
Genome-Wide Identification and Expression Analysis of m6A Methyltransferase Family in Przewalskia tangutica Maxim.


Abstract
1. Introduction
2. Results
2.1. Identifying the PtMT Genes
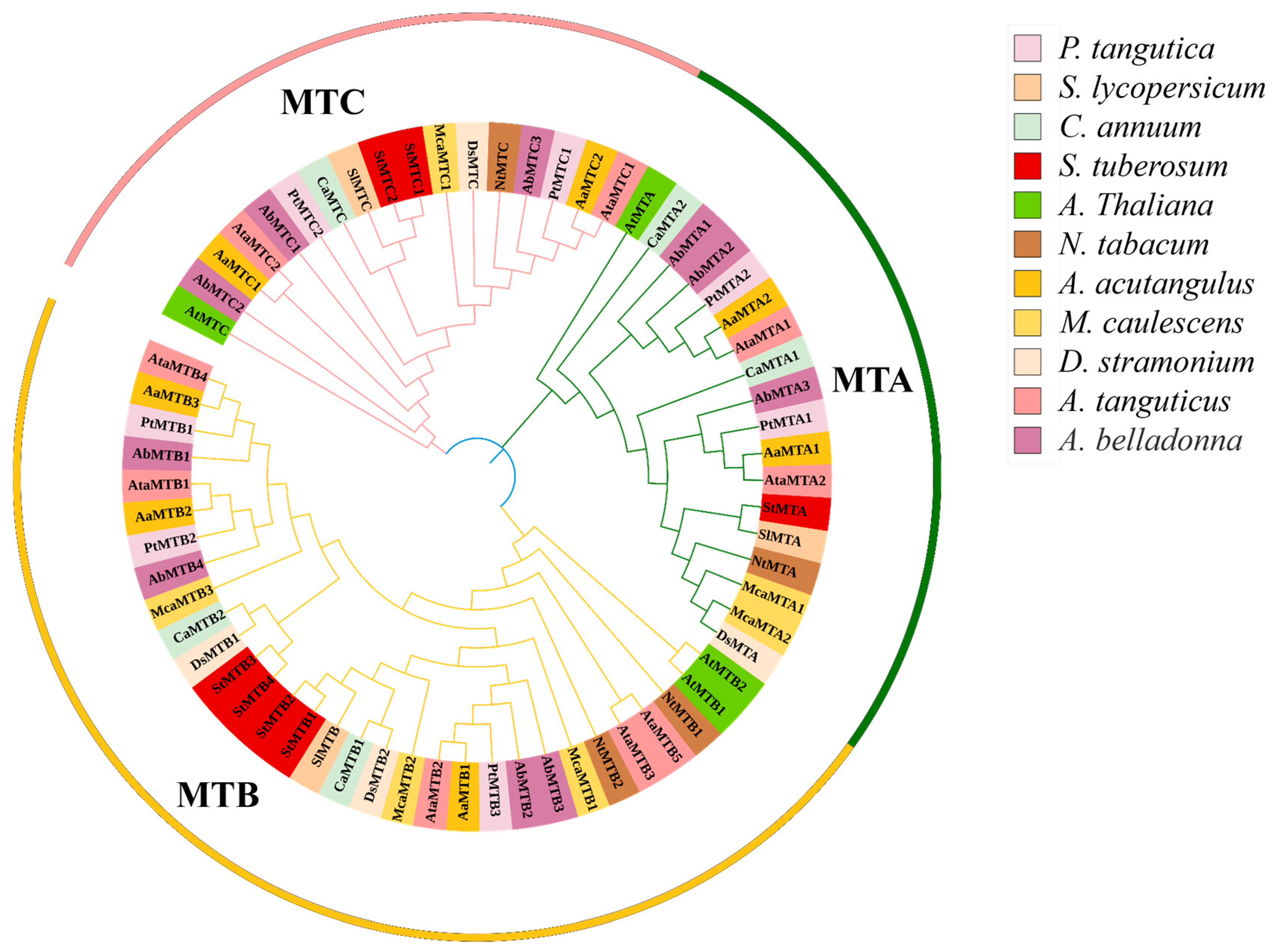
2.2. Phylogenetic Relationship Analysis
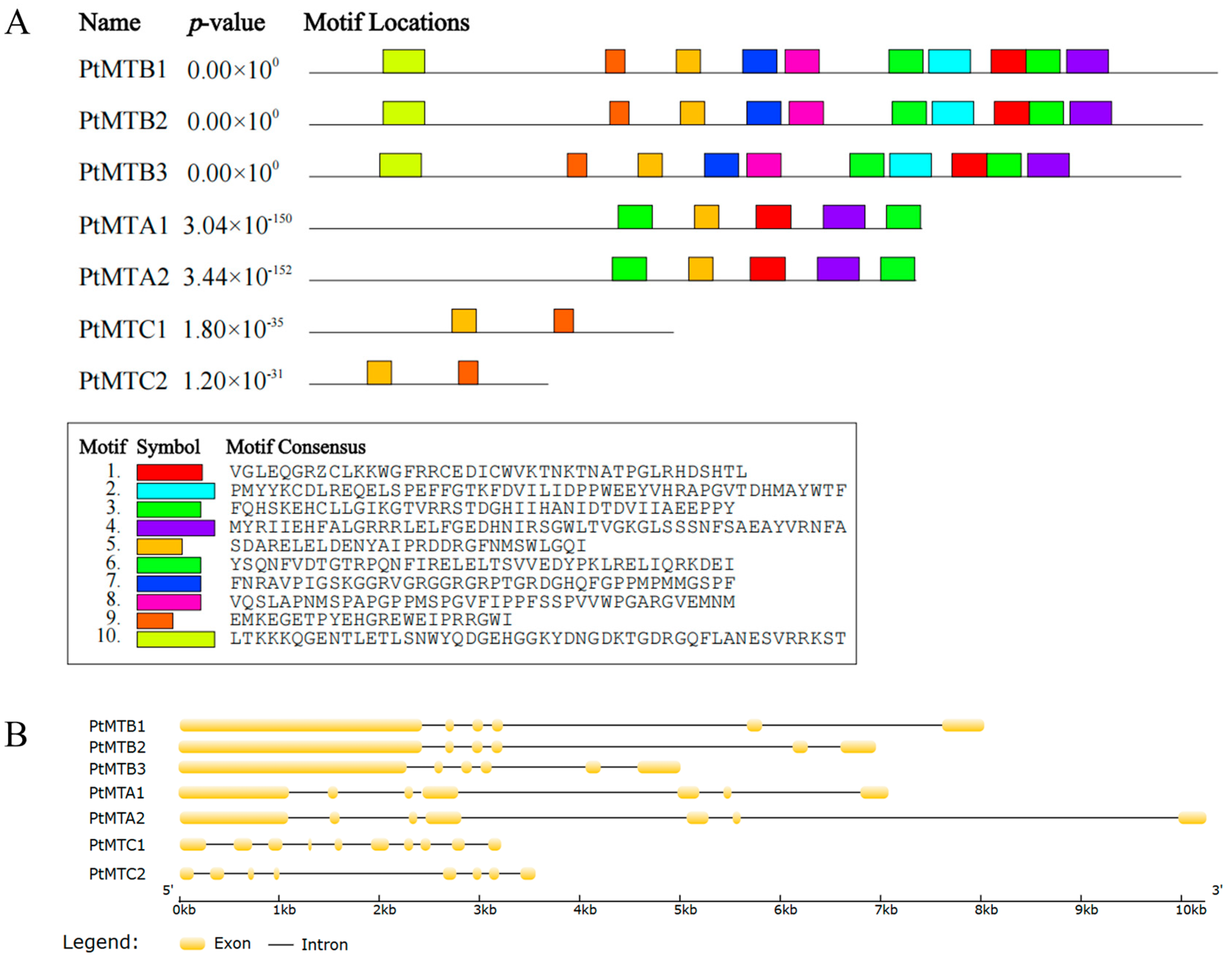
2.3. Structural Features Analysis
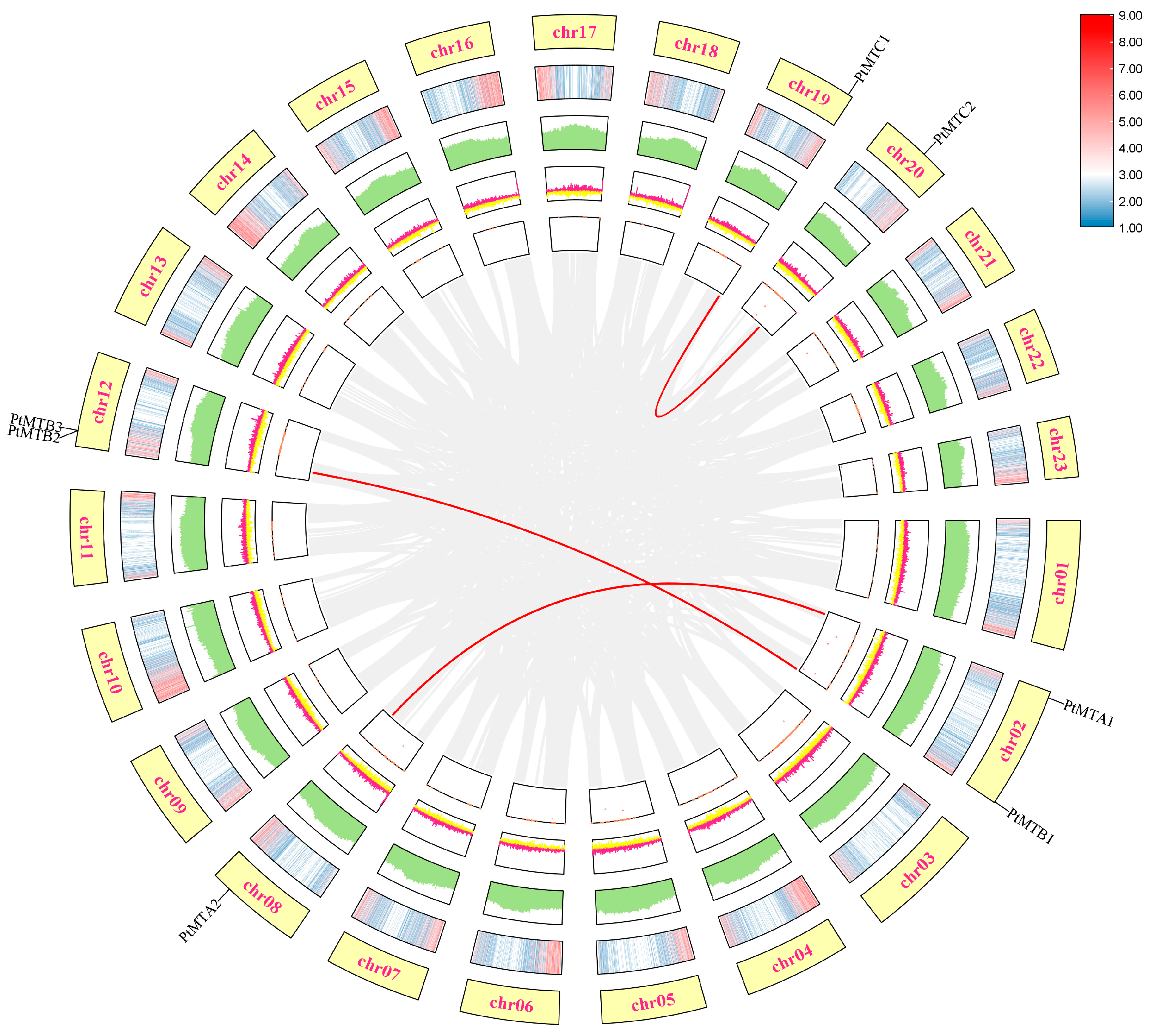
2.4. Chromosomal Location and GO Enrichment Analysis
2.5. Promoter Analysis of PtMT Genes
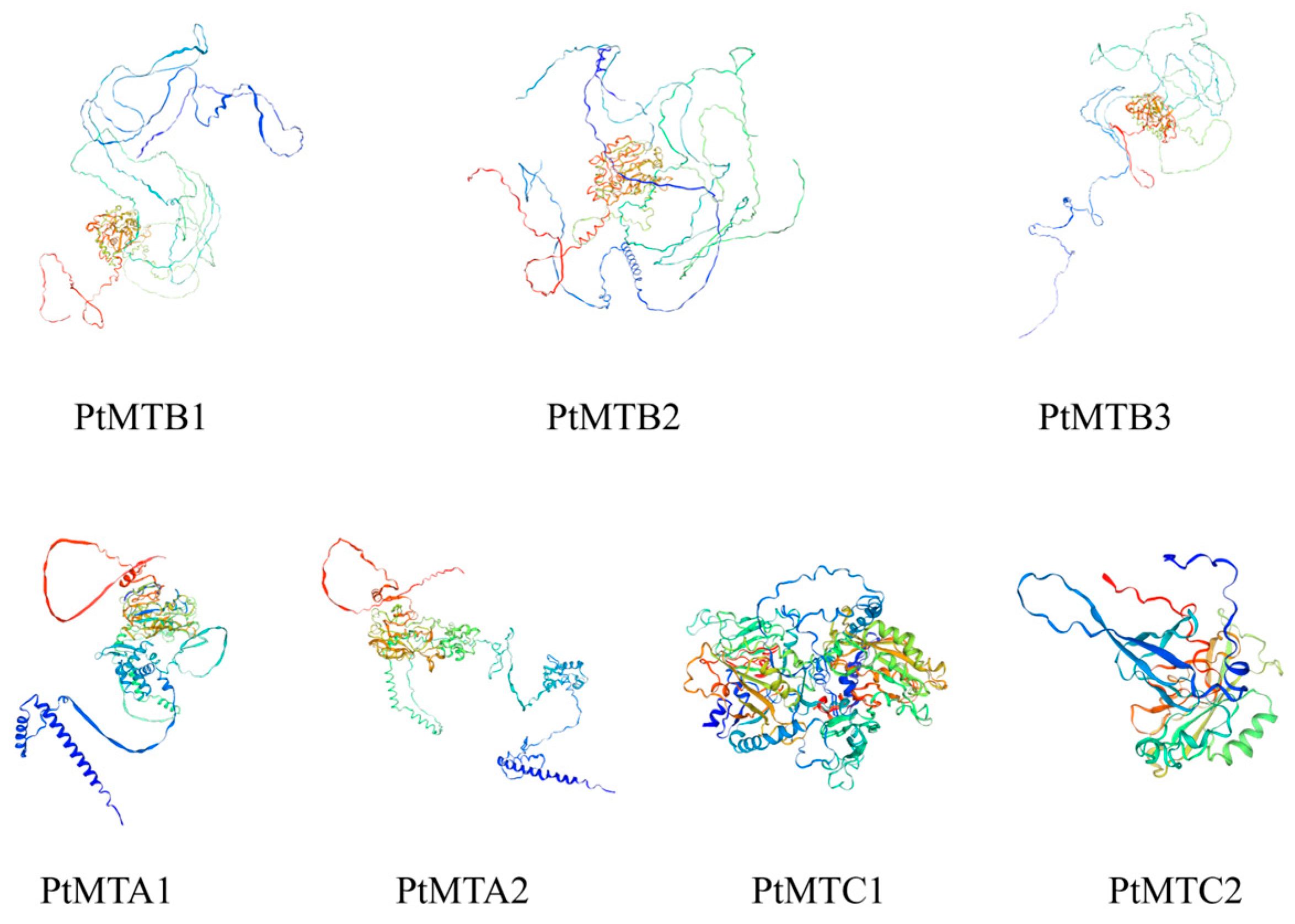
2.6. The 3D Protein Structure Analysis
2.7. Collinearity Analysis
2.8. Expression Profiles of MT-A70 Genes
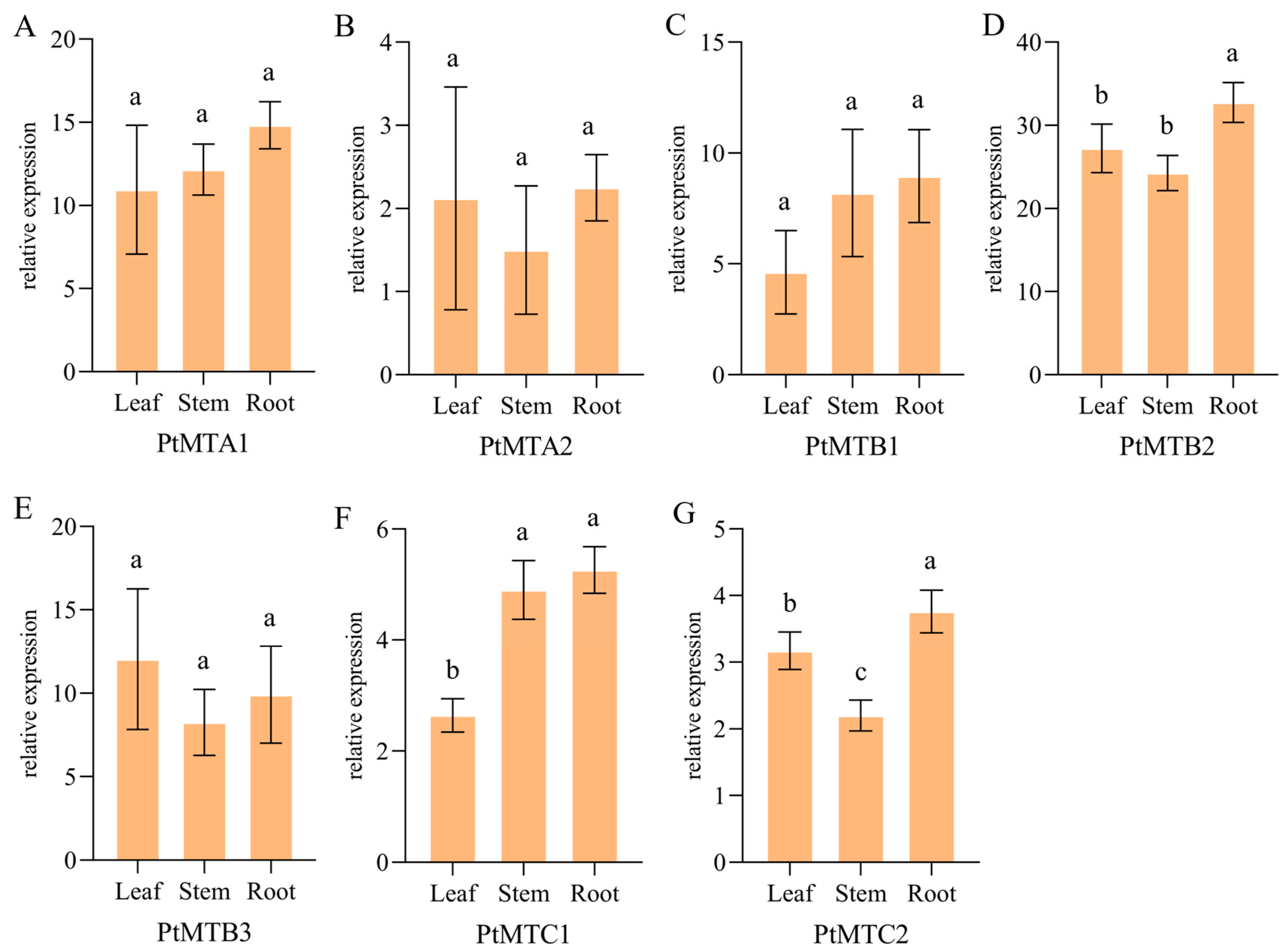
2.9. qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Identification of PtMTs Gene Family
4.2. Physicochemical Properties Analysis
4.3. Gene Structure, Conserved Motifs, and 3D Protein Analysis
4.4. Phylogenetic Analysis
4.5. Enrichment Analysis Using Gene Ontology (GO) and Cis-Regulatory Elements
4.6. Gene Expression Pattern and Collinearity Analysis
4.7. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic code and its alterations in human disease. Trends Mol. Med. 2018, 24, 886–903. [Google Scholar] [PubMed]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic modifications of mRNA and DNA in plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [PubMed]
- Zhang, T.; Shi, C.; Hu, H.; Zhang, Z.; Wang, Z.; Chen, Z.; Feng, H.; Liu, P.; Guo, J.; Lu, Q. N6-methyladenosine RNA modification promotes viral genomic RNA stability and infection. Nat. Commun. 2022, 13, 6576. [Google Scholar]
