


Current Research Trends in Glioblastoma: Focus on Receptor Tyrosine Kinases
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
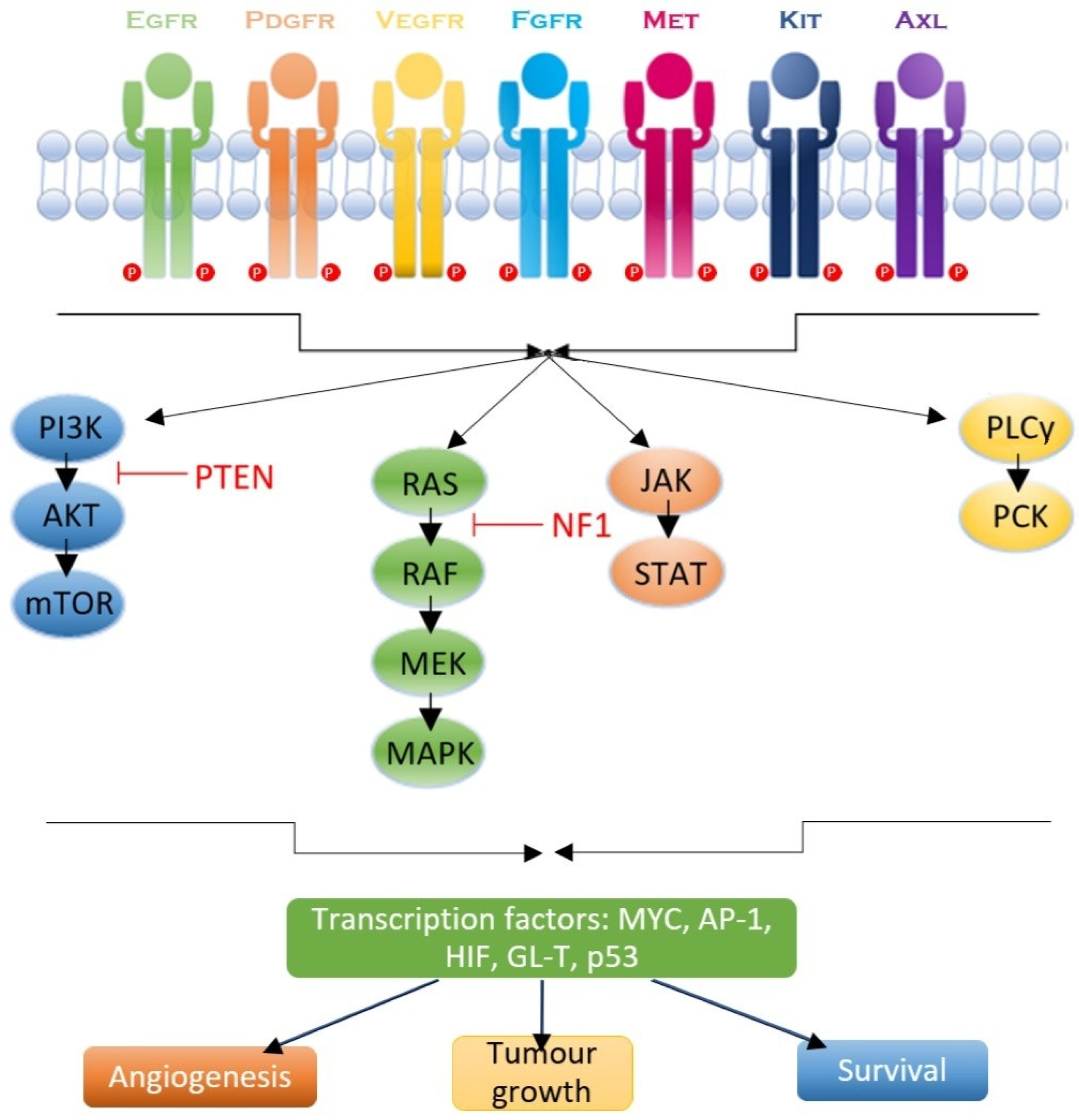
2. RTK Signaling Pathways in GBM
2.1. Epithelial Growth Factor Receptor (EGFR)
2.2. Platelet-Derived Growth Factor Receptor (PDGFR)
2.3. Vascular Endothelial Growth Factor Receptor (VEGFR)
2.4. c-MET and the Hepatocyte Growth Factor (HGF) Pathway
2.5. AXL Receptor
2.6. RTK Downstream Signaling Pathways
2.6.1. RAS/MAPK/ERK Pathway
2.6.2. JAK/STAT Pathway
2.6.3. PI3K/AKT Pathway
2.6.4. PLC/PKC Pathway
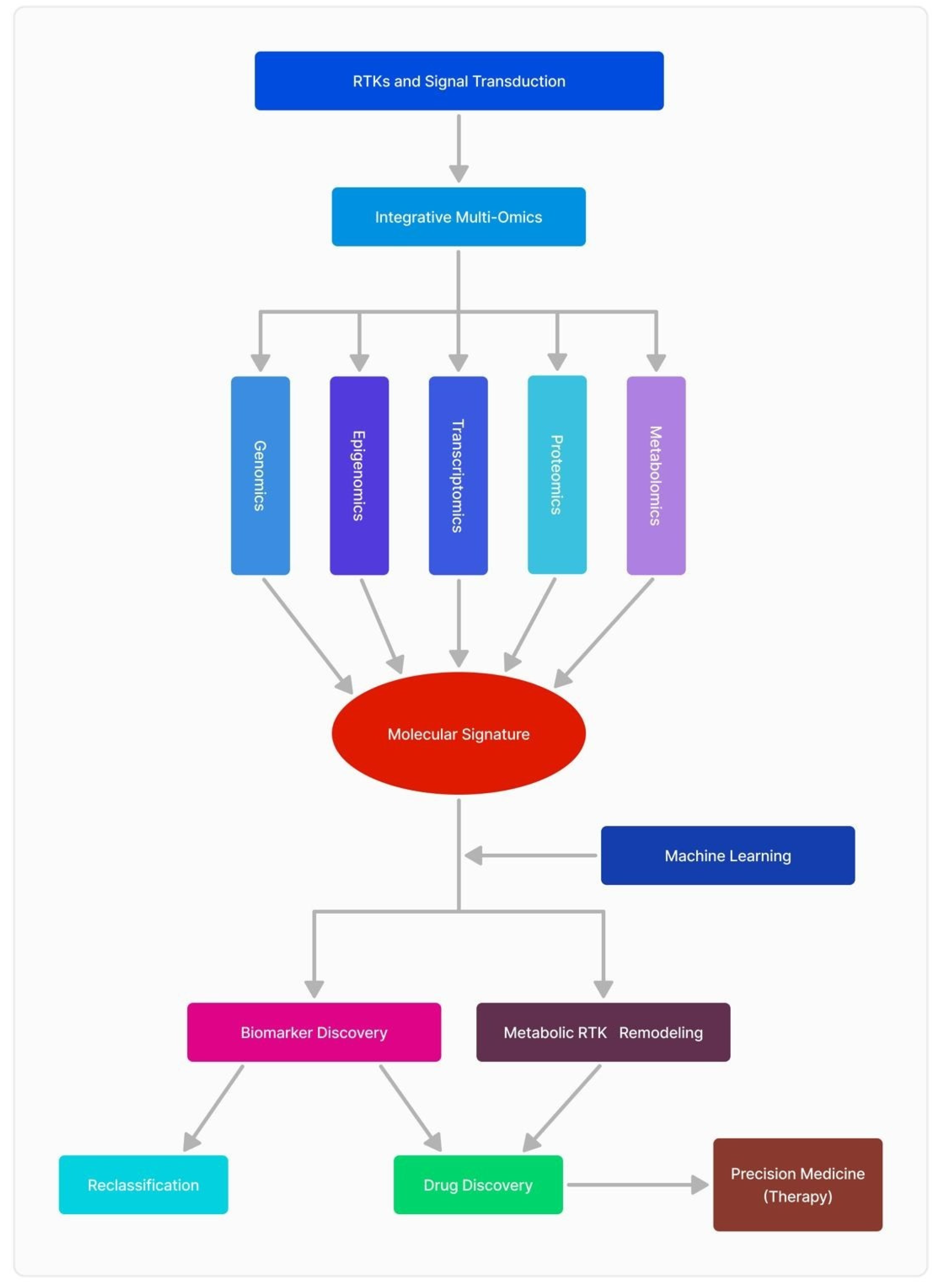
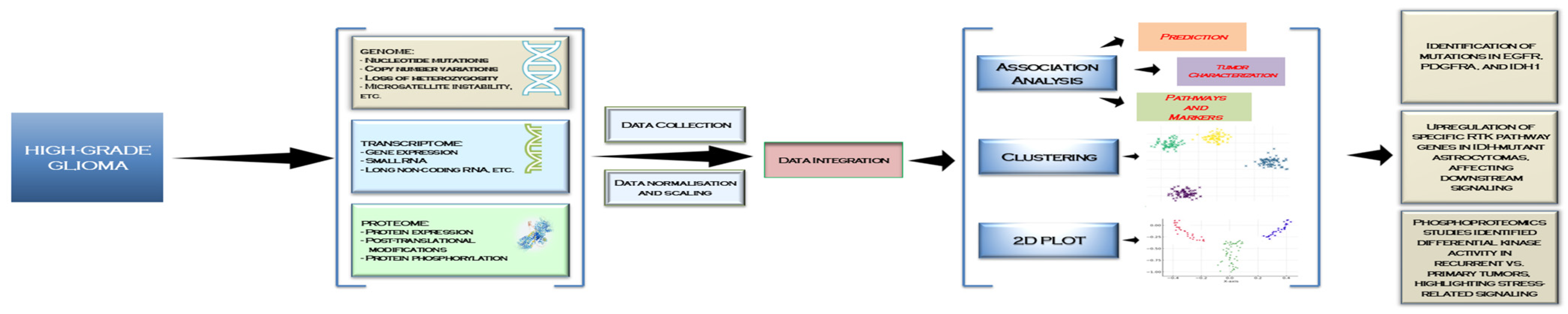
3. Recent Advances in RTK -Omics Approaches and Their Impact on Diagnosis and Therapeutic Targets in GBM
3.1. Genomics
3.2. Transcriptomics
3.3. Proteomics
3.4. Metabolomics
3.5. The Interplay of Multi-Omics Sciences and Clinical Data
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of glioblastoma multiforme–literature review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95. [Google Scholar]
- Rodriguez, S.M.B.; Staicu, G.-A.; Sevastre, A.-S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma stem cells—Useful tools in the battle against cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef]
- Sevastre, A.-S.; Costachi, A.; Tataranu, L.G.; Brandusa, C.; Artene, S.A.; Stovicek, O.; Alexandru, O.; Danoiu, S.; Sfredel, V.; Dricu, A. Glioblastoma pharmacotherapy: A multifaceted perspective of conventional and emerging treatments. Exp. Ther. Med. 2021, 22, 1408. [Google Scholar] [CrossRef]
- Koukourakis, G.V.; Kouloulias, V.; Zacharias, G.; Papadimitriou, C.; Pantelakos, P.; Maravelis, G.; Fotineas, A.; Beli, I.; Chaldeopoulos, D.; Kouvaris, J. Temozolomide with radiation therapy in high grade brain gliomas: Pharmaceuticals considerations and efficacy; a review article. Molecules 2009, 14, 1561–1577. [Google Scholar] [CrossRef]
- Daianu, O.; Georgescu, A.M.; Ciurea, M.; Alexandru, O.; Artene, S.-A.; Tataranu, L.G.; Purcaru, O.S.; Tache, D.E.; Danciulescu, M.M.; Taisescu, O.; et al. Temozolomide and targeted therapy against epidermal growth factor receptor in glioma. Int. J. Clin. Exp. Med. 2016, 9, 15249–15261. [Google Scholar]
- Kubben, P.L.; ter Meulen, K.J.; Schijns, O.E.; ter Laak-Poort, M.P.; van Overbeeke, J.J.; van Santbrink, H. Intraoperative MRI-guided resection of glioblastoma multiforme: A systematic review. Lancet Oncol. 2011, 12, 1062–1070. [Google Scholar] [CrossRef]
- Alexandru, O.; Purcaru, S.O.; Tataranu, L.G.; Lucan, L.; Castro, J.; Folcuţi, C.; Artene, S.-A.; Tuţă, C.; Dricu, A. The influence of EGFR inactivation on the radiation response in high grade glioma. Int. J. Mol. Sci. 2018, 19, 229. [Google Scholar] [CrossRef]
- Alexandru, O.; Sevastre, A.-S.; Castro, J.; Artene, S.-A.; Tache, D.E.; Purcaru, O.S.; Sfredel, V.; Tataranu, L.G.; Dricu, A. Platelet-derived growth factor receptor and ionizing radiation in high grade glioma cell lines. Int. J. Mol. Sci. 2019, 20, 4663. [Google Scholar] [CrossRef]
