

Comparative Analysis of Transcriptome Data of Wings from Different Developmental Stages of the Gynaephora qinghaiensis

Abstract
:1. Introduction
2. Results
2.1. Transcriptome Sequencing of Gynaephora qinghaiensis
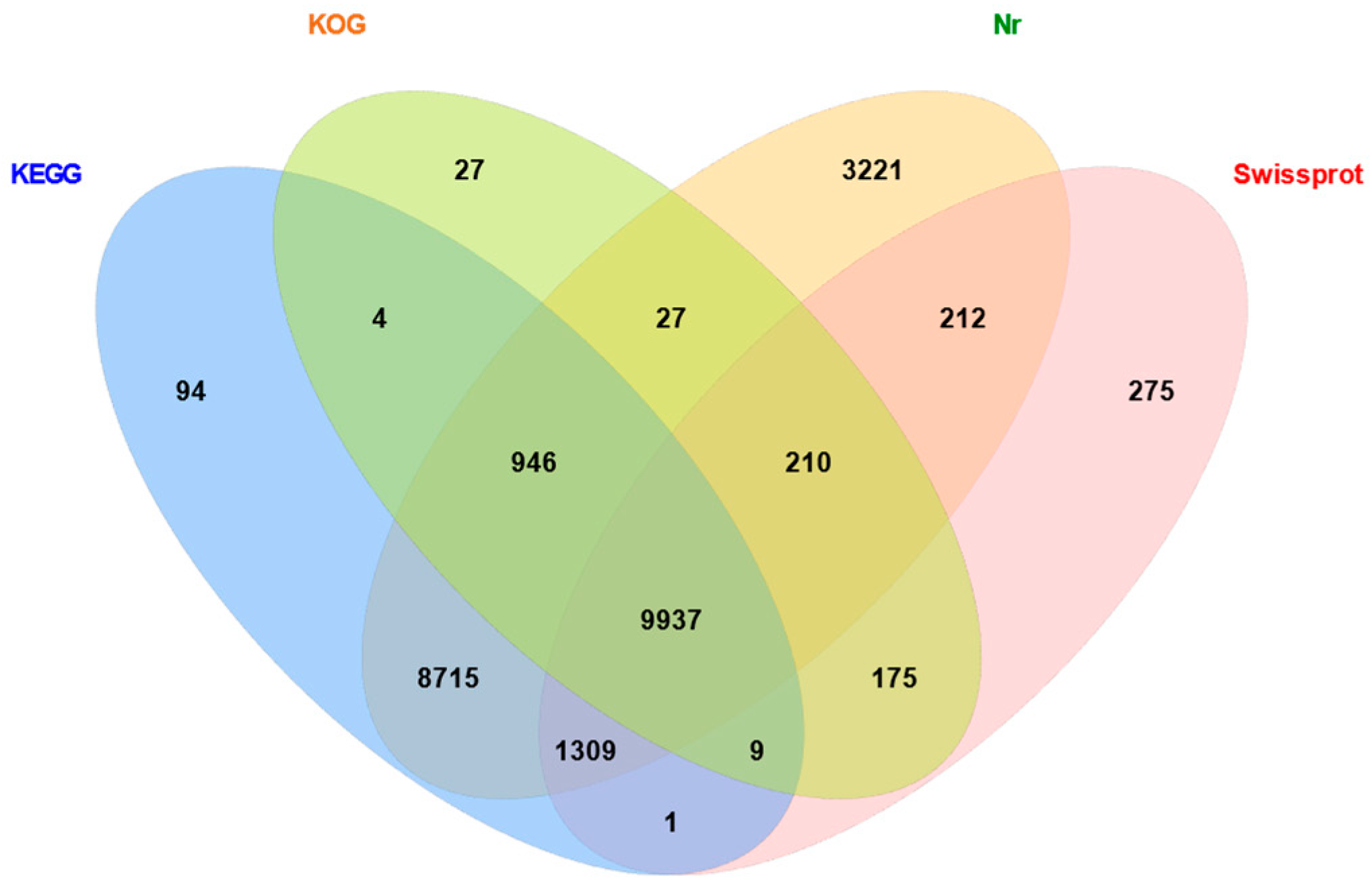
2.2. Functional Annotation of Unigenes from Gynaephora qinghaiensis
2.3. Numbers of DEGs Associated with Wing Development in Gynaephora qinghaiensis
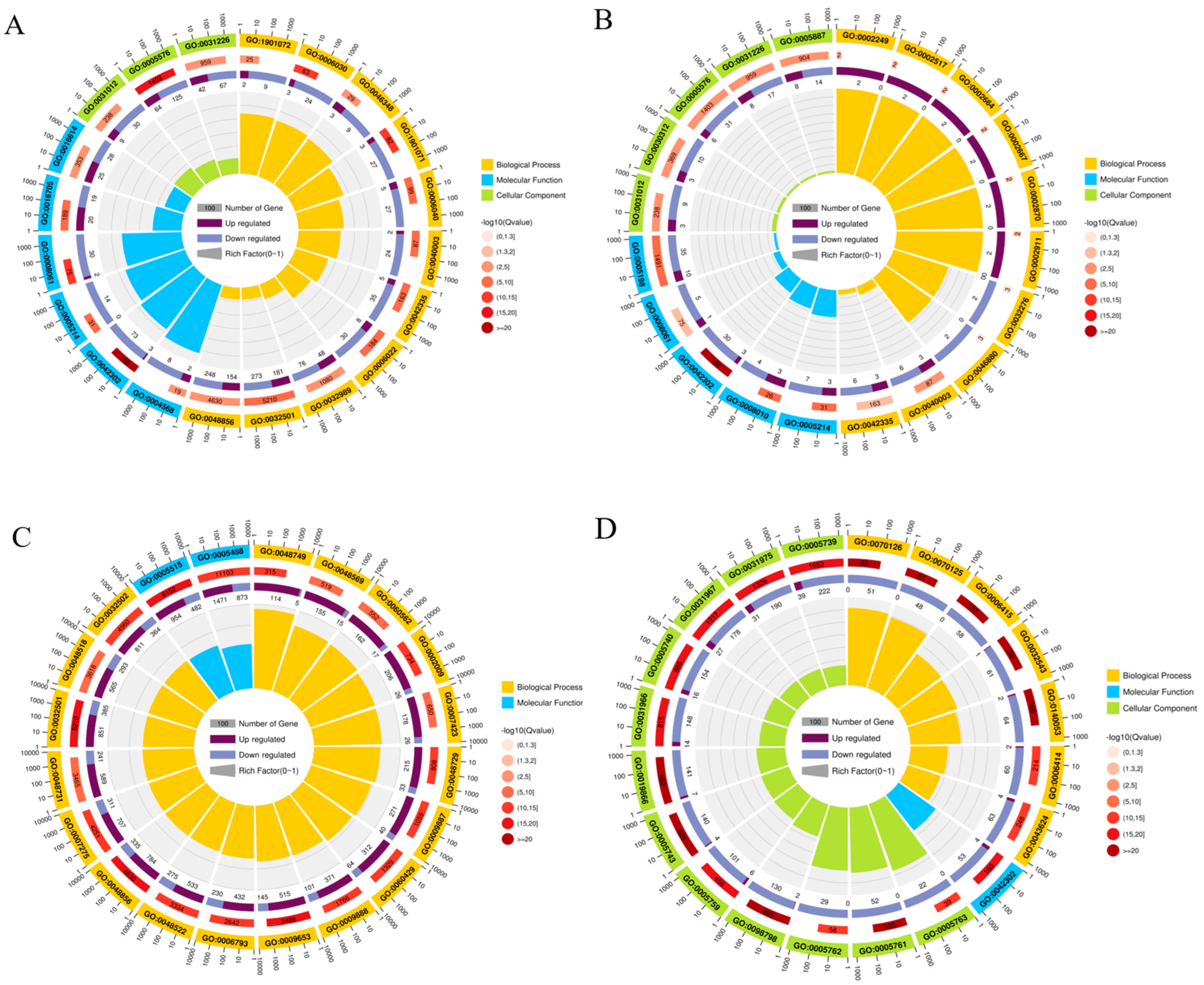
2.4. GO Enrichment Analysis of DEGs in Gynaephora qinghaiensis
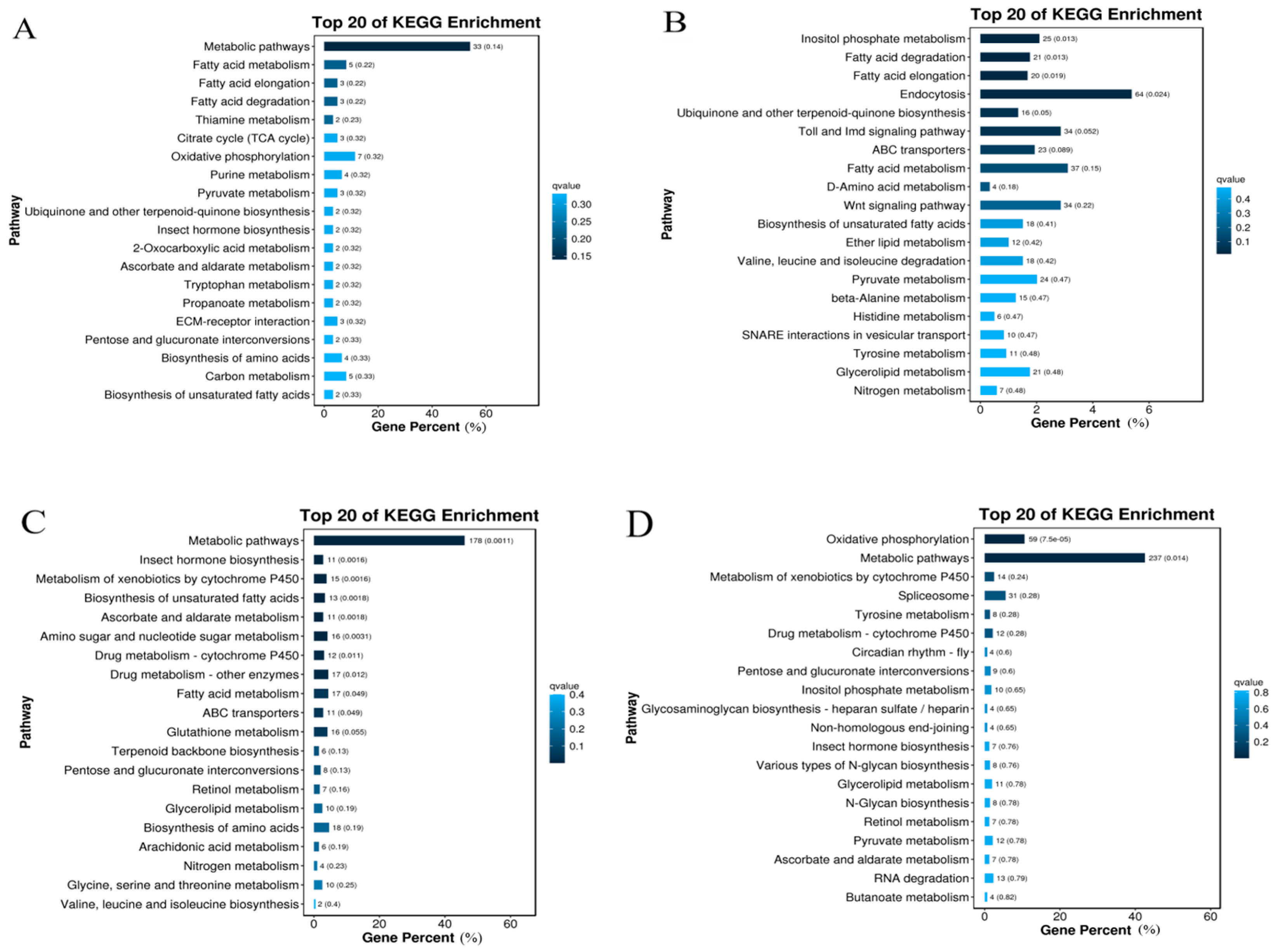
2.5. KEGG Enrichment of Gynaephora qinghaiensis DEGs
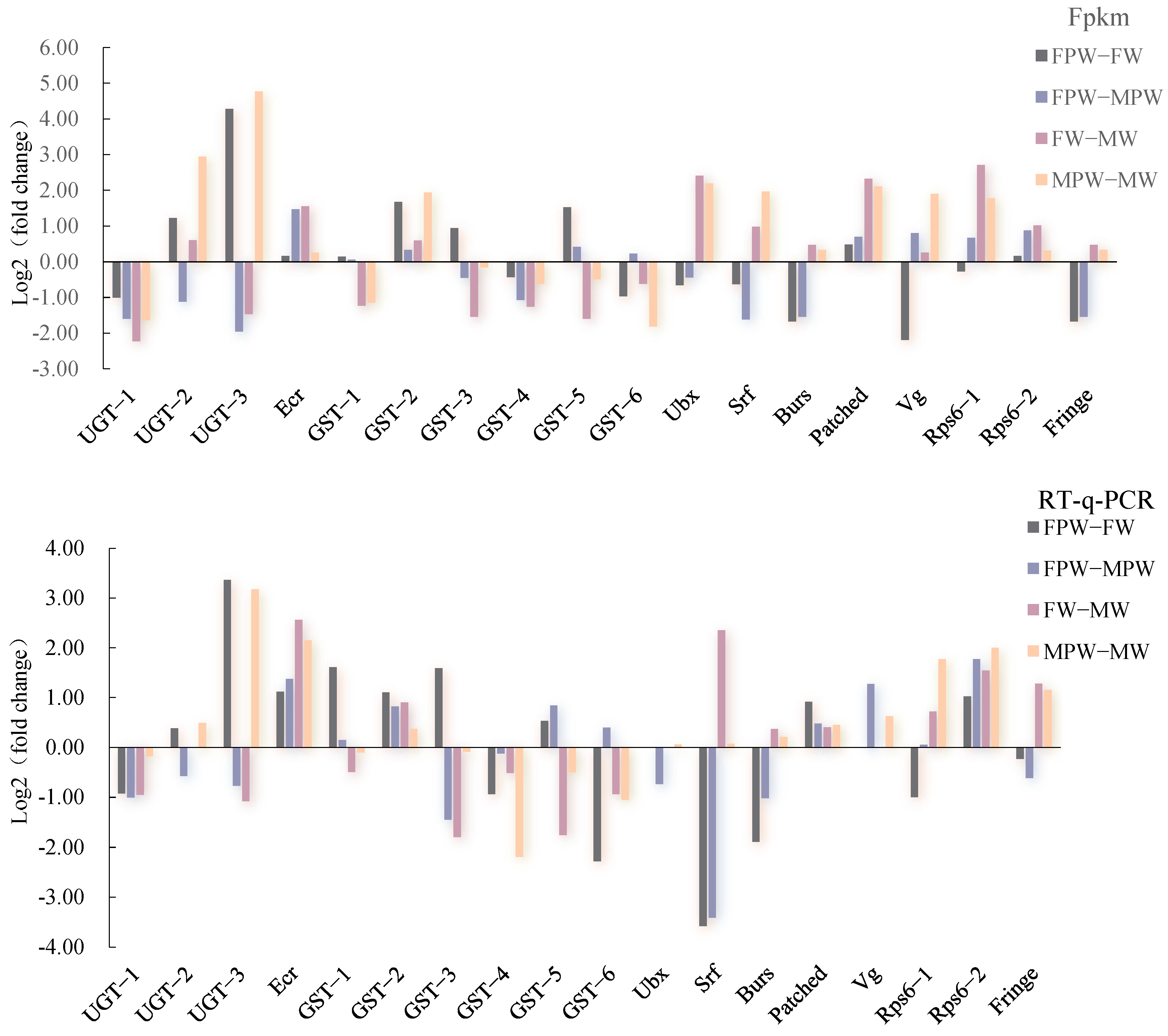
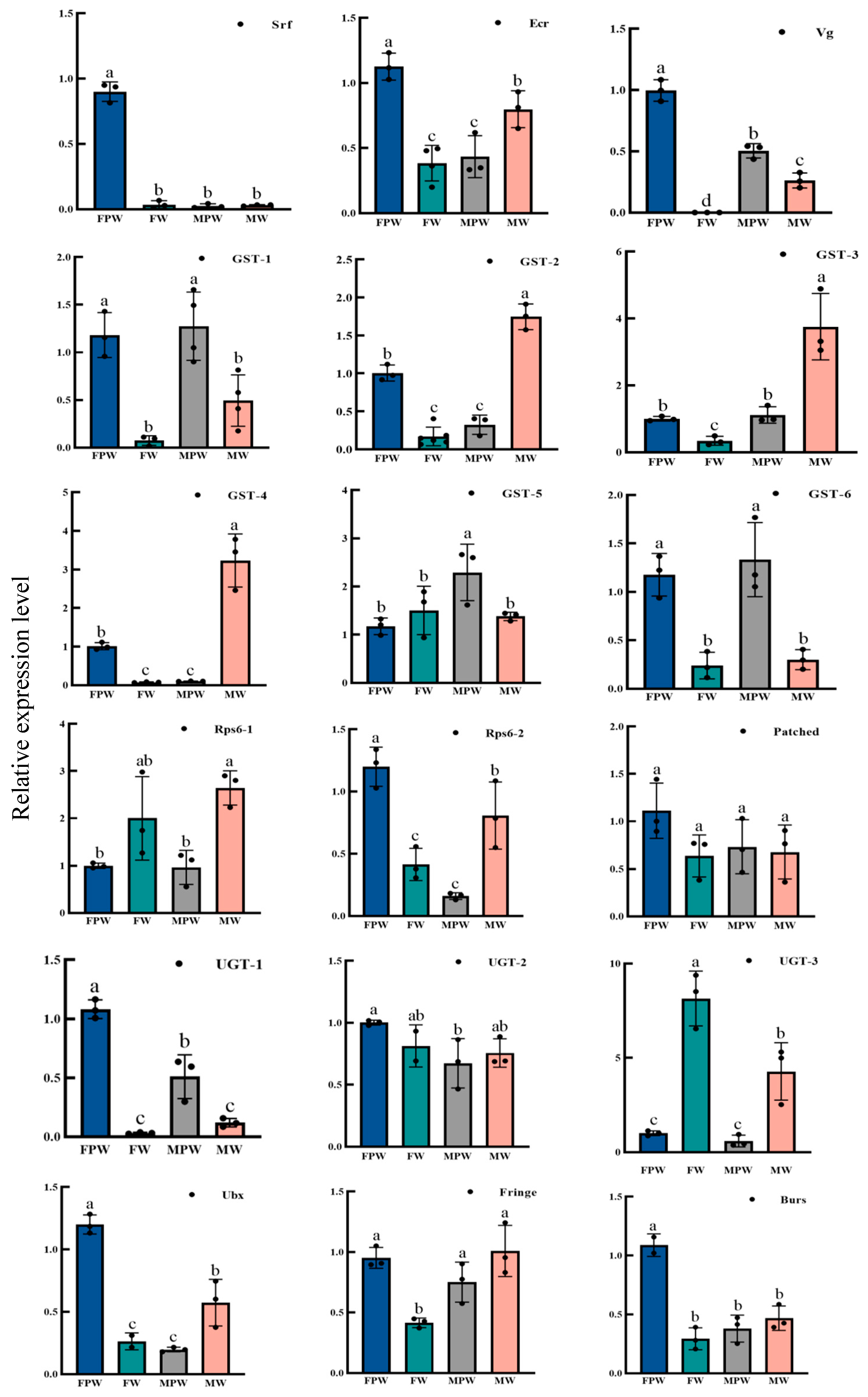
2.6. Validation and Expression Profiling of DEGs for Wing Development in the Gynaephora qinghaiensis
3. Discussion
4. Materials and Methods
4.1. Insects Materials and Sampling
4.2. Total RNA Extraction of Trichoderma reesei from Gynaephora qinghaiensis
4.3. Identification of DEGs
4.4. Screening and Functional Enrichment of DEGs
4.5. RT-qPCR Verification of DEGs
4.6. Data Processing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yuan, M.L.; Zhang, Q.L.; Wang, Z.F.; Guo, Z.L.; Bao, G.S. Molecular Phylogeny of Grassland Caterpillars (Lepidoptera: Lymantriinae: Gynaephora) Endemic to the Qinghai-Tibetan Plateau. PLoS ONE 2015, 10, e0127257. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, G.; Liu, Z.C. Number of Instars and Stadium Duration of Gynaephora menyuanensis (Lepidoptera: Lymantriidae) from Qinghai-Tibetan Plateau in China. Ann. Entomol. Soc. Am. 2006, 99, 1012–1018. [Google Scholar] [CrossRef]
- Wang, H.Z.; Zhong, X.; Zhang, G.R.; Liu, X.; Gu, L. Transcriptome characterization and gene expression analysis related to immune response in Gynaephora qinghaiensis pupae. J. Asia-Pac. Entomol. 2020, 23, 458–469. [Google Scholar] [CrossRef]
- Zhou, R.; Yin, X.C. Taxonomic studies on grassland caterpillars. Entomotaxonomia 1979, 1, 23–28. [Google Scholar]
- Bai, H.T.; Wang, W.; Wei, X.J.; Han, R.C.; Xu, C.T. Pathogenicity of entomopathogenicnematodes against Gynaephora alpherakii larvae by bioassay. J. Environ. Entomol. 2020, 42, 1269–1274. [Google Scholar]