- Luo, G.Z.; MacQueen, A.; Zheng, G.; Duan, H.; Dore, L.C.; Lu, Z.; Liu, J.; Chen, K.; Jia, G.; Bergelson, J.; et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 2014, 5, 5630. [Google Scholar]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-Methyladenosine Inhibits Local Ribonucleolytic Cleavage to Stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157. [Google Scholar]
- Arribas-Hernández, L.; Rennie, S.; Schon, M.; Porcelli, C.; Enugutti, B.; Andersson, R.; Nodine, M.D.; Brodersen, P. The YTHDF proteins ECT2 and ECT3 bind largely overlapping target sets and influence target mRNA abundance, not alternative polyadenylation. Elife 2021, 10, e72377. [Google Scholar]
- Kramer, M.C.; Janssen, K.A.; Palos, K.; Nelson, A.D.L.; Vandivier, L.E.; Garcia, B.A.; Lyons, E.; Beilstein, M.A.; Gregory, B.D. N6-methyladenosine and RNA secondary structure affect transcript stability and protein abundance during systemic salt stress in Arabidopsis. Plant direct 2020, 4, e00239. [Google Scholar]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar]
- Schibler, U.; Kelley, D.E.; Perry, R.P. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol. 1977, 115, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Levis, R.; Penman, S. 5′-terminal structures of poly(A)+ cytoplasmic messenger RNA and of poly(A)+ and poly(A)- heterogeneous nuclear RNA of cells of the dipteran Drosophila melanogaster. J. Mol. Biol. 1978, 120, 487–515. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.D.; Lane, B.G. Wheat embryo ribonucleates. XIII. Methyl-substituted nucleoside constituents and 5′-terminal dinucleotide sequences in bulk poly(AR)-rich RNA from imbibing wheat embryos. Can. J. Biochem. 1979, 57, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Haugland, R.A.; Cline, M.G. Post-transcriptional modifications of oat coleoptile ribonucleic acids. 5′-Terminal capping and methylation of internal nucleosides in poly(A)-rich RNA. Eur. J. Biochem. 1980, 104, 271–277. [Google Scholar] [CrossRef]
- Zhu, J.; An, T.; Zha, W.; Gao, K.; Li, T.; Zi, J. Manipulation of IME4 expression, a global regulation strategy for metabolic engineering in Saccharomyces cerevisiae. Acta Pharm. Sin. B 2023, 13, 2795–2806. [Google Scholar] [CrossRef]
- Hu, J.; Xu, T.; Kang, H. Crosstalk between RNA m6A modification and epigenetic factors in plant gene regulation. Plant Commun. 2024, 5, 101037. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, L.; Mu, L.; Wang, Y.; Zhao, M.; Wang, H.; Li, X.; Zhao, L.; Lin, C.; Zhang, H.; et al. Evolution and post-transcriptional regulation insights of m6A writers, erasers, and readers in plant epitranscriptome. Plant J. 2024, 120, 505–525. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Kan, L.; Grozhik, A.V.; Vedanayagam, J.; Patil, D.P.; Pang, N.; Lim, K.S.; Huang, Y.C.; Joseph, B.; Lin, C.J.; Despic, V.; et al. The m6A pathway facilitates sex determination in Drosophila. Nat. Commun. 2017, 8, 15737. [Google Scholar] [CrossRef]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [PubMed]