- Artene, S.-A.; Turcu-Stiolica, A.; Hartley, R.; Ciurea, M.E.; Daianu, O.; Brindusa, C.; Alexandru, O.; Tataranu, L.G.; Purcaru, S.O.; Dricu, A. Dendritic cell immunotherapy versus bevacizumab plus irinotecan in recurrent malignant glioma patients: A survival gain analysis. Onco Targets Ther. 2016, 9, 6669–6677. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-approved therapies for high-grade malignant gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Brada, M.; Stenning, S.; Gabe, R.; Thompson, L.C.; Levy, D.; Rampling, R.; Erridge, S.; Saran, F.; Gattamaneni, R.; Hopkins, K. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J. Clin. Oncol. 2010, 28, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma immunotherapy: A systematic review of the present strategies and prospects for advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, X.; Sander, M.; Zhang, H.; Yan, G.; Lin, Y. Oncolytic viro-immunotherapy: An emerging option in the treatment of gliomas. Front. Immunol. 2021, 12, 721830. [Google Scholar] [CrossRef]
- Serban, F.; Artene, S.-A.; Georgescu, A.M.; Purcaru, S.O.; Tache, D.E.; Alexandru, O.; Dricu, A. Epidermal growth factor, latrophilin, and seven transmembrane domain-containing protein 1 marker, a novel angiogenesis marker. Onco Targets Ther. 2015, 8, 3767–3774. [Google Scholar]
- Caccese, M.; Desideri, I.; Villani, V.; Simonelli, M.; Buglione, M.; Chiesa, S.; Franceschi, E.; Gaviani, P.; Stasi, I.; Caserta, C. REGOMA-OSS: A large, Italian, multicenter, prospective, observational study evaluating the efficacy and safety of regorafenib in patients with recurrent glioblastoma. ESMO Open 2024, 9, 102943. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Pallini, R.; D’Alessandris, Q.G.; Levi, A.; Falchetti, M.L. Regorafenib and glioblastoma: A literature review of preclinical studies, molecular mechanisms and clinical effectiveness. Expert Rev. Mol. Med. 2024, 26, e5. [Google Scholar] [CrossRef]
- Kaina, B. Temozolomide, procarbazine and nitrosoureas in the therapy of malignant gliomas: Update of mechanisms, drug resistance and therapeutic implications. J. Clin. Med. 2023, 12, 7442. [Google Scholar] [CrossRef]
- Muñoz-Mármol, A.M.; Meléndez, B.; Hernandez, A.; Sanz, C.; Domenech, M.; Arpí-Llucia, O.; Gut, M.; Esteve, A.; Esteve-Codina, A.; Parra, G. Multikinase Treatment of Glioblastoma: Evaluating the Rationale for Regorafenib. Cancers 2025, 17, 375. [Google Scholar] [CrossRef]
- Hoosemans, L.; Vooijs, M.; Hoeben, A. Opportunities and challenges of small molecule inhibitors in glioblastoma treatment: Lessons learned from clinical trials. Cancers 2024, 16, 3021. [Google Scholar] [CrossRef]
- Carapancea, M.; Alexandru, O.; Fetea, A.S.; Dragutescu, L.; Castro, J.; Georgescu, A.; Popa-Wagner, A.; Bäcklund, M.L.; Lewensohn, R.; Dricu, A. Growth factor receptors signaling in glioblastoma cells: Therapeutic implications. J. Neuro-Oncol. 2009, 92, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.-S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An overview of EGFR mechanisms and their implications in targeted therapies for glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. [Google Scholar] [CrossRef] [PubMed]
- Sevastre, A.-S.; Buzatu, I.M.; Baloi, C.; Oprita, A.; Dragoi, A.; Tataranu, L.G.; Alexandru, O.; Tudorache, S.; Dricu, A. ELTD1—An emerging silent actor in cancer drama play. Int. J. Mol. Sci. 2021, 22, 5151. [Google Scholar] [CrossRef] [PubMed]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef]
- Onciul, R.; Brehar, F.-M.; Toader, C.; Covache-Busuioc, R.-A.; Glavan, L.-A.; Bratu, B.-G.; Costin, H.P.; Dumitrascu, D.-I.; Serban, M.; Ciurea, A.V. Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas. Curr. Issues Mol. Biol. 2024, 46, 2402–2443. [Google Scholar] [CrossRef]
