


Pomiferin Induces Antiproliferative and Pro-Death Effects in High-Risk Neuroblastoma Cells by Modulating Multiple Cell Death Pathways

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
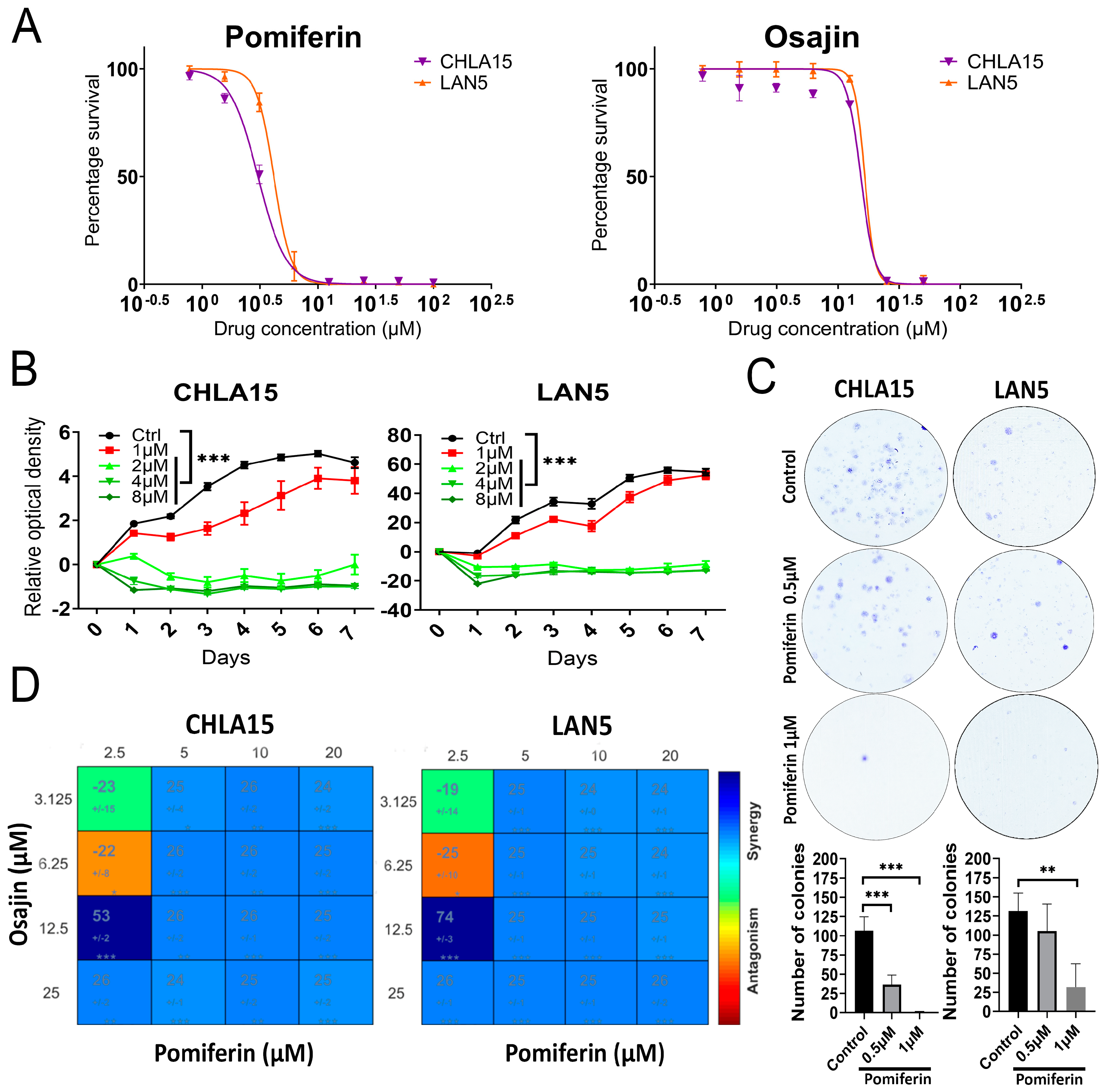
2.1. Antiproliferative Role of Pomiferin in High-Risk Neuroblastoma
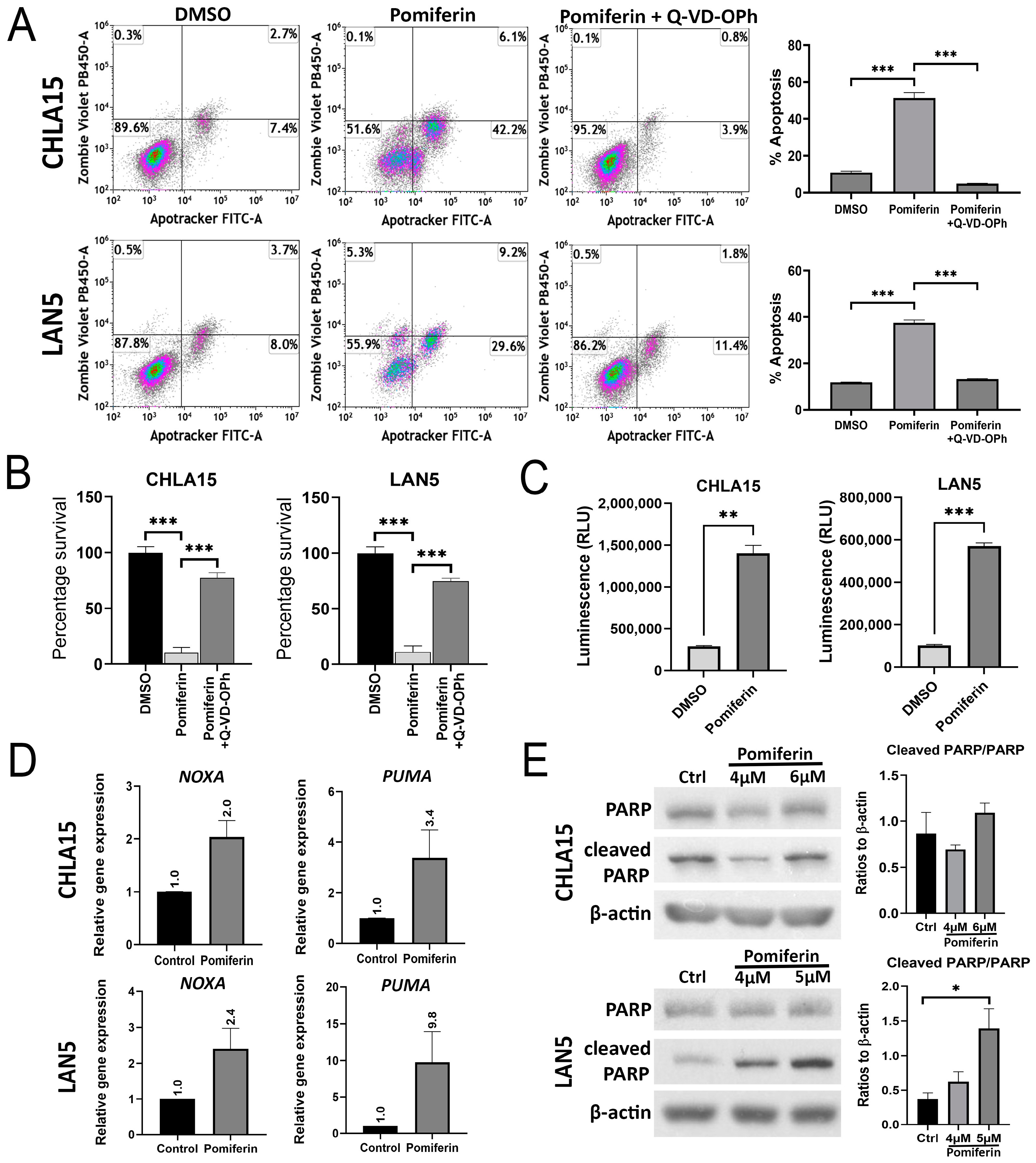
2.2. Pomiferin Triggers Apoptosis in NB
2.3. Ferroptosis Is a Key Modulator of Pomiferin-Induced Cell Death in NB
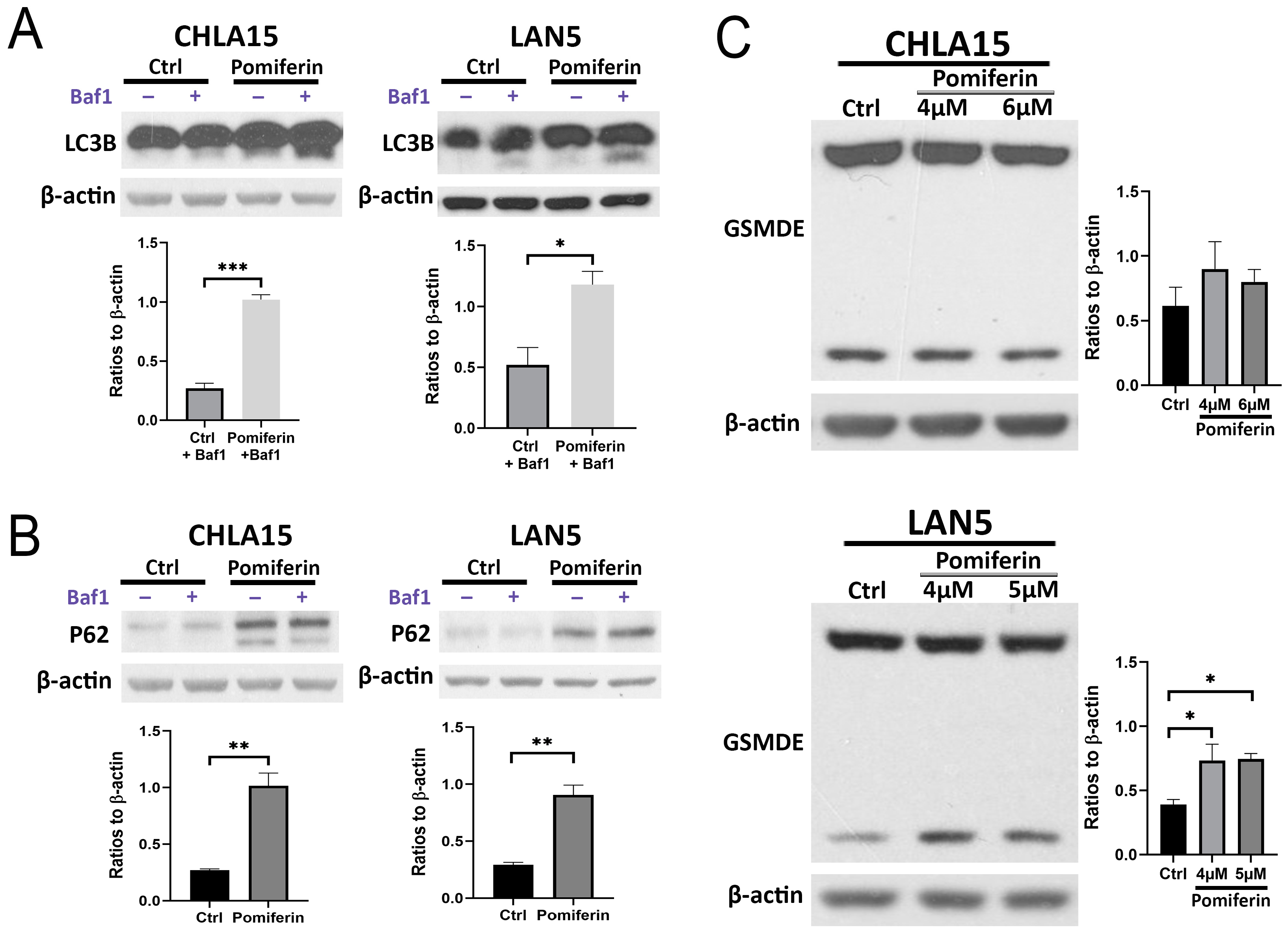
2.4. Changes in Other Cell Death Pathways in Response to Pomiferin in NB
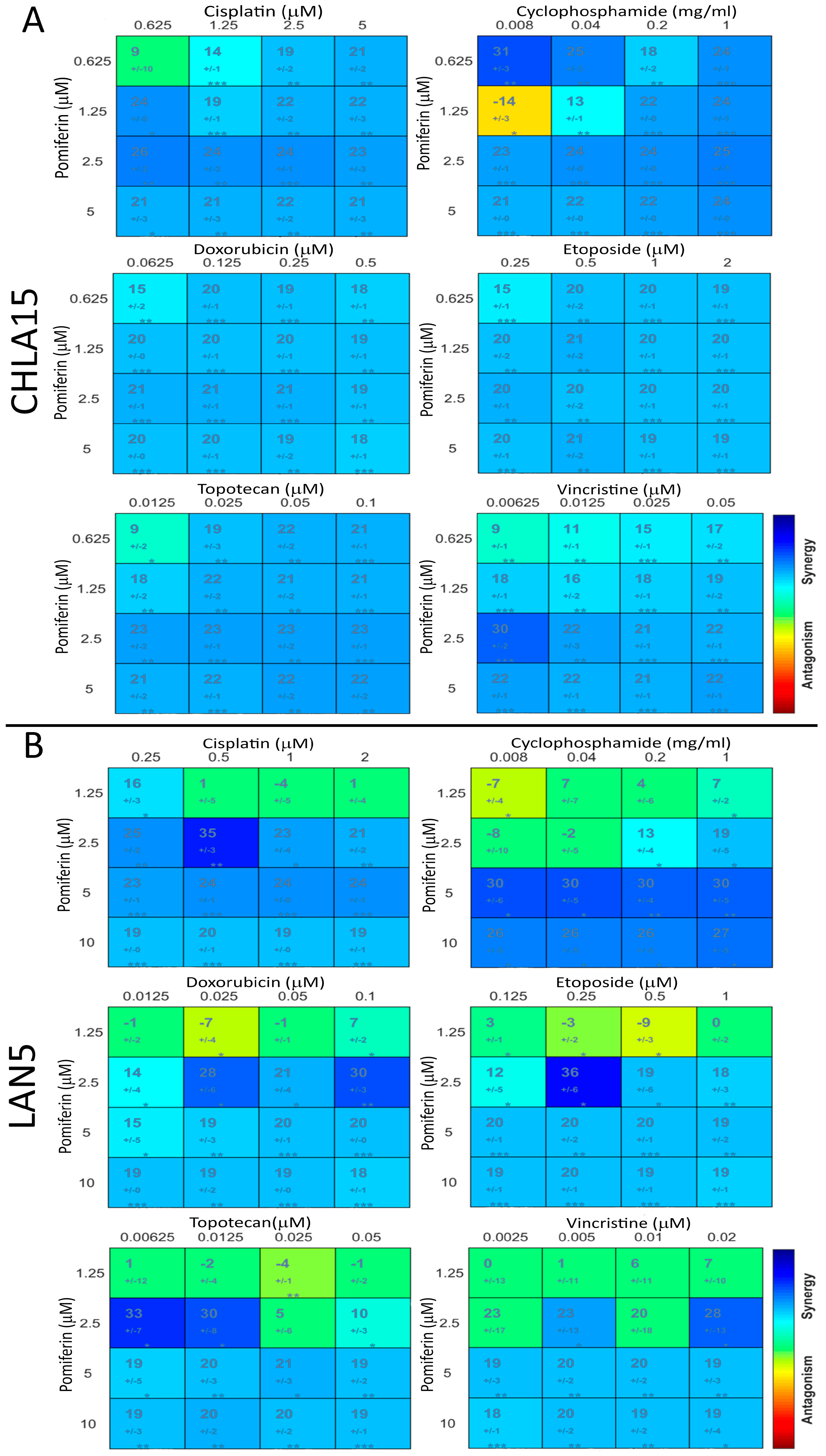
2.5. Pomiferin Synergizes with Conventional Chemotherapy Drugs
3. Discussion
4. Materials and Methods
4.1. Purification of Osajin and Pomiferin by Flash Chromatography
4.2. HPLC Analysis of Osajin and Pomiferin Fractions
4.3. Cell Lines, Chemicals, and Media
4.4. Cell Viability Assay
4.5. Proliferation Assay
4.6. Colony Formation Assay
4.7. Synergy Assay
4.8. Cell Cycle Analysis
4.9. Western Blot
4.10. Real-Time RT PCR
4.11. Apotracker Assay
4.12. Caspase 3/7 Assay
4.13. Lipid Peroxidation Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnsen, J.I.; Dyberg, C.; Wickström, M. Neuroblastoma-A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Alzrigat, M.; Rodriguez-Garcia, A.; Wang, X.; Bexelius, T.S.; Johnsen, J.I.; Arsenian-Henriksson, M.; Liaño-Pons, J.; Bedoya-Reina, O.C. Target Genes of C-MYC and MYCN with Prognostic Power in Neuroblastoma Exhibit Different Expressions during Sympathoadrenal Development. Cancers 2023, 15, 4599. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct Transcriptional MYCN/C-MYC Activities are Associated with Spontaneous Regression Or Malignant Progression in Neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X.; Li, N.; Guo, Y.; Yang, X.; Lei, Y. Therapy Resistance in Neuroblastoma: Mechanisms and Reversal Strategies. Front. Pharmacol. 2023, 14, 1114295. [Google Scholar] [CrossRef]
- Hošek, J.; Toniolo, A.; Neuwirth, O.; Bolego, C. Prenylated and Geranylated Flavonoids Increase Production of Reactive Oxygen Species in Mouse Macrophages but Inhibit the Inflammatory Response. J. Nat. Prod. 2013, 76, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Huang, G.; Hsieh, P.; Wu, H.; Huang, W. Osajin Displays Potential Antiprostate Cancer Efficacy Via Impairment of Fatty Acid Synthase and Androgen Receptor Expression. Prostate 2019, 79, 1543–1552. [Google Scholar] [CrossRef]
- Yang, R.; Hanwell, H.; Zhang, J.; Tsao, R.; Meckling, K.A. Antiproliferative Activity of Pomiferin in Normal (MCF-10A) and Transformed (MCF-7) Breast Epithelial Cells. J. Agric. Food Chem. 2011, 59, 13328–13336. [Google Scholar] [CrossRef]
- Vanco, J.; Travnicek, Z.; Hosek, J.; Malina, T.; Dvorak, Z. Copper(II) Complexes Containing Natural Flavonoid Pomiferin show Considerable in Vitro Cytotoxicity and Anti-Inflammatory Effects. Int. J. Mol. Sci. 2021, 22, 7626. [Google Scholar] [CrossRef]