- Jiménez-Ramírez, I.A.; Pijeira-Fernández, G.; Moreno-Cálix, D.M.; De-la-Peña, C. Same modification, different location: The mythical role of N6-adenine methylation in plant genomes. Planta 2022, 256, 9. [Google Scholar] [PubMed]
- Kan, L.; Ott, S.; Joseph, B.; Park, E.S.; Dai, W.; Kleiner, R.E.; Claridge-Chang, A.; Lai, E.C. A neural m6A/Ythdf pathway is required for learning and memory in Drosophila. Nat. Commun. 2021, 12, 1458. [Google Scholar] [PubMed]
- Govindan, G.; Sharma, B.; Li, Y.F.; Armstrong, C.D.; Merum, P.; Rohila, J.S.; Gregory, B.D.; Sunkar, R. mRNA N6-methyladenosine is critical for cold tolerance in Arabidopsis. Plant J. 2022, 111, 1052–1068. [Google Scholar]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar]
- Zhang, K.; Zhuang, X.; Dong, Z.; Xu, K.; Chen, X.; Liu, F.; He, Z. The dynamics of N6-methyladenine RNA modification in interactions between rice and plant viruses. Genome Biol. 2021, 22, 189. [Google Scholar]
- Zhou, L.; Tang, R.; Li, X.; Tian, S.; Li, B.; Qin, G. N6-methyladenosine RNA modification regulates strawberry fruit ripening in an ABA-dependent manner. Genome Biol. 2021, 22, 168. [Google Scholar]
- Lu, J.; Tang, X.; Quan, H.; Lan, X. An overview of research on Przewalskia tangutica Maxim., an endangered tibetan medicinal plant. Agric. Sci. Technol. 2017, 18, 2320–2325. [Google Scholar]