- Deleanu, R.; Ceafalan, L.C.; Dricu, A. Transcriptomic crosstalk between gliomas and telencephalic neural stem and progenitor cells for defining heterogeneity and targeted signaling pathways. Int. J. Mol. Sci. 2021, 22, 13211. [Google Scholar] [CrossRef]
- Carrasco-García, E.; Saceda, M.; Martínez-Lacaci, I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014, 3, 199–235. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Khan, S.; Zhang, C.; Gao, F.; Sen, S.; Wasylishen, A.R.; Zhao, Y.; Lozano, G.; Koul, D. EGFR suppresses p53 function by promoting p53 binding to DNA-PKcs: A noncanonical regulatory axis between EGFR and wild-type p53 in glioblastoma. Neuro-Oncol. 2022, 24, 1712–1725. [Google Scholar] [CrossRef]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 signaling and its inhibition in modulating tumor invasion: Experimental evidence in different metastatic cancer models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [PubMed]
- Repici, A.; Ardizzone, A.; De Luca, F.; Colarossi, L.; Prestifilippo, A.; Pizzino, G.; Paterniti, I.; Esposito, E.; Capra, A.P. Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma. Cells 2024, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF receptors in glioma. Upsala J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef]
- Cenciarelli, C.; Marei, H.E.; Felsani, A.; Casalbore, P.; Sica, G.; Puglisi, M.A.; Cameron, A.J.; Olivi, A.; Mangiola, A. PDGFRα depletion attenuates glioblastoma stem cells features by modulation of STAT3, RB1 and multiple oncogenic signals. Oncotarget 2016, 7, 53047. [Google Scholar] [CrossRef]
- Heldin, C.-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef]
- Caporarello, N.; D’Angeli, F.; Cambria, M.T.; Candido, S.; Giallongo, C.; Salmeri, M.; Lombardo, C.; Longo, A.; Giurdanella, G.; Anfuso, C.D. Pericytes in microvessels: From “mural” function to brain and retina regeneration. Int. J. Mol. Sci. 2019, 20, 6351. [Google Scholar] [CrossRef]
- Andersson, P. Mechanisms of Tumor Microenvironment in Promoting Metastasis; Karolinska Institutet: Stockholm, Sweden, 2016. [Google Scholar]
- Balaziova, E.; Vymola, P.; Hrabal, P.; Mateu, R.; Zubal, M.; Tomas, R.; Netuka, D.; Kramar, F.; Zemanova, Z.; Svobodova, K. Fibroblast activation protein expressing mesenchymal cells promote glioblastoma angiogenesis. Cancers 2021, 13, 3304. [Google Scholar] [CrossRef]
- di Tomaso, E.; London, N.; Fuja, D.; Logie, J.; Tyrrell, J.A.; Kamoun, W.; Munn, L.L.; Jain, R.K. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS ONE 2009, 4, e5123. [Google Scholar] [CrossRef]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.P. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Jenny, B.; Harrison, J.; Baetens, D.; Tille, J.C.; Burkhardt, K.; Mottaz, H.; Kiss, J.Z.; Dietrich, P.Y.; De Tribolet, N.; Pizzolato, G. Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J. Pathol. 2006, 209, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, E.; Puerto, I.; Medina, M.Á. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Ghisai, S.A.; Barin, N.; van Hijfte, L.; Verhagen, K.; de Wit, M.; van den Bent, M.J.; Hoogstrate, Y.; French, P.J. Transcriptomic analysis of EGFR co-expression and activation in glioblastoma reveals associations with its ligands. Neuro-Oncol. Adv. 2025, 7, vdae229. [Google Scholar] [CrossRef]
- Huang, P.H.; Miraldi, E.R.; Xu, A.M.; Kundukulam, V.A.; Del Rosario, A.M.; Flynn, R.A.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Mol. BioSystems 2010, 6, 1227–1237. [Google Scholar] [CrossRef]
