
Merging High-Throughput, Amplicon-Based Second and Third Generation Sequencing Data: An Integrative and Modular Data Analysis Framework for Haplotype Prediction and Output Evaluation

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
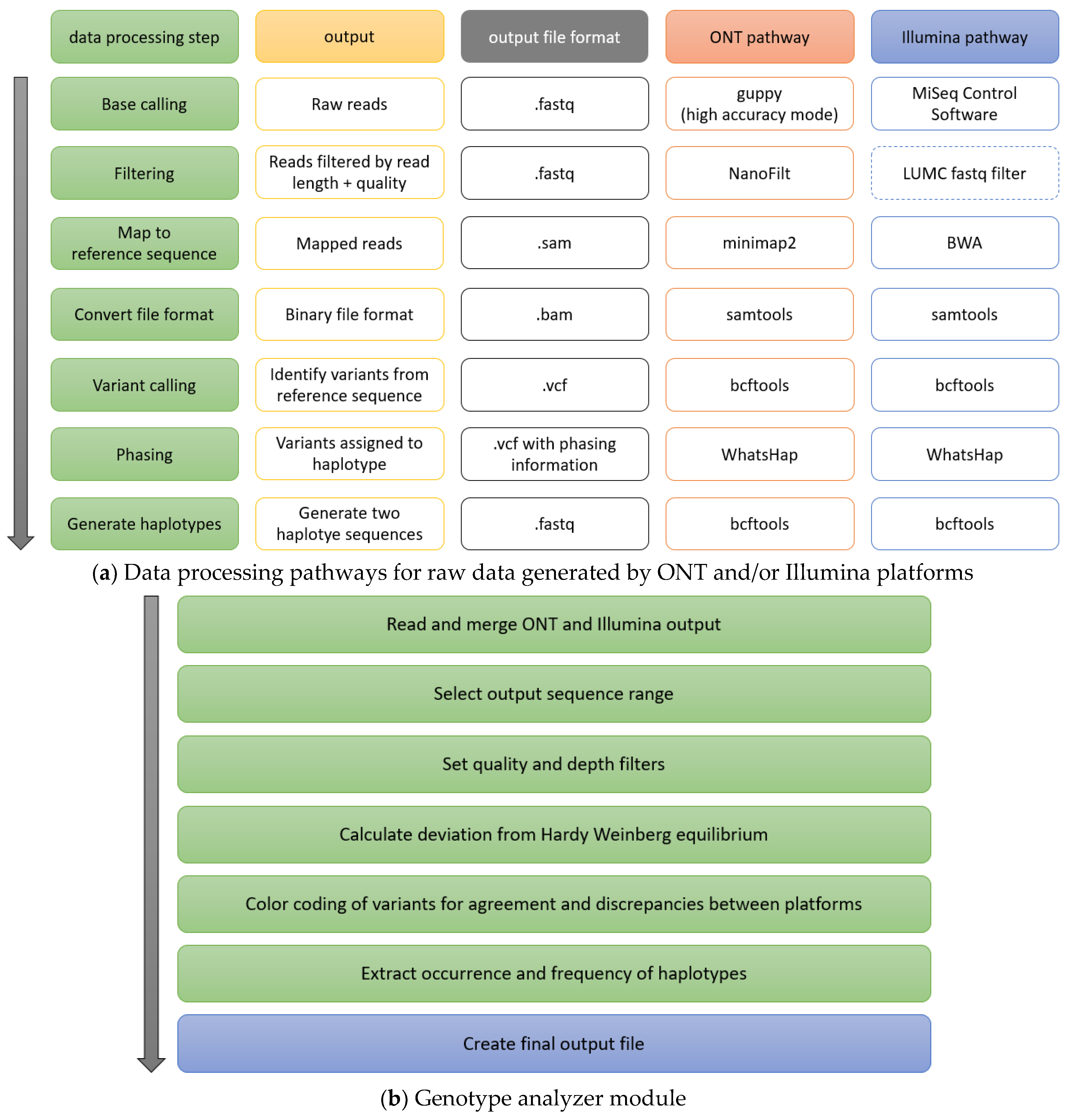
2.1. Framework
2.2. Superordinate Analysis Module
2.3. Sample Collection and Testing
2.4. DNA Extraction and Long-Range PCR
2.5. ONT Sequencing
2.6. Illumina Sequencing
3. Results
3.1. Investigated Genetic Regions
3.2. Variant Output and Phasing
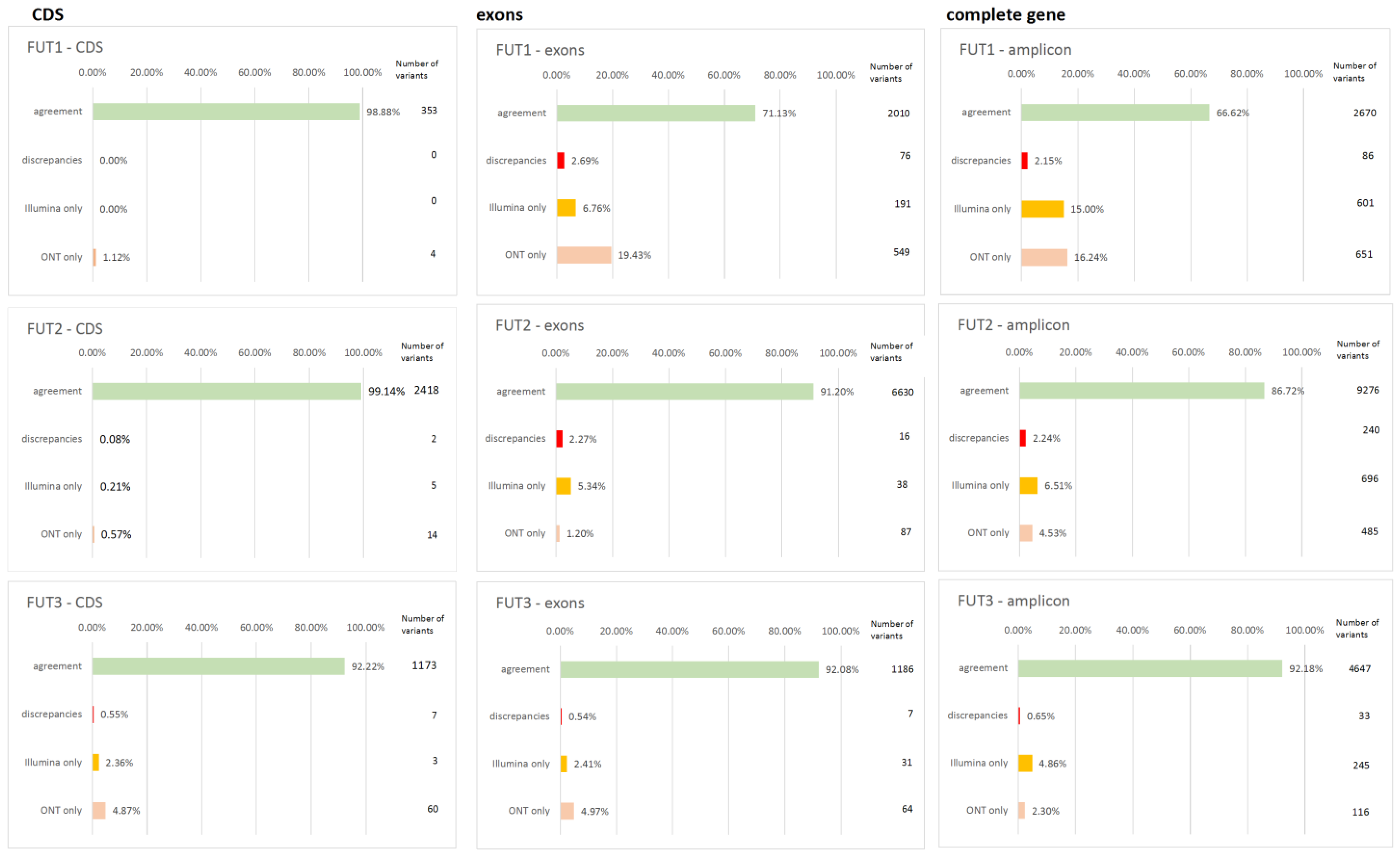
3.3. Agreement Between Second and Third Generation Platforms
3.4. Performance
3.5. Synthetic Data Framework Validation
4. Discussion
4.1. Key Results
4.2. Interpretation
4.3. Performance
4.4. Limitations
4.5. Comparison to Other Tools
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NGS | next-generation sequencing |
| ONT | Oxford Nanopore Technologies |
| WGS | whole genome sequencing |
| PCR | polymerase chain reaction |
References
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Caspar, S.M.; Dubacher, N.; Kopps, A.M.; Meienberg, J.; Henggeler, C.; Matyas, G. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin. Genet. 2018, 93, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long reads: Their purpose and place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Schmidt, R.J.; Ankala, A.; McDonald Gibson, K.; Bowser, M.; Sharma, H.; Duffy, E.; Hegde, M.; Santani, A.; Lebo, M.; et al. Navigating highly homologous genes in a molecular diagnostic setting: A resource for clinical next-generation sequencing. Genet. Med. 2016, 18, 1282–1289. [Google Scholar] [CrossRef]