- Xiao, P.; Xia, G.; He, L. The occurrence of some important tropane alkaloids in Chinese solanaceous plants. Acta Bot. Sin. 1973, 15, 187–194. [Google Scholar]
- Wu, Y.; Yang, J.; Yang, Y.; Liu, J. The genome sequence and demographic history of Przewalskia tangutica (Solanaceae), an endangered alpine plant on the Qinghai-Tibet Plateau. DNA Res. 2023, 30, dsad005. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, W.; Yang, Y.; Wilson, I.; Shao, F.; Qiu, D. Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K. Genes 2022, 13, 1018. [Google Scholar] [CrossRef]
- Tang, L.; Xue, J.; Ren, X.; Zhang, Y.; Du, L.; Ding, F.; Zhou, K.; Ma, W. Genome-Wide Identification and Expression Analysis of m6A Writers, Erasers, and Readers in Litchi (Litchi chinensis Sonn.). Genes 2022, 13, 2284. [Google Scholar] [CrossRef]
- Lin, H.; Shi, T.; Zhang, Y.; He, C.; Zhang, Q.; Mo, Z.; Pan, W.; Nie, X. Genome-Wide Identification, Expression and Evolution Analysis of m6A Writers, Readers and Erasers in Aegilops_tauschii. Plants 2023, 12, 2747. [Google Scholar] [CrossRef]
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Hasan, M.; Nishat, Z.S.; Hasan, M.S.; Hossain, T.; Ghosh, A. Identification of m6A RNA Methylation Genes in Oryza sativa and Expression Profiling in Response to Different Developmental and Environmental Stimuli. Biochem. Biophys. Rep. 2024, 38, 101677. [Google Scholar] [CrossRef]