- Chen, K.L.; Yu, X.C.; Yao, B.Q.; Ma, Z.; Wang, W.Y.; Wang, H.C.; Zhou, H.K.; Zhao, X.Q. Spatial Distribution of Gynaephora menyuanensis under Different Grazing Intensities in Alpine Meadow. Acta Agrestia Sin. 2016, 24, 191–197. [Google Scholar]
- Wang, H.Z.; Xin, Z.; Lin, H.F.; Li, S.S.; Yi, J.Q.; Zhang, G.R.; Liu, X.; Gu, L. Genetic Diversity and Population Structure of Gynaephora qinghaiensis in Yushu Prefecture, Qinghai Province Based on the Mitochondrial COI Gene. Biochem. Genet. 2021, 59, 1–17. [Google Scholar] [CrossRef]
- Yan, L.; Jiang, X.L.; Wang, G. Studies on the developmental characteristics of caterpillar larvae in the Menyuan grassland. Acta Prataculturae Sin. 2005, 14, 116–120. [Google Scholar]
- Strathdee, A.T.; Bale, J.S. Life on the edge: Insect ecology in arctic environments. Annu. Rev. Entomol. 1998, 43, 85–106. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Yuan, M.L.; Deng, X.Y.; Guo, J.; Feng, W.; Lin, L.B.; Wang, Y.J. Gene expression analysis of the tibetan grassland caterpillars(Lepidoptera: Lymantriinae: Gynaephora) in response to high-altitude stress. AIP Conf. Proc. 2019, 2079, 020003. [Google Scholar]
- Yuan, M.L.; Bao, M.H.; Zhang, Q.L.; Guo, Z.L.; Li, M.; Wang, J. Mitochondrial phylogeography of grassland caterpillars (Lepidoptera: Lymantriinae: Gynaephora) endemic to the Qinghai-Tibetan plateau. Ecol. Evol. 2024, 14, e7027. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J. The complete mitochondrial genome of Gynaephora alpherakii (Lepidoptera: Lymantriidae). Mitochondrial DNA Part A 2016, 27, 1–2270. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.X.; Ai, L.; Xu, M.J.; Chen, S.H.; Zhang, Y.N.; Guo, J.; Cai, Y.C.; Tian, L.G.; Zhang, L.L.; Zhu, X.Q.; et al. Identification and characterization of microRNAs in Trichinella spiralis by comparison with Brugia malayi and Caenorhabditis elegans. Parasitol. Res. 2011, 109, 553–558. [Google Scholar] [CrossRef]
- David, L.; Huber, W.; Granovskaia, M.; Toedling, J.; Palm, C.J.; Bofkin, L.; Jones, T.; Davis, R.W.; Steinmetz, L.M. A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. USA 2006, 103, 5320–5325. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Shimomura, K.; Hosoi, A.; Sato, Y.; Oikawa, Y.; Seino, Y.; Kuribara, T.; Yajima, S.; Tomizawa, M. Antennal transcriptome analysis of chemosensory genes in the cowpea beetle, Callosobruchus maculatus (F.). PLoS ONE 2022, 17, e0262817. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.X.; Su, R.; Ouyang, H.; Zheng, X.L.; Lu, W.; Wang, X.Y. Antennal Transcriptome Analysis and Identification of Olfactory Genes in Glenea cantor Fabricius (Cerambycidae: Lamiinae). Insects 2022, 13, 553. [Google Scholar] [CrossRef]
- Yuan, F.X.; Zhong, H.; Zhao, Y.X.; Zhan, J.L.; Wei, C.; Ying, X.L.; Gu, C.Z.; Xiao, Q.L.; Ming, J.D.; Hai, Z.Y. Integrated transcriptome and 1H NMR-based metabolome to explore the potential mechanism of Spodoptera litura in response to flupyrimin. Pestic. Biochem. Physiol. 2024, 205, 106146. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wang, C.C.; Liu, Y.; Shi, X.Y.; Gao, X. Transcriptome analysis and identification of P450 genes relevant to imidacloprid detoxification in Bradysia odoriphaga. Sci. Rep. 2018, 8, 2564. [Google Scholar] [CrossRef]
- McCulloch, G.A.; Wallis, G.P.; Waters, J.M. Do insects lose flight before they lose their wings? Population genetic structure in subalpine stoneflies. Mol. Ecol. 2009, 18, 4073–4087. [Google Scholar] [CrossRef]
- Zhang, F.; Gao, H.; Tong, J.; Zhou, J.; Ma, Y.H. Research on elytron section microstructure of four species beetles and biomimetic models. Trans. Chin. Soc. Agric. Eng. 2011, 27, 105–109. [Google Scholar]
- Wu, J.E.; Liang, A.W.; Feng, B.; Hu, Y.; Wang, F.H. Ultrastructures of the wings from the long-winged female adult of Sogatella furcifera. Acta Sci. Nat. Univ. Sunyatseni 2024, 63, 1–5. [Google Scholar]
- Hong, J.; Ye, G.Y.; Hu, C. Characterization of wing surface markings and ultrastructure of scales in four species of tigonopteryx butterfly. J. Zhejiang Univ. (Agric. Life Sci.) 1998, 24, 47–52. [Google Scholar]
- Jin, M.L.; Feng, S.; Bi, Y.D.; Lin, W.; Qing, C.L.; Jin, J.W.; Wei, D. Characterization of two Bursicon genes and their association with wing development in the brown citrus aphid, Aphis citricidus. Insect Sci. 2024, 31, 1684–1696. [Google Scholar]
- Zhang, X.B.; Liu, M.Q.; Cheng, A.D.; Bernard, M.; Zhang, J.Z.; Dong, W. Role of CYP311A1 in wing development of Drosophila melanogaster. Insect Sci. 2024, 31, 748–758. [Google Scholar] [CrossRef]
