
Deep Learning-Based Comparative Prediction and Functional Analysis of Intrinsically Disordered Regions in SARS-CoV-2



Abstract

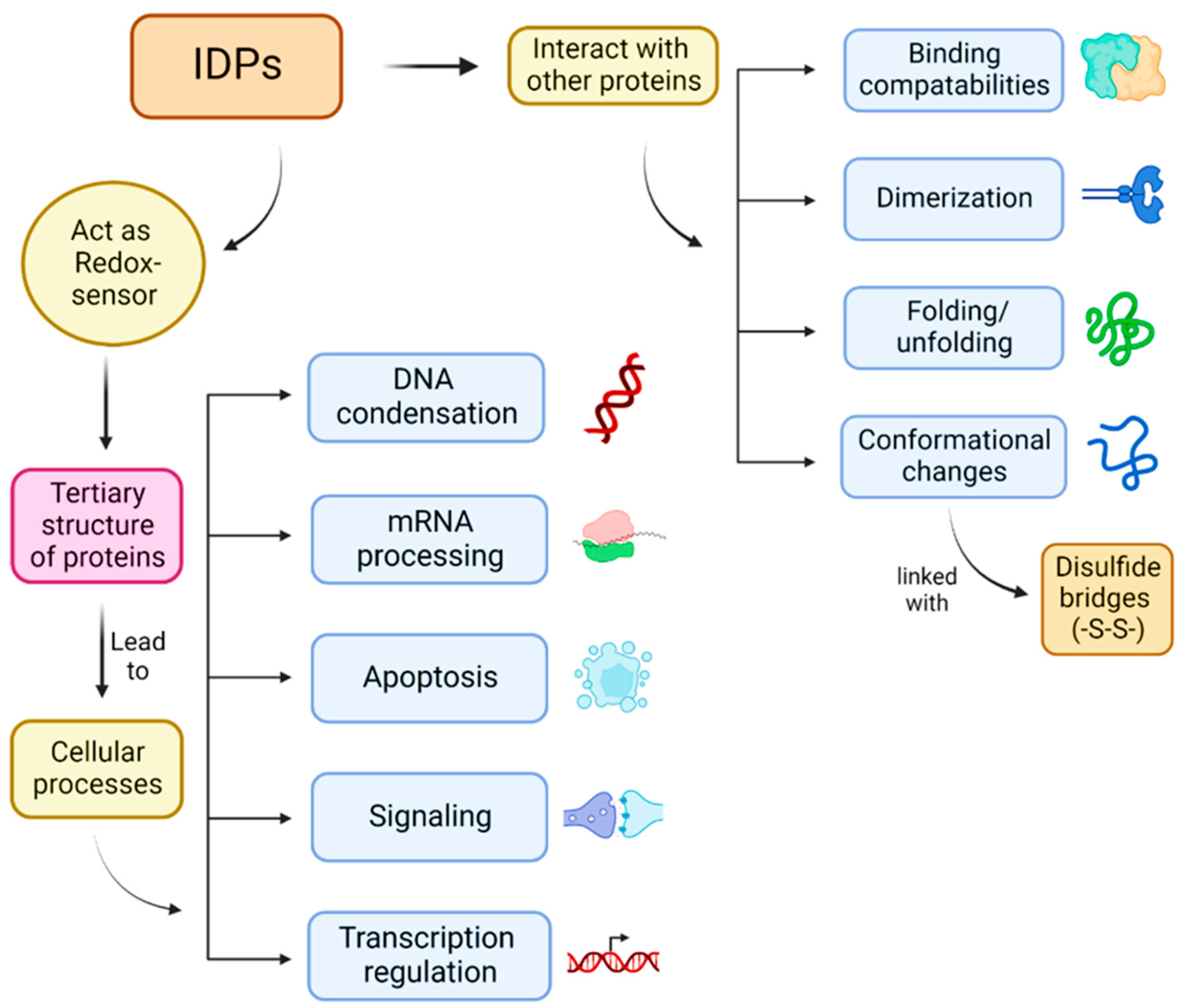
1. Introduction
2. Results and Discussion
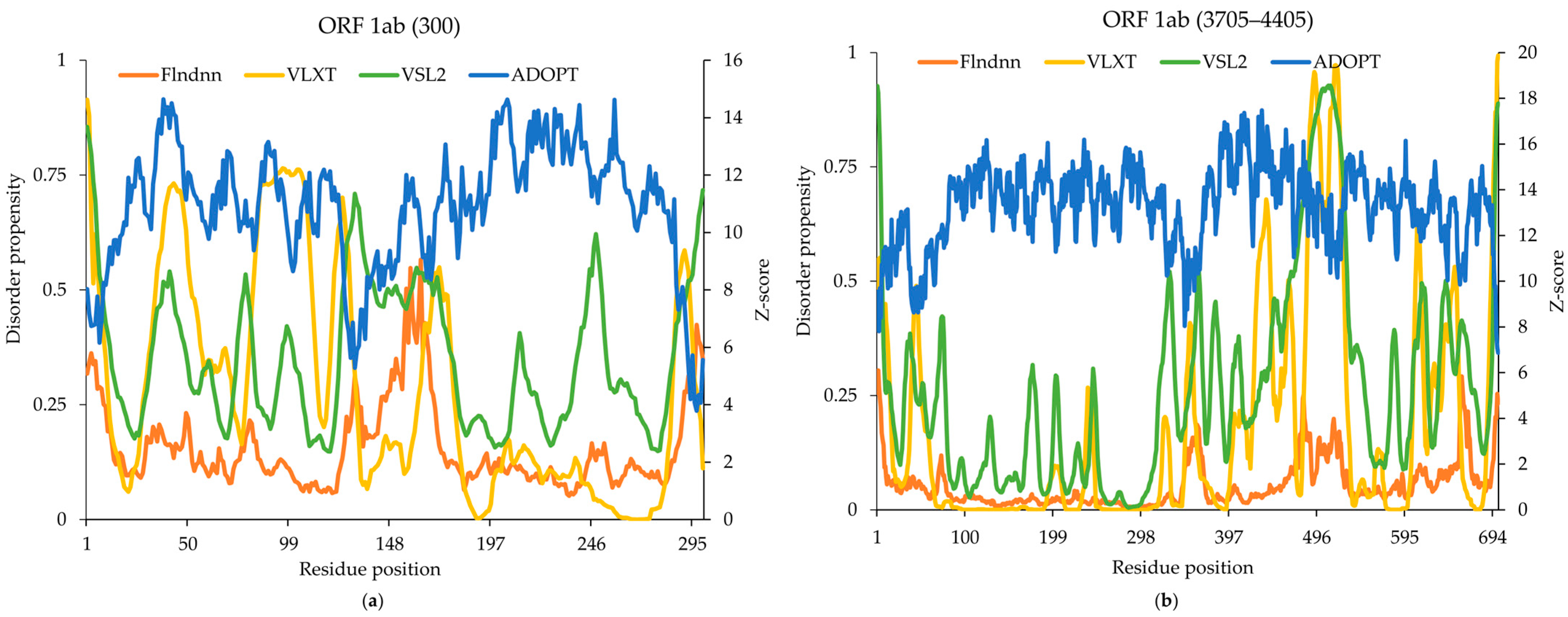
2.1. Replicase Polyprotein 1ab
2.2. NSP1 Flexible Linker Region
2.3. NSP1 Cu(II) Binding Region
2.4. RNA Binding Region in Polymerase
2.5. NSP6 Lipid Binding Region
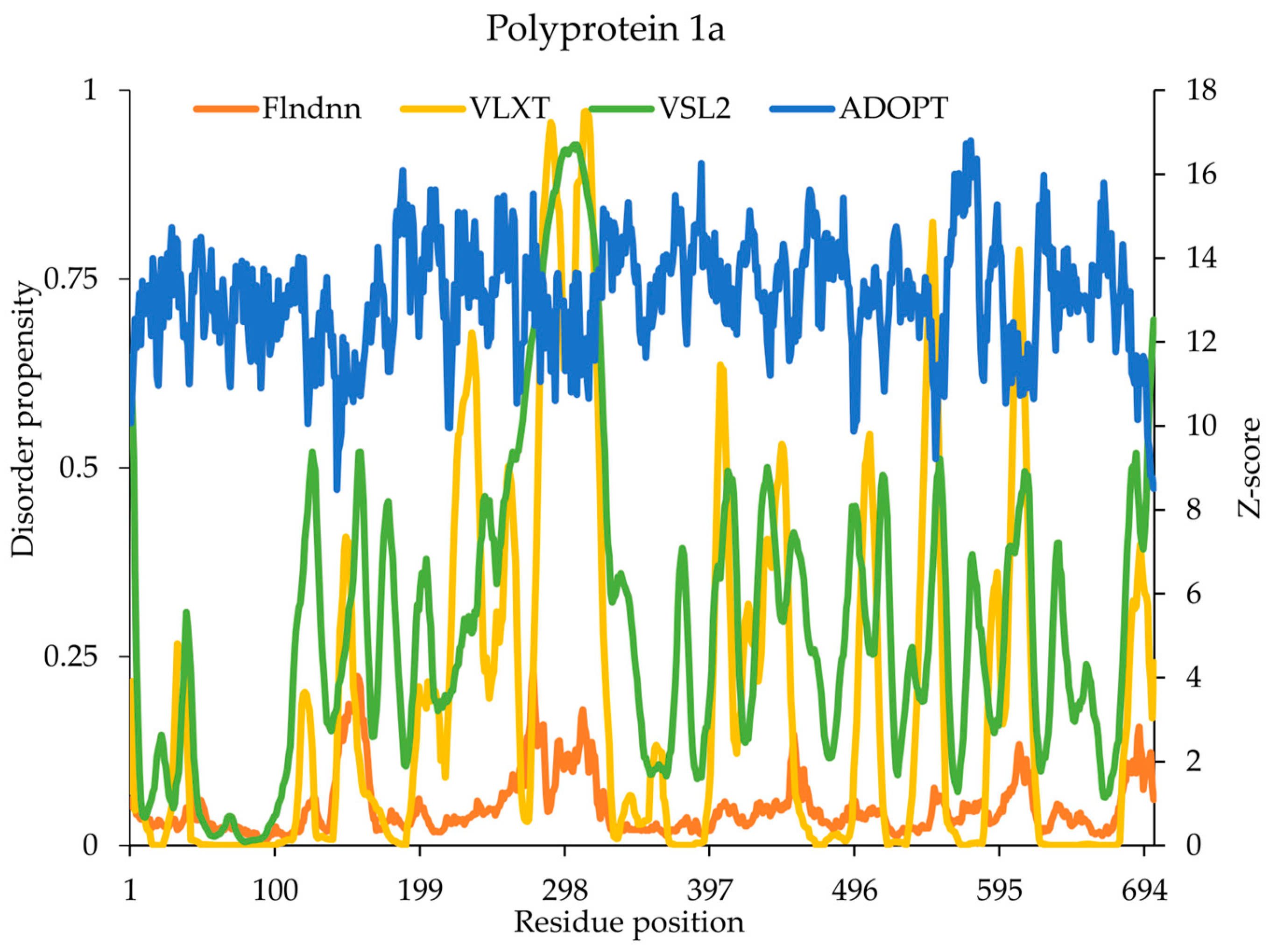
2.6. Polyprotein 1a
2.7. Spike Glycoprotein
2.8. ORF3a Protein
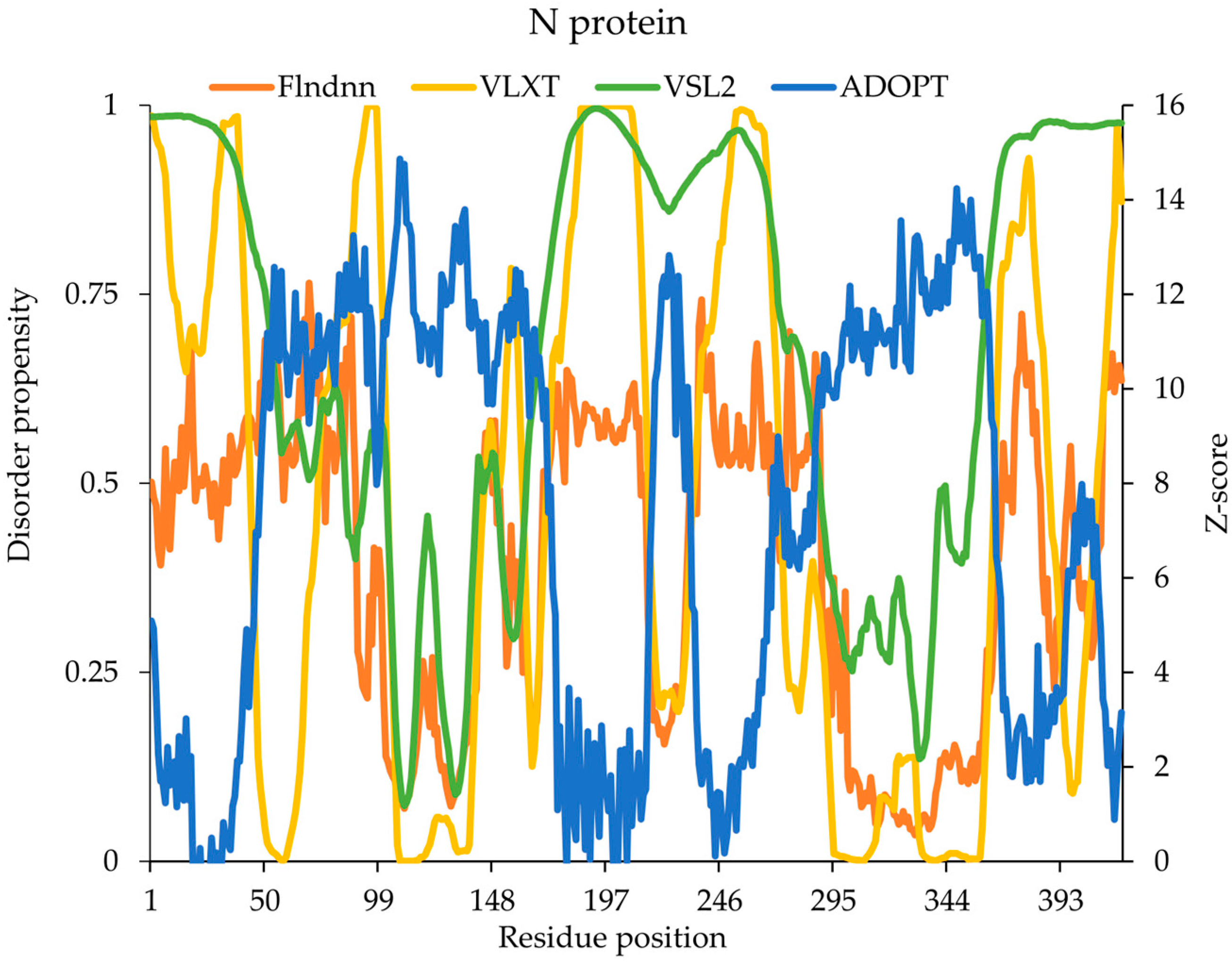
2.9. Nucleocapsid (N) Protein
2.10. Statistical Analysis
2.11. Comparative Performance of Disorder Prediction Models
2.12. Targeting IDRs in Drug Design
3. Materials and Methods
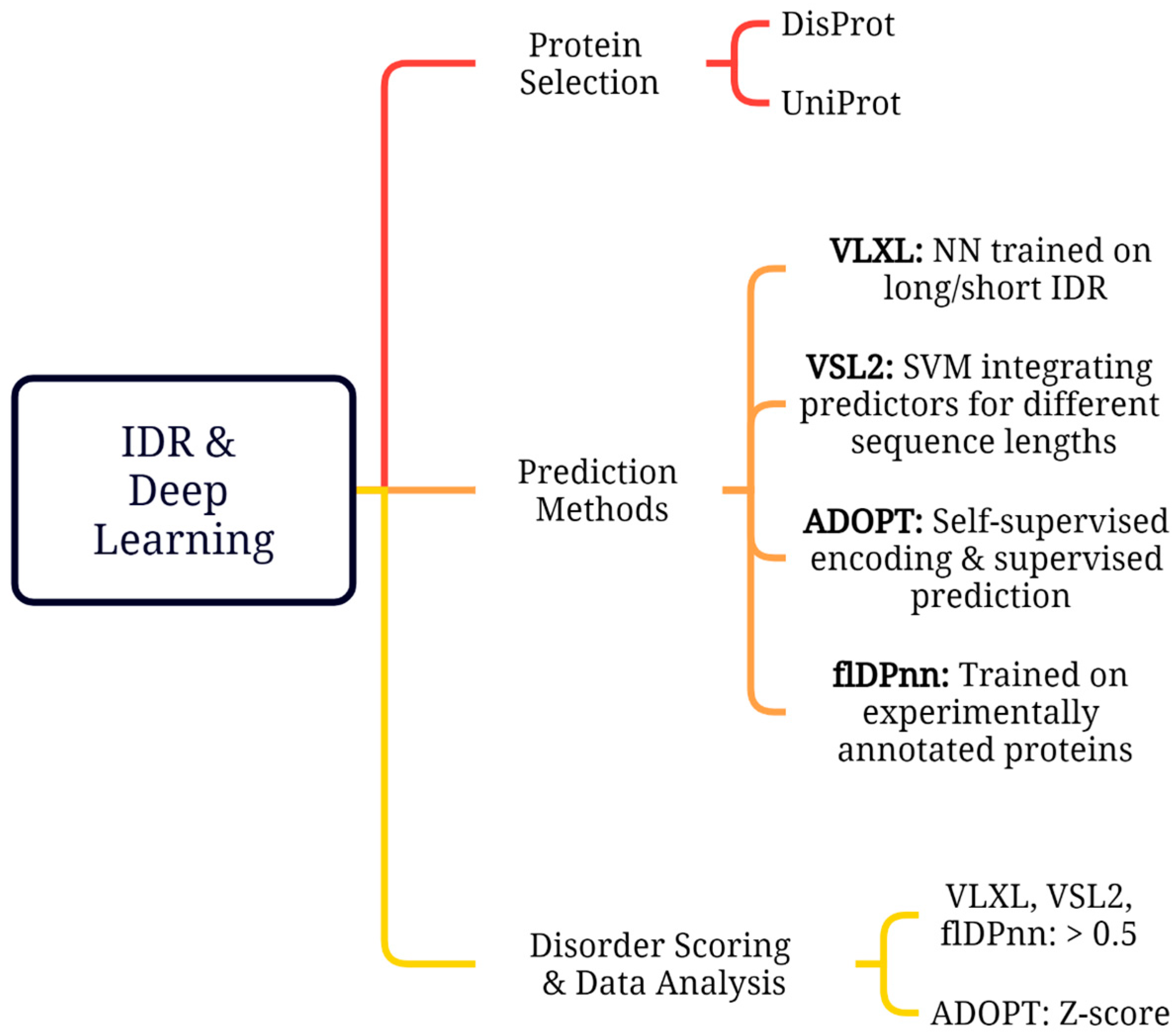
3.1. SARS-CoV-2 Protein Selection
3.2. Disorder Prediction Tools
3.3. Disorder Scoring and Data Analysis
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Todorov, G.; Uversky, V.N. A Possible Path towards Rapid Development of Live-Attenuated SARS-CoV-2 Vaccines: Plunging into the Natural Pool. Biomolecules 2020, 10, 1438. [Google Scholar] [CrossRef] [PubMed]
- Anjum, F.; Mohammad, T.; Asrani, P.; Shafie, A.; Singh, S.; Yadav, D.K.; Uversky, V.N.; Hassan, M.I. Identification of Intrinsically Disorder Regions in Non-Structural Proteins of SARS-CoV-2: New Insights into Drug and Vaccine Resistance. Mol. Cell Biochem. 