- Qu, Y.; Song, L.; Xu, S.; Yu, M.S.Y.; Kadioglu, O.; Michelangeli, F.; Law, B.Y.K.; Efferth, T.; Lam, C.W.; Wong, V.K.W. Pomiferin Targets SERCA, mTOR, and P-Gp to Induce Autophagic Cell Death in Apoptosis-Resistant Cancer Cells, and Reverses the MDR Phenotype in Cisplatin-Resistant Tumors in Vivo. Pharmacol. Res. 2023, 191, 106769. [Google Scholar] [CrossRef]
- Stokes, M.E.; Vasciaveo, A.; Small, J.C.; Zask, A.; Reznik, E.; Smith, N.; Wang, Q.; Daniels, J.; Forouhar, F.; Rajbhandari, P.; et al. Subtype-Selective Prenylated Isoflavonoids Disrupt Regulatory Drivers of MYCN-Amplified Cancers. Cell. Chem. Biol. 2024, 31, 805–819.e9. [Google Scholar] [CrossRef]
- Thomas, M.; Nguyen, T.H.; Drnevich, J.; D’Souza, A.M.; de Alarcon, P.A.; Gnanamony, M. Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma. Cancers 2024, 16, 2064. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs Over the nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Son, I.H.; Chung, I.; Lee, S.I.; Yang, H.D.; Moon, H. Pomiferin, Histone Deacetylase Inhibitor Isolated from the Fruits of Maclura Pomifera. Bioorg. Med. Chem. Lett. 2007, 17, 4753–4755. [Google Scholar] [CrossRef]
- Zhao, D.; Yao, C.; Chen, X.; Xia, H.; Zhang, L.; Liu, H.; Jiang, X.; Dai, Y.; Liu, J. The Fruits of Maclura Pomifera Extracts Inhibits Glioma Stem-Like Cell Growth and Invasion. Neurochem. Res. 2013, 38, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.; Reznik, E.; Bisikirska, B.; Polychronidou, V.; Wang, Q.; Zask, A.; Forouhar, F.; Califano, A.; Stockwell, B.R. Kinases Controlling Stability of the Oncogenic MYCN Protein. ACS Med. Chem. Lett. 2023, 14, 1664–1672. [Google Scholar] [CrossRef]
- Aztopal, N.; Cevatemre, B.; Sarimahmut, M.; Ari, F.; Dere, E.; Ozel, M.Z.; Firat, M.; Ulukaya, E. Pelargonium Quercetorum Agnew Induces Apoptosis without PARP Or Cytokeratin 18 Cleavage in Non-Small Cell Lung Cancer Cell Lines. Oncol. Lett. 2016, 12, 1429–1437. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, J.; Zhang, X. Natural Flavonoids and Ferroptosis: Potential Therapeutic Opportunities for Human Diseases. J. Agric. Food Chem. 2023, 71, 5902–5916. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; Luo, L.; Zong, D.; Li, H.; Zeng, Z.; Cui, Y.; Meng, W.; Chen, Y. Dihydroquercetin Suppresses Cigarette Smoke Induced Ferroptosis in the Pathogenesis of Chronic Obstructive Pulmonary Disease by Activating Nrf2-Mediated Pathway. Phytomedicine 2022, 96, 153894. [Google Scholar] [CrossRef]
- Lai, X.; Sun, Y.; Zhang, X.; Wang, D.; Wang, J.; Wang, H.; Zhao, Y.; Liu, X.; Xu, X.; Song, H.; et al. Honokiol Induces Ferroptosis by Upregulating HMOX1 in Acute Myeloid Leukemia Cells. Front. Pharmacol. 2022, 13, 897791. [Google Scholar] [CrossRef] [PubMed]