- Jernej, P.; Elizaveta, K.; Adam, W.H.; Igor, M. Wing buzzing as a mechanism for generating vibrational signals in psyllids (Hemiptera: Psylloidea). Insect Sci. 2024, 31, 1466–1476. [Google Scholar]
- Yu, X.; Sun, B.; Gao, X.Q.; Liu, Q.X.; Zhou, Z.Z.; Zhao, Y.H. miR-927 regulates insect wing development by targeting the Hippo pathway. Insect Sci. 2024, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Niu, J.Z.; Ding, B.Y.; Zhang, W.; Wei, D.D.; Wei, D.; Jiang, H.B.; Wang, J.J. The miR-9b microRNA mediates dimorphism and development of wing in aphids. Proc. Natl. Acad. Sci. USA 2020, 117, 8404–8409. [Google Scholar] [CrossRef]
- Broehan, G.; Kroeger, T.; Lorenzen, M.; Merzendorfer, H. Functional analysis of the ATP-binding cassette (ABC) transporter gene family of Tribolium castaneum. BMC Genom. 2013, 14, 6. [Google Scholar] [CrossRef]
- JiA, X.P.; Ye, X.Q.; Liang, L.J.; Deng, Y.M.; Sun, X.B.; Yu, J.M. High-throughput sequencing-based transcriptomics of Paspalum vaginatum, Seashore paspalum. Acta Prataculturae Sin. 2014, 23, 242–252. [Google Scholar]
- Wang, X.C.; Tan, H.L.; Chen, Z.; Meng, L.Z.; Wang, W.B.; Fan, S.C. RNA-Seq-based assembly and analysis of Fructus Forsythiae transcriptome and development of SSR molecular markers. Sci. Sin. (Vitae) 2015, 45, 301–310. [Google Scholar]
- Jacques, M.; Sylvie, F.; Mohamed, O. Insights on membrane topology and structure function of UDP-glucuronosyltransferases. Drug Metab. Rev. 2010, 42, 159–166. [Google Scholar]
- Israni, B.; Wouters, F.C.; Luck, K.; Seibel, E.; Ahn, S.J.; Paetz, C.; Reinert, M.; Vogel, H.; Erb, M.; Heckel, D.G.; et al. The Fall Armyworm Spodoptera frugiperda Utilizes Specific UDP-Glycosyltransferases to Inactivate Maize Defensive Benzoxazinoids. Front. Physiol. 2020, 11, 604754. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Ye, G. Molecular characterization of the sigma class gutathione S-transferase from Chilo. J. Econ. Entomol. 2011, 104, 2046–2053. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Pavlidi, N.; Vontas, J.; Leeuwen, V.T. The role of glutathione S-transferases (GSTs) in insecticide resistance in crop pests and disease vectors. Curr. Opin. Insect Sci. 2018, 27, 97–102. [Google Scholar] [CrossRef]