- Bryc, K.; Patterson, N.; Reich, D. A novel approach to estimating heterozygosity from low-coverage genome sequence. Genetics 2013, 195, 553–561. [Google Scholar] [CrossRef]
- Edge, P.; Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019, 10, 4660. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Naquin, D.; Gorrichon, K.; Jaszczyszyn, Y.; Ouazahrou, R.; Thermes, C.; Hernandez, C. Genomics in the long-read sequencing era. Trends Genet. 2023, 39, 649–671. [Google Scholar] [CrossRef]
- Midha, M.K.; Wu, M.C.; Chiu, K.P. Long-read sequencing in deciphering human genetics to a greater depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef]
- De Coster, W.; Weissensteiner, M.H.; Sedlazeck, F.J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 2021, 22, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Ferguson, S.; McLay, T.; Andrew, R.L.; Bruhl, J.J.; Schwessinger, B.; Borevitz, J.; Jones, A. Species-specific basecallers improve actual accuracy of nanopore sequencing in plants. Plant Methods 2022, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Poore, H.; Tanksley, P.T.; Kweon, H.; Courchesne-Krak, N.S.; Londono-Correa, D.; Mallard, T.T.; Barr, P.; Koellinger, P.D.; Waldman, I.D.; et al. Guidelines for Evaluating the Comparability of Down-Sampled GWAS Summary Statistics. Behav. Genet. 2023, 53, 404–415. [Google Scholar] [CrossRef]
