
DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese


 add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Egg Production Statistics
2.2. Summary of RNA-Seq Sequencing Data
2.3. Summary of Data from Genome-Wide Methylation Sequencing
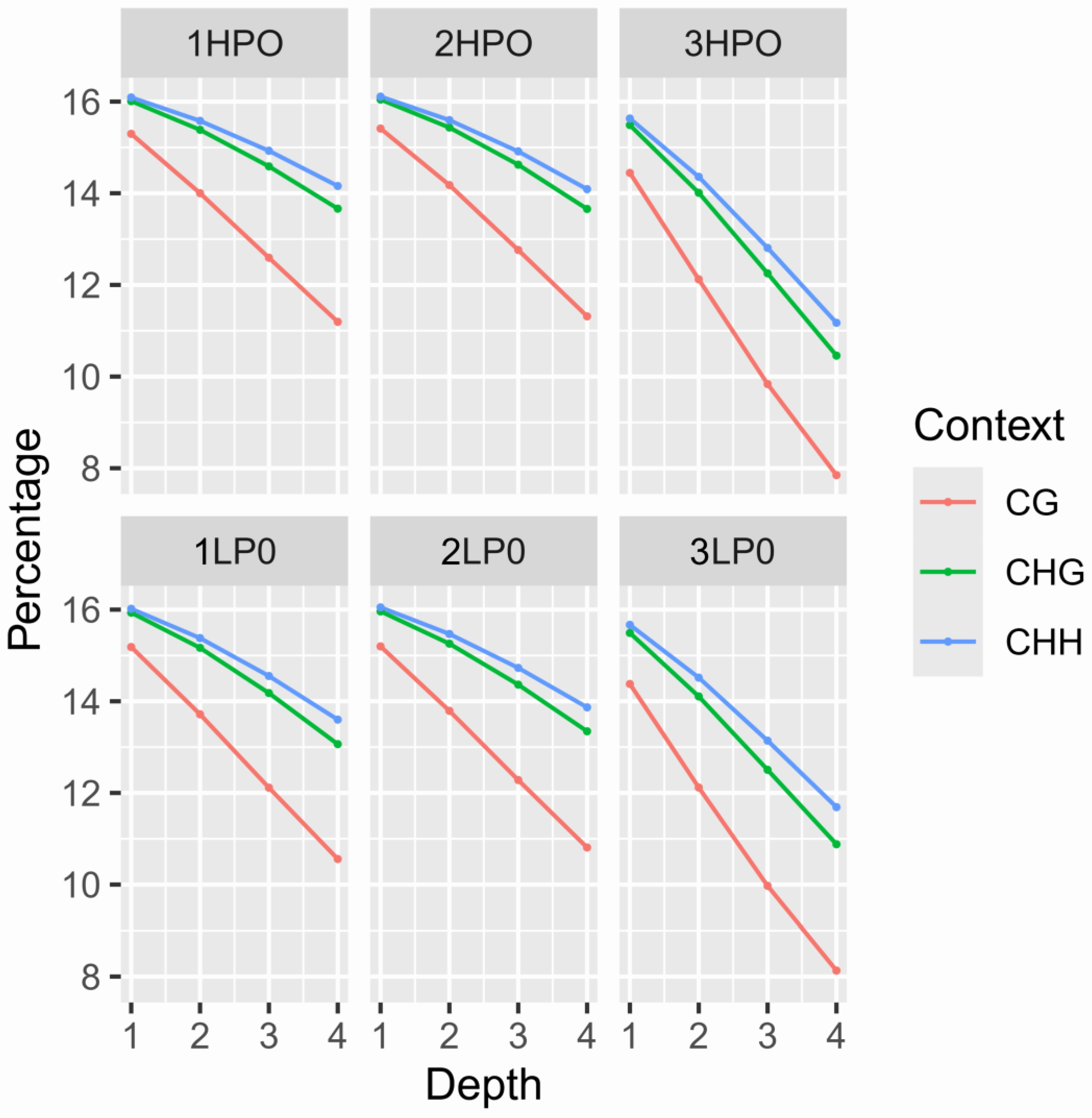
2.4. Analysis of Different Types of Methylation Levels
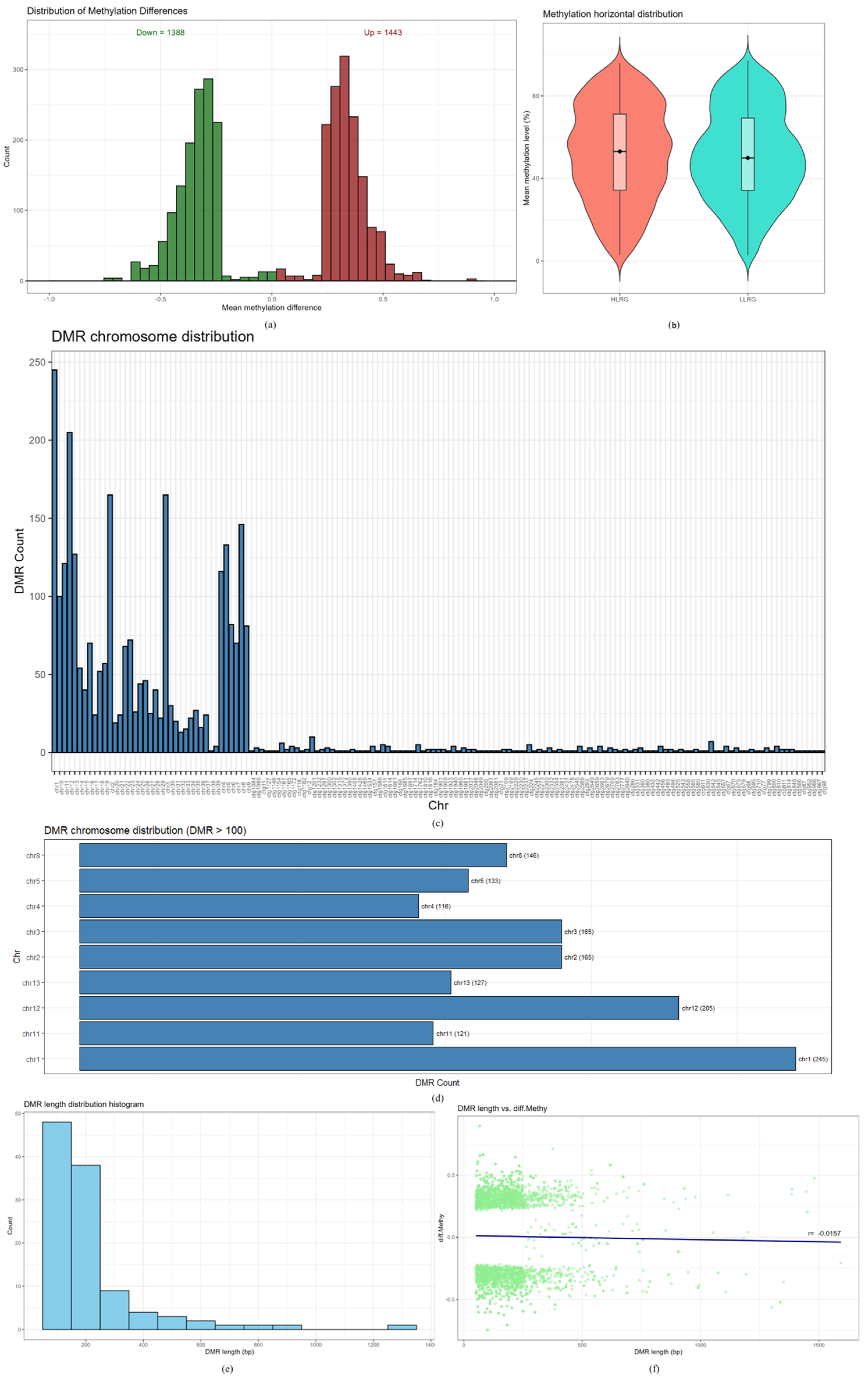
2.5. Analysis of Differential Methylation Regions (DMRs)
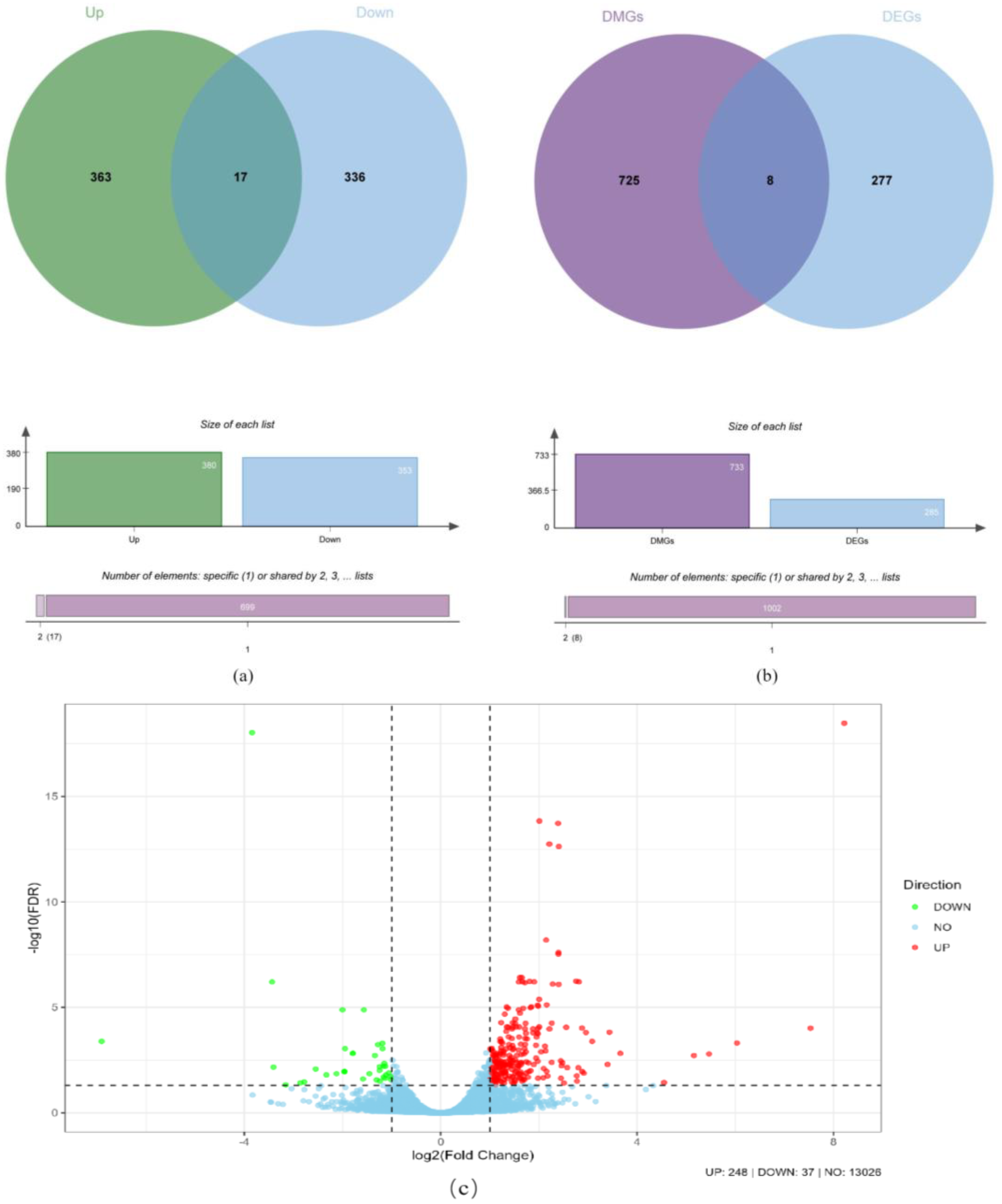
2.6. Association Analysis of Differentially Methylated Genes (DMGs)
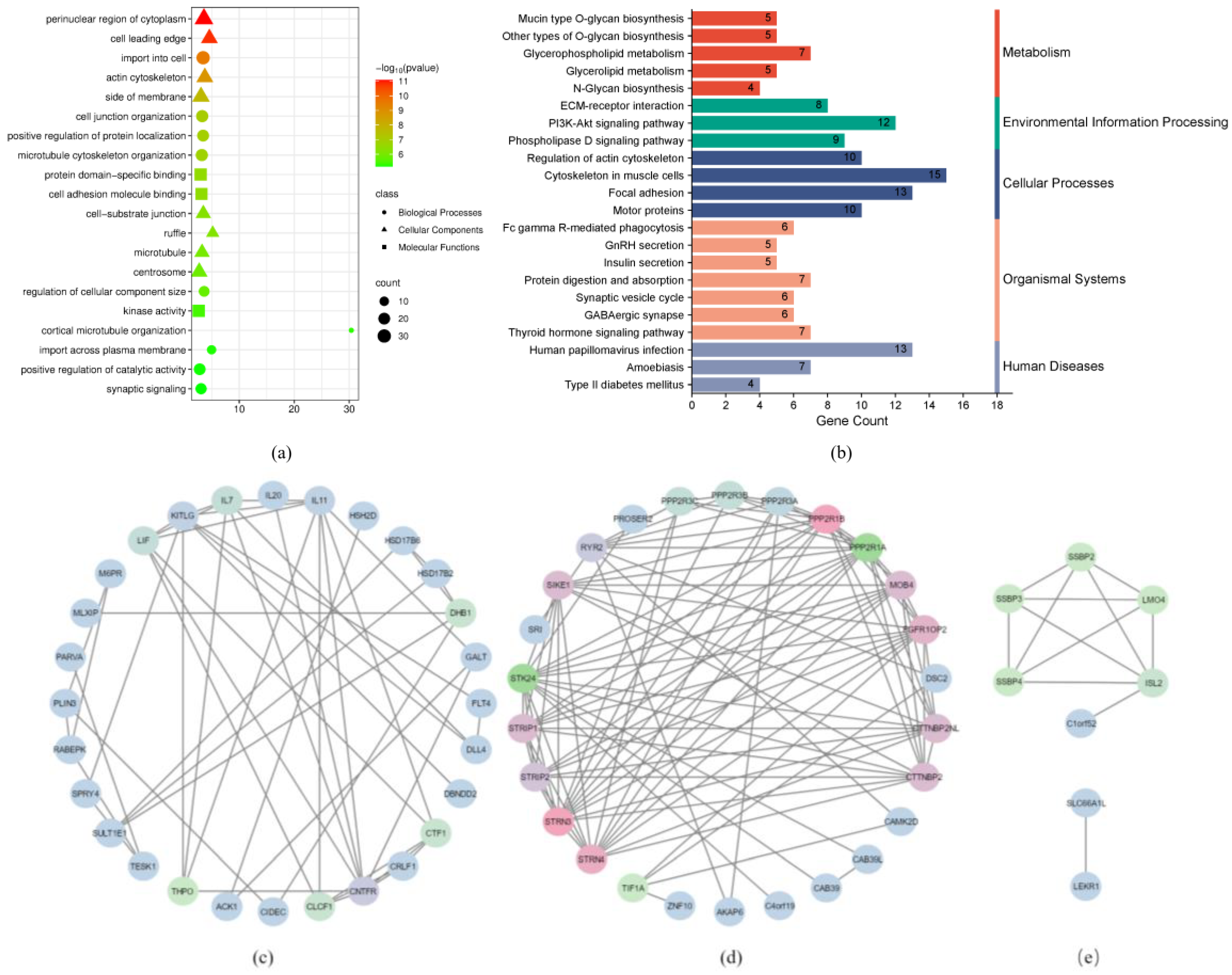
2.7. Enrichment and Network Interaction Analysis of Differentially Methylated Genes (DMGs)
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Sample Collection
4.2. Total RNA Extraction, Library Preparation, and Sequencing
4.3. DNA Extraction and Whole-Genome Bisulfite Sequencing (WGBS) Library Construction
4.4. Whole-Genome Bisulfite Sequencing (WGBS) Data Processing
4.5. Differential Methylation Regions (DMRs) Analysis
4.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
4.7. Data Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bao, Q.; Zhang, Y.; Yao, Y.; Luo, X.; Zhao, W.; Wang, J.; Xu, Q. Characteristics of the mating behavior of domesticated geese from Anser cygnoides and Anser anser. Animals 2022, 12, 2326. [Google Scholar] [CrossRef]
- Salamon, A. Fertility and hatchability in goose eggs: A review. Int. J. Poult. Sci. 2020, 19, 51–65. [Google Scholar] [CrossRef]