- Zhang, C.C.; Yang, H.; Yang, M.F.; Yu, H.P.; Zhang, W. Cloning and Expression of Bursicon Gene of Myzus persicae (Sulzer). Fujian J. Agric. Sci. 2017, 32, 1244–1250. [Google Scholar]
- Wu, W.J.; Wang, Y.; Huang, H.J.; Bao, Y.Y.; Zhang, C.X. Ecdysone receptor controls wing morphogenesis and melanization during rice planthopper metamorphosis. J. Insect Physiol. 2012, 58, 420–426. [Google Scholar] [CrossRef]
- Gehring, J.W.; Kloter, U.; Suga, H. Chapter 2 Evolution of the Hox Gene Complex from an Evolutionary Ground State. Curr. Top. Dev. Biol. 2009, 88, 35–61. [Google Scholar]
- Anastasios, P.; Michael, A. Hox gene Ultrabithorax regulates distinct sets of target genes at successive stages of Drosophila haltere morphogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2855–2860. [Google Scholar]
- Hughes, C.L.; Kaufman, T.C. Hox genes and the evolution of the arthropod body plan. Evol. Dev. 2002, 4, 459–499. [Google Scholar] [CrossRef]
- Mika, M.; Toshinobu, Y.; Teruyuki, N. Functional analysis of Ultrabithorax in the silkworm, Bombyx mori, using RNAi. Dev. Genes Evol. 2009, 219, 437–444. [Google Scholar]
- Roy, S.; Vijayraghavan, K. Homeotic genes and the regulation of myoblast migration, fusion, and fibre-specific gene expression during adult myogenesis in Drosophila. Development 1997, 124, 3333–3341. [Google Scholar] [CrossRef]
- Shore, P.; Sharrocks, A.D. The MADS-box family of transcription factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef]
- Jacques, M.; Jay, G.; Karen, G.; Mark, A.K.; Walter, J.G.; Markus, A. The Drosophila serum response factor gene is required for the formation of intervein tissue of the wing and is allelic to blistered. Development 1996, 122, 2589–2597. [Google Scholar]
- Markus, A.; Jacques, M.; Uwe, W.; Jay, G.; Urs., K.; Marilina, L.R.; Walter, J.G. The Drosophila SRF homolog is expressed in a subset of tracheal cells and maps within a genomic region required for tracheal development. Development 1994, 120, 743–753. [Google Scholar]
- Louis, G.; Jordi, C. The Drosophila homologue of SRF acts as a boosting mechanism to sustain FGF-induced terminal branching in the tracheal system. Development 2011, 138, 1269–1274. [Google Scholar]
- Somogyi, K.; Rørth, P. Evidence for Tension-Based Regulation of Drosophila MAL and SRF during Invasive Cell Migration. Dev. Cell 2004, 7, 85–93. [Google Scholar] [CrossRef]