- Liu, P.; Liu, H.; Zhao, J.; Yang, T.; Guo, S.; Chang, L.; Xiao, T.; Xu, A.; Liu, X.; Zhu, C.; et al. Genome-wide identification and functional analysis of mRNA m6A writers in soybean under abiotic stress. Front. Plant Sci. 2024, 15, 1446591. [Google Scholar] [CrossRef]
- Sheikh, A.H.; Tabassum, N.; Rawata, A.; Almeida Trapp, M.; Nawaz, K.; Hirt, H. m6A RNA Methylation Counteracts Dark-induced Leaf Senescence in Arabidopsis. Plant Physiol. 2024, 194, 2663–2678. [Google Scholar] [PubMed]
- Hu, J.; Manduzio, S.; Kang, H. Epitranscriptomic RNA Methylation in Plant Development and Abiotic Stress Responses. Front. Plant Sci. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; He, Q.; Qi, Z.; Zhang, G.; Xu, W.; Yi, T.; Wu, G.; Li, R. MTA, an RNA m6A Methyltransferase, Enhances Drought Tolerance by Regulating the Development of Trichomes and Roots in Poplar. Int. J. Mol. Sci. 2020, 21, 2462. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.; Xu, T.; Kang, H. N6-Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Yao, Y.; Zhang, H.; Wang, Y.; Gao, J.; Fan, M. Overexpression of watermelon m6A methyltransferase ClMTB enhances drought tolerance in tobacco by mitigating oxidative stress and photosynthesis inhibition and modulating stress-responsive gene expression. Plant Physiol. Biochem. 2021, 168, 340–352. [Google Scholar]
- Cao, J.; Huang, C.; Liu, J.; Li, C.; Liu, X.; Zheng, Z.; Hou, L.; Huang, J.; Wang, L.; Zhang, Y.; et al. Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton. Int. J. Mol. Sci. 2022, 23, 14111. [Google Scholar] [CrossRef]