- El-Hanoun, A.M.; Attia, Y.A.; Gad, H.A.M.; Abdella, M.M. Effect of different managerial systems on productive and reproductive traits, blood plasma hormones and serum biochemical constituents of geese. Animals 2012, 6, 1795–1802. [Google Scholar] [CrossRef]
- Akhtar, M.F.; Shafiq, M.; Ali, I. Improving gander reproductive efficacy in the context of globally sustainable goose production. Animals 2022, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Gumułka, M.; Rozenboim, I. Mating activity of domestic geese ganders (Anser anser f. domesticus) during breeding period in relation to age, testosterone and thyroid hormones. Anim. Reprod. Sci. 2013, 3, 183–190. [Google Scholar]
- Shi, Z.D.; Tian, Y.B.; Wu, W.; Wang, Z.Y. Controlling reproductive seasonality in the geese: A review. World’s Poultry Sci. J. 2008, 3, 343–355. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes. Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Hunzicker-Dunn, M.; Mayo, K. Gonadotropin signaling in the ovary. In Knobil and Neill’s Physiology of Reproduction, 1st ed.; Knobil, E., Neill, J., Eds.; Elsevier: New York, NY, USA, 2015; pp. 895–946. [Google Scholar]
- Yang, Y.Z.; Yao, Y.; Cao, Z.F.; Gu, T.T.; Xu, Q.; Chen, G.H. Histological characteristics of follicles and reproductive hormone secretion during ovarian follicle development in laying geese. Poultry Sci. 2019, 98, 6063–6070. [Google Scholar] [CrossRef]