- Medjkane, S.; Cristina, P.S.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef]
- Ekaterina, L.; Eleanor, T.; Magdalene, M.; Anne, V.; Tan, S.J.; Adam, F.; Matthias, K.; Daimark, B. Drosophila Pico and Its Mammalian Ortholog Lamellipodin Activate Serum Response Factor and Promote Cell Proliferation. Dev. Cell 2008, 15, 680–690. [Google Scholar]
- Thompson, B.J. Mal/SRF is dispensable for cell proliferation in Drosophila. PLoS ONE 2010, 5, e10077. [Google Scholar] [CrossRef]
- Fristrom, D.; Gotwals, P.; Eaton, S.; Kornberg, T.B.; Sturtevant, M.; Bier, E.; Fristrom, J.W. Blistered: A gene required for vein/intervein formation in wings of Drosophila. Development 1994, 120, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Clark-Hachtel, C.M.; Linz, D.M.; Tomoyasu, Y. Insights into insect wing origin provided by functional analysis of vestigial in the red flour beetle, Tribolium castaneum. Proc. Natl. Acad. Sci. USA 2013, 110, 16951–16956. [Google Scholar] [CrossRef]
- Parker, J.; Struhl, G. Control of Drosophila wing size by morphogen range and hormonal gating. Proc. Natl. Acad. Sci. USA 2020, 117, 31935–31944. [Google Scholar] [CrossRef]
- Goto, S.; Hayashi, S. Specification of the embryonic limb primordium by graded activity of Decapentaplegic. Development 1997, 124, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Goto, S.; Eto, K.; Hayashi, S. EGF receptor attenuates Dpp signaling and helps to distinguish the wing and leg cell fates in Drosophila. Development 2000, 127, 3769–3776. [Google Scholar] [CrossRef]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef]
- Iadevaia, V.; Huo, Y.; Zhang, Z.; Foster, L.J.; Proud, C.G. Roles of the mammalian target of rapamycin, mTOR, in controlling ribosome biogenesis and protein synthesis. Biochem. Soc. Trans. 2012, 40, 168–172. [Google Scholar] [CrossRef]
- Panin, V.M.; Irvine, K.D. Modulators of Notch signaling. Semin. Cell Dev. Biol. 1998, 9, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Moloney, D.J.; Panin, V.M.; Johnston, S.H.; Chen, J.; Shao, L.; Wilson, R.; Wang, Y.; Stanley, P.; Irvine, K.D.; Haltiwanger, R.S.; et al. Fringe is a glycosyltransferase that modifies Notch. Nature 2000, 406, 369–375. [Google Scholar] [CrossRef]