2022, 477, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Bondos, S.E.; Huang, F.; Oldfield, C.J. Intrinsically Disordered Proteins and Multicellular Organisms. Semin. Cell Dev. Biol. 2015, 37, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional Anthology of Intrinsic Disorder. 1. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef]
- Uversky, V.N. New Technologies to Analyse Protein Function: An Intrinsic Disorder Perspective. F1000Res. 2020, 9, F1000 Faculty Rev-101. [Google Scholar] [CrossRef]
- Quaglia, F.; Salladini, E.; Carraro, M.; Minervini, G.; Tosatto, S.C.E.; Le Mercier, P. SARS-CoV-2 Variants Preferentially Emerge at Intrinsically Disordered Protein Sites Helping Immune Evasion. FEBS J. 2022, 289, 4240–4250. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; MacKens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Mishra, P.M.; Verma, N.C.; Rao, C.; Uversky, V.N.; Nandi, C.K. Intrinsically Disordered Proteins of Viruses: Involvement in the Mechanism of Cell Regulation and Pathogenesis. Prog. Mol. Biol. Transl. Sci. 2020, 174, 1–78. [Google Scholar]
- Tompa, P. Intrinsically Disordered Proteins: A 10-Year Recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- van Roey, K.; Gibson, T.J.; Davey, N.E. Motif Switches: Decision-Making in Cell Regulation. Curr. Opin. Struct. Biol. 2012, 22, 378–385. [Google Scholar] [CrossRef]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an Intrinsically Disordered Protein by Phosphorylation as a Regulatory Switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Koide, A.; Noval, M.G.; Panchenko, T.; Romero, L.A.; Teng, K.W.; Tada, T.; Landau, N.R.; Stapleford, K.A.; Koide, S. The ACE2-Binding Interface of SARS-CoV-2 Spike Inherently Deflects Immune Recognition. J. Mol. Biol. 2021, 433, 166748. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Khan, M.T.; Zeb, M.T.; Ahsan, H.; Ahmed, A.; Ali, A.; Akhtar, K.; Malik, S.I.; Cui, Z.; Ali, S.; Khan, A.S.; et al. SARS-CoV-2 Nucleocapsid and Nsp3 Binding: An in Silico Study. Arch. Microbiol. 2021, 203, 59–66. [Google Scholar] [CrossRef]
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-Cov-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef]
- Schiavina, M.; Pontoriero, L.; Uversky, V.N.; Felli, I.C.; Pierattelli, R. The Highly Flexible Disordered Regions of the SARS-CoV-2 Nucleocapsid N Protein within the 1–248 Residue Construct: Sequence-Specific Resonance Assignments through NMR. Biomol. NMR Assign. 2021, 15, 219–227. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Z.; Zhao, L.; Li, S. Novel Strategies for Drug Discovery Based on Intrinsically Disordered Proteins (IDPs). Int. J. Mol. Sci. 2011, 12, 3205–3219. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural Biology of SARS-CoV-2 and Implications for Therapeutic Development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Morales, M.; Ravanfar, R.; Oyala, P.H.; Gray, H.B.; Winkler, J.R. Copper(II) Binding to the Intrinsically Disordered C-Terminal Peptide of SARS-CoV-2 Virulence Factor Nsp1. Inorg. Chem. 2022, 61, 8992–8996. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, P.; Saumya, K.U.; Giri, R. Investigating the Conformational Dynamics of SARS-CoV-2 NSP6 Protein with Emphasis on Non-Transmembrane 91–112 & 231–290 Regions. Microb. Pathog. 2021, 161, 105236. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Kumar, P.; Kumar, A.; Bhardwaj, T.; Garg, N.; Giri, R. Conformational Dynamics of 13 Amino Acids Long NSP11 of SARS-CoV-2 under Membrane Mimetics and Different Solvent Conditions. Microb. Pathog. 2021, 158, 105041. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-NCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antiviral Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM Structure of the SARS-CoV-2 3a Ion Channel in Lipid Nanodiscs. bioRxiv 2021. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe Acute Respiratory Syndrome Coronavirus ORF3a Protein Activates the NLRP3 Inflammasome by Promoting TRAF3-Dependent Ubiquitination of ASC. FASEB J. 2019, 33, 8865. [Google Scholar] [CrossRef]
- Pontoriero, L.; Schiavina, M.; Korn, S.M.; Schlundt, A.; Pierattelli, R.; Felli, I.C. NMR Reveals Specific Tracts within the Intrinsically Disordered Regions of the SARS-CoV-2 Nucleocapsid Protein Involved in RNA Encountering. Biomolecules 2022, 12, 929. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Holehouse, A.S.; Hall, K.B.; Stuchell-Brereton, M.D.; Soranno, A. The Disordered N-Terminal Tail of SARS-CoV-2 Nucleocapsid Protein Forms a Dynamic Complex with RNA. Nucleic Acids Res. 2024, 52, 2609–2624. [Google Scholar] [CrossRef]