- Corpas, M.; Fatumo, S.; Schneider, R. How not to be a bioinformatician. Source Code Biol. Med. 2012, 7, 3. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Martin, M.; Patterson, M.; Garg, S.; O Fischer, S.; Pisanti, N.; Klau, G.W.; Schöenhuth, A.; Marschall, T. WhatsHap: Fast and accurate read-based phasing. bioRxiv 2016, 085050. [Google Scholar] [CrossRef]
- Leiden University Medical Center. LUMC/fastq-filter. Available online: https://github.com/LUMC/fastq-filter (accessed on 2 October 2023).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Marschall, T.; Pisanti, N.; Van Iersel, L.; Stougie, L.; Klau, G.W.; Schönhuth, A. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 2015, 22, 498–509. [Google Scholar] [CrossRef]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Ball, S.P.; Tongue, N.; Gibaud, A.; Le Pendu, J.; Mollicone, R.; Gérard, G.; Oriol, R. The human chromosome 19 linkage group FUT1 (H), FUT2 (SE), LE, LU, PEPD, C3, APOC2, D19S7 and D19S9. Ann. Hum. Genet. 1991, 55, 225–233. [Google Scholar] [CrossRef]
- Oriol, R.; Candelier, J.J.; Mollicone, R. Molecular genetics of H. Vox Sang. 2000, 78 (Suppl. 2), 105–108. [Google Scholar] [CrossRef] [PubMed]
- Mollicone, R.; Cailleau, A.; Oriol, R. Molecular genetics of H, Se, Lewis and other fucosyltransferase genes. Transfus. Clin. Biol. 1995, 2, 235–242. [Google Scholar] [CrossRef]
- Phan, L.; Jin, Y.; Zhang, H.; Qiang, W.; Shekhtman, E.; Shao, D.; Revoe, D.; Villamarin, R.; Ivanchenko, E.; Kimura, M.; et al. ALFA: Allele Frequency Aggregator. Available online: www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/ (accessed on 6 May 2024).