- Zweidler-McKay, A.P.; Pear, S.W. Notch and T cell malignancy. Semin. Cancer Biol. 2004, 14, 329–340. [Google Scholar] [CrossRef]
- Johoston, L.A.; Edgar, B.A. Wingless and Notch regulate cell cycle ar-rest in the developing Drosophila wing. Nature 1998, 394, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Correia, T.; Papayannopoulos, V.; Panin, V.; Woronoff, P.; Jiang, J.; Vogt, T.F.; Irvine, K.D. Molecular genetic analysis of the glycosyltransferase Fringe in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 6404–6409. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Part | Sample ID | Clean Data/Article | Mapping Rate | Q20(%) | Q30(%) | Clean Data/bp |
|---|---|---|---|---|---|---|
| Female pupa | FPW1 | 40,171,912 | 84.41% | 98.22% | 96.59% | 5,986,819,607 |
| FPW2 | 57,571,540 | 85.97% | 97.66% | 94.48% | 8,583,049,456 | |
| FPW3 | 51,834,670 | 84.96% | 98.07% | 96.36% | 7,717,310,487 | |
| Female wing buds | FW1 | 46,961,368 | 84.81% | 97.79% | 95.16% | 7,002,870,012 |
| FW2 | 42,698,294 | 83.52% | 98.10% | 96.42% | 6,362,782,499 | |
| FW3 | 36,469,654 | 83.35% | 98.20% | 96.54% | 5,432,263,013 | |
| Male pupa | MPW1 | 45,752,948 | 86.25% | 98.12% | 96.45% | 6,807,876,453 |
| MPW2 | 48,991,512 | 85.91% | 98.15% | 96.49% | 7,298,753,786 | |
| MPW3 | 38,033,428 | 84.32% | 98.27% | 96.74% | 5,668,575,333 | |
| Male wings | MW1 | 55,372,384 | 85.86% | 97.74% | 94.75% | 8,267,356,555 |
| MW2 | 36,205,752 | 85.60% | 98.14% | 96.47% | 5,361,072,666 | |
| MW3 | 38,739,102 | 86.18% | 98.16% | 96.50% | 5,747,113,609 |
| Pairs | Up | Down | Total |
|---|---|---|---|
| FPW-vs-MPW | 166 | 311 | 477 |
| FW-vs-MW | 4592 | 2935 | 7527 |
| FPW-vs-FW | 1474 | 1390 | 2864 |
| MPW-vs-MW | 1551 | 1754 | 3305 |
| Primer Name | Primer Sequence (5′–3′) | Product Size |
|---|---|---|
| Burs | F:TTGCCAAGAATCTGGTGAG | 138 |
| R:TCCTCAATACCGCCACAT | 100 | |
| Ecr | F:ACGAGGATTCCGACTTACCAT | 149 |
| R:TTGGCGAATCCTTGTAGACCT | 123 | |
| Fringe | F:GCAAGTGGTGGCAGATTCATCA | 104 |
| R:GCCAGTAGCTTCATTTGTTCCA | 104 | |
| GST1 | F:CGACCAACGTCTATACTTCGACA | 108 |
| R:AGAGCATCCTCGATCTTAGCTT | 108 | |
| GST2 | F:CACAACATACCATTCCGAT | 116 |
| R:AAGAGAATCATCTTCACCGTA | 127 | |