- Korbecki, J.; Bosiacki, M.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Biosynthesis and significance of fatty acids, glycerophospholipids, and triacylglycerol in the processes of glioblastoma tumorigenesis. Cancers 2023, 15, 2183. [Google Scholar] [CrossRef]
- Yalaza, C.; Ak, H.; Cagli, M.S.; Ozgiray, E.; Atay, S.; Aydin, H.H. R132H mutation in IDH1 gene is associated with increased tumor HIF1-alpha and serum VEGF levels in primary glioblastoma multiforme. Ann. Clin. Lab. Sci. 2017, 47, 362–364. [Google Scholar]
- Zheng, S.; Tao, W. Identification of novel transcriptome signature as a potential prognostic biomarker for anti-angiogenic therapy in glioblastoma multiforme. Cancers 2021, 13, 1013. [Google Scholar] [CrossRef]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR axis revisited: Implications for cancer therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, P.; Fatteh, M.; Kamson, D.O.; Balan, A.; Chang, M.; Tao, J.; Blakeley, J.; Investigators, J.H.M.T.B.; Canzoniero, J.; Grossman, S.A. Druggable genomic landscapes of high-grade gliomas. Front. Med. 2023, 10, 1254955. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Yuan, Y.; Lin, Y.; Wang, Y.-X.; Zhou, J.; Gai, Q.-J.; Zhang, L.; Mao, M.; Yao, X.-X.; Qin, Y. ERBB3, IGF1R, and TGFBR2 expression correlate with PDGFR expression in glioblastoma and participate in PDGFR inhibitor resistance of glioblastoma cells. Am. J. Cancer Res. 2018, 8, 792. [Google Scholar] [PubMed]
- Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-López, L.; Tristante, E.; Carballo-Santana, M.; García-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, Á.; Saceda, M. PDGFR and IGF-1R inhibitors induce a G2/M arrest and subsequent cell death in human glioblastoma cell lines. Cells 2018, 7, 131. [Google Scholar] [CrossRef]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Huang, B.; Park, M. Aberrant MET receptor tyrosine kinase signaling in glioblastoma: Targeted therapy and future directions. Cells 2024, 13, 218. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, T.T.; Shang, E.; Mela, A.; Humala, N.; Mahajan, A.; Zhao, J.; Shu, C.; Torrini, C.; Sanchez-Quintero, M.J. MET inhibition elicits PGC1α-dependent metabolic reprogramming in glioblastoma. Cancer Res. 2020, 80, 30–43. [Google Scholar] [CrossRef]
- Cheng, P.; Phillips, E.; Kim, S.-H.; Taylor, D.; Hielscher, T.; Puccio, L.; Hjelmeland, A.B.; Lichter, P.; Nakano, I.; Goidts, V. Kinome-wide shRNA screen identifies the receptor tyrosine kinase AXL as a key regulator for mesenchymal glioblastoma stem-like cells. Stem Cell Rep. 2015, 4, 899–913. [Google Scholar] [CrossRef]
- Onken, J.; Vajkoczy, P.; Torka, R.; Hempt, C.; Patsouris, V.; Heppner, F.L.; Radke, J. Phospho-AXL is widely expressed in glioblastoma and associated with significant shorter overall survival. Oncotarget 2017, 8, 50403. [Google Scholar] [CrossRef]
- Sadahiro, H.; Kang, K.-D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; deCarvalho, A.C.; Mikkelsen, T.; Lehman, N.L.; Calvert, V.; Espina, V.; Liotta, L.A.; Petricoin III, E.F. Glioblastoma cell enrichment is critical for analysis of phosphorylated drug targets and proteomic–genomic correlations. Cancer Res. 2014, 74, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhu, S.; Zhang, Y.; Wang, J. Interplay of VEGFa and MMP2 regulates invasion of glioblastoma. Tumor Biol. 2014, 35, 11879–11885. [Google Scholar] [CrossRef]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A. EMT-and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef]