- Galkina, S.; Vishnevskaya, M. 23rd International Colloquium on Animal Cytogenetics and Genomics (23 ICACG) June 9–12, 2018, Saint-Petersburg, Russia. Compar. Cytogenet. 2018, 12, 299. [Google Scholar] [CrossRef]
- Chapelle, V.; Silvestre, F. Population epigenetics: The extent of DNA methylation variation in wild animal populations. Epigenomes 2022, 6, 31. [Google Scholar] [CrossRef]
- Ju, X.; Wang, Z.; Cai, D.; Bello, S.F.; Nie, Q. DNA methylation in poultry: A review. J. Anim. Sci. Biotechnol. 2023, 14, 138. [Google Scholar]
- Klughammer, J.; Romanovskaia, D.; Nemc, A.; Posautz, A.; Seid, C.A.; Schuster, L.C.; Bock, C. Comparative analysis of genome-scale, base-resolution DNA methylation profiles across 580 animal species. Nat. Commun. 2023, 14, 232. [Google Scholar]
- Elango, N. Evolutionary Impacts of DNA Methylation on Vertebrate Genomes; Georgia Institute of Technology: Atlanta, GA, USA, 2008. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar]
- Wutz, A. Epigenetic regulation of stem cells: The role of chromatin in cell differentiation. Transcr. Transl. Regul. Stem Cells 2013, 307–328. [Google Scholar]
- Deniz, Ö.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Padeken, J.; Zeller, P.; Gasser, S.M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015, 31, 12–19. [Google Scholar] [PubMed]
- Mifsud, K.R.; Gutièrrez-Mecinas, M.; Trollope, A.F.; Collins, A.; Saunderson, E.A.; Reul, J.M. Epigenetic mechanisms in stress and adaptation. Brain Behav. Immun. 2011, 7, 1305–1315. [Google Scholar] [CrossRef]