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Farrell, C.M.; Feldgarden, M.; Fine, A.M.; Funk, K.; et al. Database resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Res. 2023, 51, D29–D38. [Google Scholar] [CrossRef] [PubMed]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Ono, Y.; Hamada, M.; Asai, K. PBSIM3: A simulator for all types of PacBio and ONT long reads. NAR Genom. Bioinform. 2022, 4, lqac092. [Google Scholar] [CrossRef]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Royo, J.L. Hardy Weinberg Equilibrium Disturbances in Case-Control Studies Lead to Non-Conclusive Results. Cell J. 2021, 22, 572–574. [Google Scholar] [CrossRef]
- Chen, B.; Cole, J.W.; Grond-Ginsbach, C. Departure from Hardy Weinberg Equilibrium and Genotyping Error. Front. Genet. 2017, 8, 167. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore adaptive sampling: A tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Ewald, D.R.; Sumner, S.C.J. Blood type biochemistry and human disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 517–535. [Google Scholar] [CrossRef]
- Donta, A.; Gorakshakar, A.C.; Ghosh, K. Divergence in phenotyping and genotyping analysis of the Lewis histo-blood group system. Transfus. Med. 2021, 31, 129–135. [Google Scholar] [CrossRef]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef] [PubMed]
- Betschart, R.O.; Thiéry, A.; Aguilera-Garcia, D.; Zoche, M.; Moch, H.; Twerenbold, R.; Zeller, T.; Blankenberg, S.; Ziegler, A. Comparison of calling pipelines for whole genome sequencing: An empirical study demonstrating the importance of mapping and alignment. Sci. Rep. 2022, 12, 21502. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Ganesamoorthy, D.; Elliott, A.G.; Zhang, H.; Cooper, M.A.; Coin, L.J.M. Streaming algorithms for identification of pathogens and antibiotic resistance potential from real-time MinION(TM) sequencing. Gigascience 2016, 5, 32. [Google Scholar] [CrossRef]
- Tyson, J.R.; O’Neil, N.J.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | |||
|---|---|---|---|
| Definition | FUT1 [bp] | FUT2 [bp] | FUT3 [bp] |
| CDS | 1197 | 1132 | 1185 |
| exons-plus | 5022 | 3614 | 3162 |
| complete gene | 7747 | 10,380 | 8961 |
| CDS | Exons-Plus | Complete Gene | ||||
| ONT | Illumina | ONT | Illumina | ONT | Illumina | |
| FUT1 | ||||||
| # SNVs (all/heterozygous) | 357/308 | 352/304 | 2614/2172 | 2251/1944 | 3377/2780 | 3316/2872 |
| # indels (all/heterozygous) | 0/0 | 0/0 | 21/18 | 17/16 | 38/32 | 29/28 |
| % phased | 100 | 98.7 | 99.1 | 87.7 | 98.5 | 86.7 |
| FUT2 | ||||||
| # SNVs (all/heterozygous) | 2430/1835 | 2425/1830 | 6836/4752 | 7101/5098 | 9873/7110 | 9970/7331 |
| # indels (all/heterozygous) | 4/0 | 0/0 | 46/21 | 82/81 | 128/81 | 242/240 |
| % phased | 100 | 98.1 | 99.5 | 97.0 | 98.6 | 92.8 |
| FUT3 | ||||||
| # SNVs (all/heterozygous) | 1244/660 | 1210/648 | 1257/673 | 1224/662 | 4787/3276 | 4856/3412 |
| # indels (all/heterozygous) | 0/0 | 0/0 | 2/2 | 0/0 | 11/11 | 69/68 |
| % phased | 99.2 | 79.0 | 99.0 | 79.0 | 99.4 | 89.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mink, S.; Attenberger, C.; Busch, Y.; Kiefer, J.; Peter, W.; Cadamuro, J.; Steiert, T.A.; Franke, A.; Gassner, C. Merging High-Throughput, Amplicon-Based Second and Third Generation Sequencing Data: An Integrative and Modular Data Analysis Framework for Haplotype Prediction and Output Evaluation. Int. J. Mol. Sci. 2025, 26, 3443. https://doi.org/10.3390/ijms26073443
Mink S, Attenberger C, Busch Y, Kiefer J, Peter W, Cadamuro J, Steiert TA, Franke A, Gassner C. Merging High-Throughput, Amplicon-Based Second and Third Generation Sequencing Data: An Integrative and Modular Data Analysis Framework for Haplotype Prediction and Output Evaluation. International Journal of Molecular Sciences. 2025; 26(7):3443. https://doi.org/10.3390/ijms26073443
Chicago/Turabian StyleMink, Sylvia, Christian Attenberger, Yannik Busch, Johanna Kiefer, Wolfgang Peter, Janne Cadamuro, Tim A. Steiert, Andre Franke, and Christoph Gassner. 2025. "Merging High-Throughput, Amplicon-Based Second and Third Generation Sequencing Data: An Integrative and Modular Data Analysis Framework for Haplotype Prediction and Output Evaluation" International Journal of Molecular Sciences 26, no. 7: 3443. https://doi.org/10.3390/ijms26073443
APA StyleMink, S., Attenberger, C., Busch, Y., Kiefer, J., Peter, W., Cadamuro, J., Steiert, T. A., Franke, A., & Gassner, C. (2025). Merging High-Throughput, Amplicon-Based Second and Third Generation Sequencing Data: An Integrative and Modular Data Analysis Framework for Haplotype Prediction and Output Evaluation. International Journal of Molecular Sciences, 26(7), 3443. https://doi.org/10.3390/ijms26073443