- Burel-Vandenbos, F.; Ngo-Mai, M.; Dadone, B.; Di Mauro, I.; Gimet, S.; Saada-Bouzid, E.; Bourg, V.; Almairac, F.; Fontaine, D.; Virolle, T. MET immunolabelling is a useful predictive tool for MET gene amplification in glioblastoma. Neuropathol. Appl. Neurobiol. 2017, 43, 252–266. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A. Axl and growth arrest–specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef]
- Kim, H.I.; Lee, S.J.; Choi, Y.-J.; Kim, M.J.; Kim, T.Y.; Ko, S.-G. Quercetin induces apoptosis in glioblastoma cells by suppressing Axl/IL-6/STAT3 signaling pathway. Am. J. Chin. Med. 2021, 49, 767–784. [Google Scholar] [CrossRef]
- Oprita, A.; Baloi, S.-C.; Staicu, G.-A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.-S. Updated insights on EGFR signaling pathways in glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Allahverdi, A.; Arefian, E.; Soleimani, M.; Ai, J.; Yousefi-Ahmadipour, A.; Babaei, A.; Islam, M.S.; Ebrahimi-Barough, S. Involvement of EGFR, ERK-1, 2 and AKT-1, 2 activity on human glioma cell growth. Asian Pac. J. Cancer Prev. 2020, 21, 3469. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The role and therapeutic targeting of JAK/STAT signaling in glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef]
- Luwor, R.B.; Stylli, S.S.; Kaye, A.H. The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 907–911. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440. [Google Scholar] [CrossRef]
- Margolis, B.; Rhee, S.; Felder, S.; Mervic, M.; Lyall, R.; Levitzki, A.; Ullrich, A.; Zilberstein, A.; Schlessinger, J. EGF induces tyrosine phosphorylation of phospholipase C-II: A potential mechanism for EGF receptor signaling. Cell 1989, 57, 1101–1107. [Google Scholar] [CrossRef]
- Reyland, M.E. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front. Biosci. (Landmark Ed.) 2009, 14, 2386. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Fan, F.; Zhang, H.; Dai, Z.; Zhang, Y.; Xia, Z.; Cao, H.; Yang, K.; Hu, S.; Guo, Y.; Ding, F. A comprehensive prognostic signature for glioblastoma patients based on transcriptomics and single cell sequencing. Cell. Oncol. 2021, 44, 917–935. [Google Scholar] [CrossRef]
- Vastrad, B.; Vastrad, C.; Godavarthi, A.; Chandrashekar, R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med. Oncol. 2017, 34, 182. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Ashizawa, T.; Komiyama, M.; Miyata, H.; Oshita, C.; Omiya, M.; Iizuka, A.; Kume, A.; Sugino, T.; Hayashi, N. YKL-40 downregulation is a key factor to overcome temozolomide resistance in a glioblastoma cell line. Oncol. Rep. 2014, 32, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Nejad, R.; Karabork, M.; Ekinci, C.; Solaroglu, I.; Aldape, K.D.; Zadeh, G. Sox2: Regulation of expression and contribution to brain tumors. CNS Oncol. 2016, 5, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wei, C.; Wang, C.; Li, F.; Wang, Z.; Xiong, J.; Zhou, Y.; Li, S.; Liu, X.; Yang, G. TIMP1/CHI3L1 facilitates glioma progression and immunosuppression via NF-κB activation. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167041. [Google Scholar] [CrossRef]