- Zhao, C.P.; Ji, G.Y.; Carrillo, J.A.; Li, Y.K.; Tian, F.; Baldwin, R.L.; Zan, L.S.; Song, J. The profiling of DNA methylation and its regulation on divergent tenderness in Angus beef cattle. Front. Genet. 2020, 11, 939. [Google Scholar]
- Luo, R.S.; Dai, X.L.; Zhang, L.; Li, G.P.; Zheng, Z. Genome-wide DNA methylation patterns of muscle and tail-fat in DairyMeade sheep and Mongolian sheep. Animals 2022, 12, 1399. [Google Scholar] [CrossRef]
- Wang, K.; Wu, P.X.; Wang, S.J.; Ji, X.; Chen, D.; Jiang, A.A.; Xiao, W.H.; Gu, Y.R.; Jiang, Y.Z.; Zeng, Y.S.; et al. Genome-wide DNA methylation analysis in Chinese Chenghua and Yorkshire pigs. BMC Genom. Data 2021, 22, 21. [Google Scholar]
- Zhu, G.; Mao, Y.; Zhou, W.; Jiang, Y. Dynamic changes in the follicular transcriptome and promoter DNA methylation pattern of steroidogenic genes in chicken follicles throughout the ovulation cycle. PLoS ONE 2015, 10, e0146028. [Google Scholar] [CrossRef]
- Sagvekar, P.; Kumar, P.; Mangoli, V.; Desai, S.; Mukherjee, S. DNA methylome profiling of granulosa cells reveals altered methylation in genes regulating vital ovarian functions in polycystic ovary syndrome. Clin. Epigenet. 2019, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, X.; Chimbaka, I.M.; Qin, N.; Xu, X.; Liswaniso, S.; Gonzalez, J.M. Transcriptome analysis of ovarian follicles reveals potential pivotal genes associated with increased and decreased rates of chicken egg production. Front. Genet. 