterns representative of the majority of the data, which is particularly important when dealing with genomic data characterized by high dimensionality and complex interactions. These adjustments aim to refine the training dataset and statistical assumptions of the model, ultimately resulting in more robust and reliable predictions. Furthermore, integrating these strategies within the GBLUP framework offers an opportunity to adapt this widely used genomic prediction method to varying data qualities and environmental conditions, addressing persistent challenges in plant breeding programs.
However, our results combining the GBLUP method with quantile mapping and outlier detection techniques did not meet expectations. In terms of Pearson’s correlation, across all datasets and within each individual dataset, the GBLUP method proved to be the most effective, consistently achieving higher correlations than the alternative approaches. This superior performance of GBLUP is further supported by its ability to minimize errors, as evidenced by lower NRMSE values. Compared to other methods, including any outlier detection method, quantile mapping, and resulting combinations of quantile mapping with outlier detection techniques, GBLUP consistently delivers more accurate predictions, reaffirming its reliability and robustness in the context of breeding programs.
Our results emphasize the benefits and robustness of the GBLUP method, which remains one of the most popular approaches for genomic prediction. Its popularity stems from several key factors. Firstly, GBLUP is computationally efficient and relatively simple to implement, making it accessible for a wide range of breeding programs. Secondly, it leverages genomic relationships to predict breeding values, effectively capturing additive genetic effects, which are crucial for many quantitative traits. Additionally, GBLUP is grounded in a solid statistical framework, offering reliable and interpretable results. Its ability to handle high-dimensional genomic data without overfitting further contributes to its widespread use. Moreover, the compatibility of GBLUP with extensions, such as the incorporation of environmental covariates or non-additive effects, enhances its adaptability to complex breeding scenarios. These advantages collectively solidify the position of GBLUP as a cornerstone method in genomic prediction.
Finally, we want to emphasize that our results are specific to the datasets used in this study, which reflect genetic and environmental conditions. The observed lack of improvement in predictive accuracy when combining GBLUP with quantile mapping and outlier detection techniques may be influenced by the nature of the datasets, such as their size, genetic architecture, or level of noise. While these combinations did not outperform the conventional GBLUP method in this context, it is important to acknowledge that their effectiveness could vary under different circumstances. For instance, in datasets with pronounced outliers or non-normal distributions, quantile mapping and outlier removal may play a more significant role in improving model performance. Additionally, these techniques might offer advantages in scenarios in which specific traits exhibit strong non-linear patterns or in which genotype-by-environment interactions are highly complex. Therefore, while our findings reaffirm the robustness of the standard GBLUP method, they also suggest the need for further exploration of these combinations across diverse datasets to fully understand their potential.
This study evaluates the impact of quantile mapping and outlier detection on the accuracy of genomic predictions using GBLUP. However, confidence intervals for accuracy metrics, such as Pearson’s correlation and root means square error, were not computed, which limits the ability to assess the statistical uncertainty associated with the observed improvements. Additionally, formal hypothesis testing, such as paired statistical tests to compare GBLUP with and without these enhancements, was not conducted. While the study primarily focused on practical predictive improvements rather than statistical inference, future research should incorporate bootstrapping or cross-validation techniques to estimate confidence intervals and apply appropriate statistical tests, such as paired t-tests or Wilcoxon signed-rank tests, to determine whether the observed differences are statistically significant. Implementing these approaches would strengthen the robustness of the conclusions and provide a clearer understanding of the reliability and generalizability of the proposed methods across different datasets and breeding populations.
Furthermore, computational time was not systematically evaluated, which is an important factor when implementing these methods in large-scale genomic selection programs. Future studies should assess the trade-off between improved prediction accuracy and the additional computational cost associated with quantile mapping and outlier detection, particularly in large datasets where efficiency is a key consideration. Implementing these approaches would strengthen the robustness of the conclusions and provide a clearer understanding of the reliability, scalability, and generalizability of the proposed methods across different datasets and breeding populations.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}