- Shapovalov, V.; Kopanitsa, L.; Pruteanu, L.-L.; Ladds, G.; Bailey, D.S. Transcriptomics-based phenotypic screening supports drug discovery in human glioblastoma cells. Cancers 2021, 13, 3780. [Google Scholar] [CrossRef]
- Atanaki, F.F.; Mirsadeghi, L.; Manesh, M.R.; Kavousi, K. Integrative analysis of single-cell transcriptomic and multilayer signaling networks in glioma reveal tumor progression stage. Front. Genet. 2024, 15, 1446903. [Google Scholar] [CrossRef]
- Zhai, X.-H.; Xiao, J.; Yu, J.-K.; Sun, H.; Zheng, S. Novel sphingomyelin biomarkers for brain glioma and associated regulation research on the PI3K/Akt signaling pathway. Oncol. Lett. 2019, 18, 6207–6213. [Google Scholar] [CrossRef]
- Pooladi, M.; Abad, S.; Hashemi, M. Proteomics analysis of human brain glial cell proteome by 2D gel. Indian J. Cancer 2014, 51, 159–162. [Google Scholar]
- Jayaram, S.; Gupta, M.K.; Raju, R.; Gautam, P.; Sirdeshmukh, R. Multi-omics data integration and mapping of altered kinases to pathways reveal gonadotropin hormone signaling in glioblastoma. OMICS 2016, 20, 736–746. [Google Scholar] [CrossRef]
- Jaroch, K.; Modrakowska, P.; Bojko, B. Glioblastoma metabolomics—In vitro studies. Metabolites 2021, 11, 315. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [PubMed]
- Hesse, F.; Wright, A.J.; Somai, V.; Bulat, F.; Kreis, F.; Brindle, K.M. Imaging glioblastoma response to radiotherapy using 2H magnetic resonance spectroscopy measurements of fumarate metabolism. Cancer Res. 2022, 82, 3622–3633. [Google Scholar] [CrossRef] [PubMed]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, K.; Bock, H.C.; Helms, G.; Jung, K.; Wrede, A.; Buhk, J.-H.; Giese, A.; Frahm, J.; Strik, H.; Dechent, P. Untreated glioblastoma multiforme: Increased myo-inositol and glutamine levels in the contralateral cerebral hemisphere at proton MR spectroscopy. Radiology 2009, 253, 805–812. [Google Scholar] [CrossRef]
- Fontanilles, M.; Heisbourg, J.-D.; Daban, A.; Di Fiore, F.; Pépin, L.-F.; Marguet, F.; Langlois, O.; Alexandru, C.; Tennevet, I.; Ducatez, F. Metabolic remodeling in glioblastoma: A longitudinal multi-omics study. Acta Neuropathol. Commun. 2024, 12, 162. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Seifert, C.; Balz, E.; Herzog, S.; Korolev, A.; Gaßmann, S.; Paland, H.; Fink, M.A.; Grube, M.; Marx, S.; Jedlitschky, G. PIM1 inhibition affects glioblastoma stem cell behavior and kills glioblastoma stem-like cells. Int. J. Mol. Sci. 2021, 22, 11126. [Google Scholar] [CrossRef]
- Martínez, A.H.; Madurga, R.; García-Romero, N.; Ayuso-Sacido, Á. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 2022, 527, 66–79. [Google Scholar] [CrossRef]
- Chakraborty, S.; Sharma, G.; Karmakar, S.; Banerjee, S. Multi-OMICS approaches in cancer biology: New era in cancer therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167120. [Google Scholar] [CrossRef]
- Liu, J.; Cao, S.; Imbach, K.J.; Gritsenko, M.A.; Lih, T.-S.M.; Kyle, J.E.; Yaron-Barir, T.M.; Binder, Z.A.; Li, Y.; Strunilin, I. Multi-scale signaling and tumor evolution in high-grade gliomas. Cancer Cell 2024, 42, 1217–1238.e19. [Google Scholar] [CrossRef]
- Alom, M.W.; Jibon, M.D.K.; Faruqe, M.O.; Rahman, M.S.; Akter, F.; Ali, A.; Rahman, M.M. Integrated Gene Expression Data-Driven Identification of Molecular Signatures, Prognostic Biomarkers, and Drug Targets for Glioblastoma. BioMed Res. Int. 2024, 2024, 6810200. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Receptor/RTK | Mutation Frequency | Functional Consequences | Recent Findings |