- Vasović, L.M.; Pavlović-Lažetić, G.M.; Kovačević, J.J.; Beljanski, M.V.; Uversky, V.N. Intrinsically Disordered Proteins and Liquid–Liquid Phase Separation in SARS-CoV-2 Interactomes. J. Cell Biochem. 2023, 125, e30502. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 Nucleocapsid Protein Phase-separates with RNA and with Human HnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef] [PubMed]
- Salma, P.; Chhatbar, C.; Seshadri, S. Intrinsically Unstructured Proteins: Potential Targets for Drug Discovery. Am. J. Infect. Dis. 2009, 5, 126–134. [Google Scholar] [CrossRef]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.J.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational Drug Design via Intrinsically Disordered Protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting Intrinsically Disordered Proteins Involved in Cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-P53 Protein-Protein Interaction to Reactivate P53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the Disordered C Terminus of PTP1B with an Allosteric Inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A Small Molecule Blocking Oncogenic Protein EWS-FLI1 Interaction with RNA Helicase A Inhibits Growth of Ewing’s Sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Iconaru, L.I.; Das, S.; Nourse, A.; Shelat, A.A.; Zuo, J.; Kriwacki, R.W. Small Molecule Sequestration of the Intrinsically Disordered Protein, P27Kip1, Within Soluble Oligomers. J. Mol. Biol. 