- Liebl, M.P.; Meister, S.C.; Frey, L.; Hendrich, K.; Klemmer, A.; Wohlfart, B.; Untucht, C.; Nuber, J.; Pohl, C.; Lakics, V. Robust LC3B Lipidation Analysis by Precisely Adjusting Autophagic Flux. Sci. Rep. 2022, 12, 79. [Google Scholar]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef] [PubMed]
- Kist, M.; Vucic, D. Cell Death Pathways: Intricate Connections and Disease Implications. EMBO J. 2021, 40, e106700. [Google Scholar] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar]
- Pouliou, M.; Koutsi, M.A.; Champezou, L.; Giannopoulou, A.; Vatsellas, G.; Piperi, C.; Agelopoulos, M. MYCN Amplifications and Metabolic Rewiring in Neuroblastoma. Cancers 2023, 15, 4803. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective Gene Dependencies in MYCN-Amplified Neuroblastoma Include the Core Transcriptional Regulatory Circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. P53 in Ferroptosis Regulation: The New Weapon for the Old Guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar]
- Setiawan, S.A.; Liu, W.Z.; Weng, P.; Lee, C.; Yadav, V.K.; Hardianti, M.S.; Yeh, C.; Chao, T. Synergistic Disruption of BTK and BCL-2 Causes Apoptosis while Inducing Ferroptosis in Double-Hit Lymphoma. Eur. J. Pharmacol. 2023, 943, 175526. [Google Scholar]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell. Chem. Biol. 2020, 27, 448–462. [Google Scholar]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Massenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the Key Ferroptosis-Surveilling Systems Hypersensitizes Mice to Tubular Necrosis during Acute Kidney Injury. Nat. Commun. 