2021, 12, 622751. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, X.Z.; A, G.; Song, T.; Wu, M.; Yan, Z.H.; Xiao, M.; Jiang, W.B.; Gao, Y.X.; Wang, H.W.; et al. Identification of DNA methylation differences in pituitary tissues of Sichuan White Geese using whole-genome bisulfite sequencing (WGBS). Biology 2025, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, K.; Liu, X.; Ren, M.; She, D.; Li, S. The hematopoietic function, histological characteristics, and transcriptome profiling of Wanxi white geese ovary during nesting and late-laying stages. Poultry Sci. 2025, 104, 104764. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Song, Y.; Li, T.; Zhang, S.; Huang, L.; Zhang, S.; Zhang, J. Comparative transcriptome profiling of ovary tissue between black muscovy duck and white muscovy duck with high- and low-egg production. Genes 2020, 12, 57. [Google Scholar] [CrossRef]
- Zhang, D.L.; Gan, Y.J.; Le, L.; Pu, L. Epigenetic regulation and breeding application of yield traits in maize. Bull. Biotechnol. 2023, 39, 31–42. [Google Scholar]
- Sundaresan, S.; Weiss, J.; Bauer-Dantoin, A.C.; Jameson, J.L. Expression of ryanodine receptors in the pituitary gland: Evidence for a role in gonadotropin-releasing hormone signaling. Endocrinology 1997, 138, 2056–2065. [Google Scholar] [CrossRef]