2021, 433, 167120. [Google Scholar] [CrossRef]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia Fusion Target AF9 Is an Intrinsically Disordered Transcriptional Regulator That Recruits Multiple Partners via Coupled Folding and Binding. Structure 2013, 21, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, B.; Lan, W.; Santofimia-Castaño, P.; Zhou, Z.; Velázquez-Campoy, A.; Abián, O.; Peng, L.; Neira, J.L.; Xia, Y.; Iovanna, J.L. Design of Inhibitors of the Intrinsically Disordered Protein Nupr1: Balance between Drug Affinity and Target Function. Biomolecules 2021, 11, 1453. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Jia, Y.; Zhang, Y. Protein Structure Prediction. Int. J. Mod. Phys. B 2018, 32, 1840009. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins Struct. Funct. Genet. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. Ser. Workshop Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-Dependent Prediction of Protein in Intrinsic Disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef]
- Redl, I.; Fisicaro, C.; Dutton, O.; Hoffmann, F.; Henderson, L.; Owens, B.M.J.; Heberling, M.; Paci, E.; Tamiola, K. ADOPT: Intrinsic Protein Disorder Prediction through Deep Bidirectional Transformers. NAR Genom. Bioinform. 2023, 5, lqad041. [Google Scholar] [CrossRef]
- Hu, G.; Katuwawala, A.; Wang, K.; Wu, Z.; Ghadermarzi, S.; Gao, J.; Kurgan, L. FlDPnn: Accurate Intrinsic Disorder Prediction with Putative Propensities of Disorder Functions. Nat. Commun. 2021, 12, 4438. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt | SARS-CoV-2 Proteins | Length | Disorder Content |