2021, 12, 4402–4406. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cai, X.; Kang, R.; Liu, J.; Tang, D. Autophagy-Dependent Ferroptosis in Cancer. Antioxid. Redox Signal. 2023, 39, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tsvetkov, A.S.; Shen, H.; Isidoro, C.; Ktistakis, N.T.; Linkermann, A.; Koopman, W.J.H.; Simon, H.; Galluzzi, L.; Luo, S.; et al. International Consensus Guidelines for the Definition, Detection, and Interpretation of Autophagy-Dependent Ferroptosis. Autophagy 2024, 20, 1213–1246. [Google Scholar] [CrossRef] [PubMed]
- Furman, W.L.; McCarville, B.; Shulkin, B.L.; Davidoff, A.; Krasin, M.; Hsu, C.; Pan, H.; Wu, J.; Brennan, R.; Bishop, M.W.; et al. Improved Outcome in Children with Newly Diagnosed High-Risk Neuroblastoma Treated with Chemoimmunotherapy: Updated Results of a Phase II Study using hu14.18K322A. J. Clin. Oncol. 2022, 40, 335–344. [Google Scholar] [CrossRef]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC Inhibitors: Targets for Tumor Therapy, Immune Modulation and Lung Diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar]
- Hwang, H.; Winkler-Moser, J.; Tisserat, B.; Harry-O’kuru, R.E.; Berhow, M.A.; Liu, S.X. Antioxidant Activity of Osage Orange Extract in Soybean Oil and Fish Oil during Storage. J. Am. Oil Chem. Soc. 2021, 98, 73–87. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable Transcript Quantification by Real-Time Reverse Transcriptase-Polymerase Chain Reaction in Primary Neuroblastoma using Normalization to Averaged Expression Levels of the Control Genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnanamony, M.; Thomas, M.; Nguyen, T.H.; Brownstein, K.; de Alarcon, P.A. Pomiferin Induces Antiproliferative and Pro-Death Effects in High-Risk Neuroblastoma Cells by Modulating Multiple Cell Death Pathways. Int. J. Mol. Sci. 2025, 26, 3600. https://doi.org/10.3390/ijms26083600
Gnanamony M, Thomas M, Nguyen TH, Brownstein K, de Alarcon PA. Pomiferin Induces Antiproliferative and Pro-Death Effects in High-Risk Neuroblastoma Cells by Modulating Multiple Cell Death Pathways. International Journal of Molecular Sciences. 2025; 26(8):3600. https://doi.org/10.3390/ijms26083600
Chicago/Turabian StyleGnanamony, Manu, Maria Thomas, Thu Hien Nguyen, Korey Brownstein, and Pedro A. de Alarcon. 2025. "Pomiferin Induces Antiproliferative and Pro-Death Effects in High-Risk Neuroblastoma Cells by Modulating Multiple Cell Death Pathways" International Journal of Molecular Sciences 26, no. 8: 3600. https://doi.org/10.3390/ijms26083600
APA StyleGnanamony, M., Thomas, M., Nguyen, T. H., Brownstein, K., & de Alarcon, P. A. (2025). Pomiferin Induces Antiproliferative and Pro-Death Effects in High-Risk Neuroblastoma Cells by Modulating Multiple Cell Death Pathways. International Journal of Molecular Sciences, 26(8), 3600. https://doi.org/10.3390/ijms26083600