- Wang, L.; Yang, C.; Shan, Q.; Zhao, M.; Yu, J.; Li, Y.-F. Transcriptome-wide profiling of mRNA N6-methyladenosine modification in rice panicles and flag leaves. Genomics 2023, 115, 110542. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar]
- Kloczkowski, A.; Ting, K.L.; Jernigan, R.L.; Garnier, J. Combining the GOR V algorithm with evolutionary information for protein secondary structure prediction from amino acid sequence. Proteins 2002, 49, 154–166. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Galaxy Team. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar]
- Chen, C.; Wu, Y.; Xia, R. A painless way to customize Circos plot: From data preparation to visualization using TBtools. Imeta 2022, 1, e35. [Google Scholar] [PubMed]
- Li, X.; Zhang, T.; Jiang, L.; Fan, G. Evaluation of Suitable Reference Genes for Quantitative Real-Time PCR in Various Tissues of Apocynum venetum. Genes 2024, 15, 231. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Length (bp) | Length (aa) | MW (kDa) | pI | Instability Index | Aliphatic Index | Grand Average of Hydropathicity (GRAVY) | Subcellular Localization Predicted |
|---|---|---|---|---|---|---|---|---|
| PtMTA1 | 2217 | 738 | 81.83 | 6.72 | 42.05 | 78.86 | −0.461 | chloroplast |
| PtMTA2 | 2196 | 731 | 80.63 | 6.47 | 41.33 | 79.48 | −0.391 | chloroplast |
| PtMTB1 | 3285 | 1094 | 122.96 | 6.16 | 57.55 | 48.39 | −1.155 | nucleus |