|---|---|---|---|
| P0DTC1 | Replicase polyprotein 1a | 4405 | 0.27% |
| P0DTC2 | Spike glycoprotein (S) | 1273 | 27.10% |
| P0DTC3 | ORF3a protein | 275 | 26.91% |
| P0DTC9 | Nucleocapsid (N) protein | 419 | 51.07% |
| P0DTD1 | Replicase polyprotein 1ab | 7096 | 4.61% |
| Model | URL | Dataset | Algorithm | Properties | AUC | Evaluation Metrics | References |
|---|---|---|---|---|---|---|---|
| PONDR®VLXT | http://www.pondr.com/ (accessed on 25 September 2024) | Data consist of 8 and 7 long disordered regions from X-ray crystallography and NMR, respectively. | Three feedforward neural networks: VL1, XN, and XC | Less sensitive | 0.757 | ACC = 69.0 ± 0.9 | [44,45] |
| PONDR®VSL2 | http://www.pondr.com/ (accessed on 25 September 2024) | Sequences of varying lengths of X-ray crystallographic data (short and long length IDRs) | Combination of neural networks and SVM | 81% accuracy reported | 0.905 | ACC = 82.3 ± 1.1 | [46] |
| ADOPT | https://github.com/PeptoneLtd/ADOPT (accessed on 15 October 2024) | Datasets (CheZoD 1325 and CheZoD 117) | Deep bidirectional transformer (ESM library) and supervised predictor | Fast, accurate, and highly sensitive | 0.964 | MCC = 0.799 | [47] |
| flDPnn | http://biomine.cs.vcu.edu/servers/flDPnn/ (accessed on 7 November 2024) | Annotated 745 proteins from the DisProt 7.0 database | Deep learning model | Fast | 0.814 | MCC = 0.370 | [48] |