| GST3 | F:GGCCGAGTCTCTATACCCT | 116 |
| R:AGGCAACTTCTTTAACTCTGA | 115 | |
| GST4 | F:TGATTACGAACGTACCGCTGAC | 122 |
| R:AAGTGGTCTGCTGCCGCATAA | 93 | |
| GST5 | F:GCAACCAACCTTCTCCTCGGAATAG | 126 |
| R:AGCCGCTATGTACTCCTTCACTCTT | 123 | |
| GST6 | F:CGCTGGTGATAAGCTAACACT | 149 |
| R:CCAGCTCGAACCATTTGACA | 100 | |
| Patched | F:TCGATGCTGCTAGTATTCCC | 108 |
| R:GCTCAGAAATCTTAGGTCGTT | 138 | |
| Rps6-1 | F:TACATAATGCTGTCCGGCAGA | 100 |
| R:GACCGCCATATACCACTCGAAC | 149 | |
| Rps6-2 | F:AAGGTGCTGCCGCTGTTCGT | 123 |
| R:ACATCATCTTCACTTGCCAGTCTAGGT | 104 | |
| Srf | F:CACCGACTACCAGCGAGCA | 104 |
| R:TTCATCTCCACTGTCGCCTGA | 108 | |
| Ubx | F:CCAGTGCAGCATCAACCCA | 108 |
| R:ATACGTTTGTCTTCCTCGTCT | 116 | |
| UGT1 | F:CTCTGTATGCCGATCAAGC | 127 |
| R:GATCCGACAACAAATTACGAA | 116 | |
| UGT2 | F:AACTCCATACTCTTATGACGGATGAGC | 115 |
| R:AGGATATGTTGGCTGTAGGTGACTTC | 122 | |
| UGT3 | F:GAAGTGCTTGCAGGCTTATCTGTAGTA | 93 |
| R:AATGGCGGTATAGACGAAGACATGAC | 126 | |
| Vg | F:GTGGTGTTCACGCACTACTCG | 123 |
| R:CCATCGGCACACTCTCTTTAGG | 149 | |
| Rps15 | F:GTTGGCTCTCTATTGTAGGTATC R:AGGCTTGTATGTGACTGA | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, G.; Zhou, Y.; Han, H.; Liu, Z.; Lai, Y.; Gao, S. Comparative Analysis of Transcriptome Data of Wings from Different Developmental Stages of the Gynaephora qinghaiensis. Int. J. Mol. Sci. 2025, 26, 3562. https://doi.org/10.3390/ijms26083562
Kou G, Zhou Y, Han H, Liu Z, Lai Y, Gao S. Comparative Analysis of Transcriptome Data of Wings from Different Developmental Stages of the Gynaephora qinghaiensis. International Journal of Molecular Sciences. 2025; 26(8):3562. https://doi.org/10.3390/ijms26083562
Chicago/Turabian StyleKou, Guixiang, Yuantao Zhou, Haibing Han, Zhanling Liu, Youpeng Lai, and Shujing Gao. 2025. "Comparative Analysis of Transcriptome Data of Wings from Different Developmental Stages of the Gynaephora qinghaiensis" International Journal of Molecular Sciences 26, no. 8: 3562. https://doi.org/10.3390/ijms26083562
APA StyleKou, G., Zhou, Y., Han, H., Liu, Z., Lai, Y., & Gao, S. (2025). Comparative Analysis of Transcriptome Data of Wings from Different Developmental Stages of the Gynaephora qinghaiensis. International Journal of Molecular Sciences, 26(8), 3562. https://doi.org/10.3390/ijms26083562