| PtMTB2 | 3231 | 1076 | 120.82 | 6.14 | 58.2 | 48.84 | −1.163 | nucleus |
| PtMTB3 | 3153 | 1050 | 117.53 | 6.77 | 56.36 | 50.99 | −1.102 | nucleus |
| PtMTC1 | 1320 | 439 | 50.13 | 7.1 | 49.4 | 81.3 | −0.389 | nucleus |
| PtMTC2 | 867 | 288 | 32.54 | 5.12 | 45.79 | 83.26 | −0.239 | cytoplasm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, X.; Hu, X.; Zhen, K.; Meng, J.; Du, H.; Cao, X.; Zhou, D. Genome-Wide Identification and Expression Analysis of m6A Methyltransferase Family in Przewalskia tangutica Maxim. Int. J. Mol. Sci. 2025, 26, 3593. https://doi.org/10.3390/ijms26083593
Ye X, Hu X, Zhen K, Meng J, Du H, Cao X, Zhou D. Genome-Wide Identification and Expression Analysis of m6A Methyltransferase Family in Przewalskia tangutica Maxim. International Journal of Molecular Sciences. 2025; 26(8):3593. https://doi.org/10.3390/ijms26083593
Chicago/Turabian StyleYe, Xing, Xingqiang Hu, Kun Zhen, Jing Meng, Heyan Du, Xueye Cao, and Dangwei Zhou. 2025. "Genome-Wide Identification and Expression Analysis of m6A Methyltransferase Family in Przewalskia tangutica Maxim." International Journal of Molecular Sciences 26, no. 8: 3593. https://doi.org/10.3390/ijms26083593
APA StyleYe, X., Hu, X., Zhen, K., Meng, J., Du, H., Cao, X., & Zhou, D. (2025). Genome-Wide Identification and Expression Analysis of m6A Methyltransferase Family in Przewalskia tangutica Maxim. International Journal of Molecular Sciences, 26(8), 3593. https://doi.org/10.3390/ijms26083593