| Disorder Level | Z-Score Range | Description |
|---|---|---|
| Fully Disordered | Z-score < 3 | High degree of disorder, lacking a stable 3D structure |
| Partially Disordered | 3 ≤ 8 | Moderate disorder, some degree of structure but still flexible |
| Flexible | 8 ≤ 11 | Some degree of secondary structure but still dynamic |
| Structured | Z-score ≥ 11 | Well-defined 3D structure, such as alpha-helices or beta-sheets |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilyas, S.; Manan, A.; Lee, D. Deep Learning-Based Comparative Prediction and Functional Analysis of Intrinsically Disordered Regions in SARS-CoV-2. Int. J. Mol. Sci. 2025, 26, 3411. https://doi.org/10.3390/ijms26073411
Ilyas S, Manan A, Lee D. Deep Learning-Based Comparative Prediction and Functional Analysis of Intrinsically Disordered Regions in SARS-CoV-2. International Journal of Molecular Sciences. 2025; 26(7):3411. https://doi.org/10.3390/ijms26073411
Chicago/Turabian StyleIlyas, Sidra, Abdul Manan, and Donghun Lee. 2025. "Deep Learning-Based Comparative Prediction and Functional Analysis of Intrinsically Disordered Regions in SARS-CoV-2" International Journal of Molecular Sciences 26, no. 7: 3411. https://doi.org/10.3390/ijms26073411
APA StyleIlyas, S., Manan, A., & Lee, D. (2025). Deep Learning-Based Comparative Prediction and Functional Analysis of Intrinsically Disordered Regions in SARS-CoV-2. International Journal of Molecular Sciences, 26(7), 3411. https://doi.org/10.3390/ijms26073411