|---|---|---|---|
| EGFR | 40–60% of GBM cases show amplification or mutation | Activates the PI3K/AKT, RAS/RAF/MEK, and JAK/STAT pathways, driving proliferation and therapy resistance | EGFR suppresses p53 by promoting DNA-PKcs binding, leading to resistance [28] |
| PDGFR | Amplifications in the proneural subtype of GBM | Promotes angiogenesis, tumor cell migration, and ECM remodeling | PDGFRβ+ pericytes attract macrophages, facilitating immune evasion [29] |
| VEGFR | VEGFR1 and VEGFR2 overexpressed in 60–80% of GBM samples | Drives hypoxia-induced vascular proliferation and therapy resistance | Tumor-educated platelets show increased VEGFR1 and VEGFR2 expression, correlating with disease progression [30] |
| MET | Amplifications in 30% of GBM, exon 14 skipping mutations | Enhances migration, invasion, and MAPK pathway activation | METΔ7–8 mutations and PTPRZ1-MET fusions increase MAPK signaling and drive aggressive phenotypes [31] |
| AXL | Overexpressed in GBM, correlating with poor prognosis | Enhances immune evasion, therapy resistance, and invasion | AXL activation in glutamine-rich environments increases micropinocytosis, aiding GBM survival [32] |
| NF1 | Mutated in 18% of GBM cases | Loss of function mutations disrupt RAS regulation, leading to unchecked proliferation | NF1 mutations associated with aggressive GBM subtypes and poor prognosis [33]. |
| RTK | Genomics | Transcriptomics | Proteomics | Metabolomics |
|---|---|---|---|---|
| EGFR | EGFR amplifications and EGFRvIII mutations drive tumor aggressiveness. PIK3CA mutations cause disruption in the PI3K pathway, contributing to recurrence [45]. | Transcriptomic analysis identifies betacellulin (BTC) and epiregulin (EREG) as key regulators of EGFR in GBM, influencing its activation and mutation sensitivity, refining EGFR-targeted therapies [46]. | EGFR overexpression and PTEN downregulation promote tumor growth and resistance. Phosphorylation (Y1068, Y1173) and PI3K/AKT signaling enhance cell survival and migration [47]. | Activation of EGFR leads to reprogramming of lipid metabolism and glycolysis, enhancing energy production and tumor survival. Studies show elevated glycerophospholipids [48]. |
| VEGFR | IDH1 R132H mutations in GBM are linked to increased HIF-1 alpha and VEGF levels, suggesting a role in tumor progression via hypoxia pathways [49]. | SOCS3-VEGFA-TEK transcriptomic signature for GBM prognosis, linking SOCS3 expression to VEGFA-driven neovascularization and response to anti-angiogenic therapy [50]. | VEGFR phosphorylation at key sites (Y951, Y1175) activates angiogenesis and cell survival pathways. Interactions with neuropilin enhance signaling [51]. | VEGFR signaling promotes glycolysis, fatty acid oxidation, and mitochondrial biogenesis, supporting tumor survival under low-oxygen conditions [52]. |
| PDGFR | PDGFR amplifications and mutations in the proneural subtype drive tumor progression by altering extracellular matrix (ECM) remodeling and promoting invasion [53]. | PDGFR is enriched in the proneural subtype of GBM, affecting migration, adhesion, and immune evasion. Altered transcriptional networks support these processes [54]. | PDGFR inhibition with JNJ disrupts phosphorylation, halting GBM growth via mitotic arrest and caspase-dependent apoptosis. Combined IGF-1R/PDGFR blockade enhances therapeutic efficacy [55]. | Metabolic coupling between tumor and stromal cells promotes lactate production and aerobic glycolysis, supporting tumor invasiveness [56]. |
| MET | MET amplifications, exon 14 skipping, and gene fusions (e.g., TPR-MET, PTPRZ1-MET) lead to persistent kinase activity and poor prognosis in GBM [57]. | c-MET inhibition induces significant transcriptomic changes, including increased PGC1α expression regulated by cAMP response elements binding protein, driving oxidative metabolism in GBM [58]. | c-MET inhibition drives mitochondrial fusion and reactive oxygen species production in GBM, revealing a shift in protein expression linked to oxidative metabolism [58]. | c-MET inhibition induces metabolic reprogramming in GBM, enhancing oxidative phosphorylation and fatty acid oxidation, along with increased acyl-carnitines and anaplerosis [58]. |
| AXL | AXL overexpression is associated with epithelial–mesenchymal transition (EMT), enhancing immune evasion and metastasis in GBM [32]. | Transcriptomic analysis and shRNA screening identified AXL as a therapeutic target, highlighting its role in MES GSC survival in GBM [59]. | Through protein expression analysis, P-AXL patterns in GBM were linked to survival, indicating its potential as a therapeutic target [60]. | PROS1/AXL signaling in GSCs triggers metabolic reprogramming, including enhanced oxidative phosphorylation and fatty acid oxidation, supporting GBM growth [61]. |
| HER2 | HER2 overexpression is linked to therapy resistance and aggressive GBM phenotypes, contributing to tumor progression and poor prognosis [45]. | HER2 gene expression changes induced by NK-92/5.28.z therapy, identifying immune response and tumor progression pathways [62]. | Laser capture microdissection–based proteomic analysis improves HER2 detection in glioblastoma, ensuring accurate tumor-specific data for targeted therapy decisions [63]. | HER2-driven metabolic changes in glycolysis, oxidative phosphorylation, and amino acid metabolism influenced by CAR NK cell therapy [62]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcan, E.N.; Duta, C.; Staicu, G.A.; Artene, S.A.; Alexandru, O.; Costachi, A.; Pirvu, A.S.; Tache, D.E.; Stoian, I.; Popescu, S.O.; et al. Current Research Trends in Glioblastoma: Focus on Receptor Tyrosine Kinases. Int. J. Mol. Sci. 2025, 26, 3503. https://doi.org/10.3390/ijms26083503
Barcan EN, Duta C, Staicu GA, Artene SA, Alexandru O, Costachi A, Pirvu AS, Tache DE, Stoian I, Popescu SO, et al. Current Research Trends in Glioblastoma: Focus on Receptor Tyrosine Kinases. International Journal of Molecular Sciences. 2025; 26(8):3503. https://doi.org/10.3390/ijms26083503
Chicago/Turabian StyleBarcan, Edmond Nicolae, Carmen Duta, Georgiana Adeline Staicu, Stefan Alexandru Artene, Oana Alexandru, Alexandra Costachi, Andreea Silvia Pirvu, Daniela Elise Tache, Irina Stoian, Stefana Oana Popescu, and et al. 2025. "Current Research Trends in Glioblastoma: Focus on Receptor Tyrosine Kinases" International Journal of Molecular Sciences 26, no. 8: 3503. https://doi.org/10.3390/ijms26083503
APA StyleBarcan, E. N., Duta, C., Staicu, G. A., Artene, S. A., Alexandru, O., Costachi, A., Pirvu, A. S., Tache, D. E., Stoian, I., Popescu, S. O., Tataranu, L. G., & Dricu, A. (2025). Current Research Trends in Glioblastoma: Focus on Receptor Tyrosine Kinases. International Journal of Molecular Sciences, 26(8), 3503. https://doi.org/10.3390/ijms26083503