- Mota, L.F.; Carvajal, A.B.; Bernardes, P.A.; Buzanskas, M.E.; Baldi, F.; Lôbo, R.B.; Munari, D.P. Integrating genome-wide association study and pathway analysis reveals physiological aspects affecting heifer early calving defined at different ages in Nelore cattle. Genomics 2022, 114, 110395. [Google Scholar] [CrossRef]
- Méar, L.; Sutantiwanichkul, T.; Östman, J.; Damdimopoulou, P.; Lindskog, C. Spatial proteomics for further exploration of missing proteins: A case study of the ovary. J. Proteome Res. 2022, 22, 1071–1079. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, J.; Wen, J.; Wang, Y.; Niu, W.; Teng, Z.; Zhao, T.; Dai, Y.; Zhang, Y.; Wang, C.; et al. CDC42 controls the activation of primordial follicles by regulating PI3K signaling in mouse oocytes. BMC Biol. 2018, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, C.; Liu, S.; Luo, J.; Chen, Z.; Zhang, C.; Xi, L. RNA sequencing and differential expression reveals the effects of serial oestrus synchronisation on ovarian genes in dairy goats. Reprod. Fertil. Dev. 2018, 30, 1622–1633. [Google Scholar]
- Niederreither, K.; Remboutsika, E.; Gansmuller, A.; Losson, R.; Dollé, P. Expression of the transcriptional intermediary factor TIF1α during mouse development and in the reproductive organs. Mech. Dev. 1999, 88, 111–117. [Google Scholar] [PubMed]
- Lin, N.; Lin, J.; Plosch, T.; Sun, P.; Zhou, X. An oxidative stress-related gene signature in granulosa cells is associated with ovarian aging. Oxid. Med. Cell. Longev. 2022, 2022, 1070968. [Google Scholar]
- Yan, C.-Y.; Wu, F.-Y.; Sun, F.; Fang, Y.; Zhang, R.-J.; Zhang, C.-R.; Zhang, C.-X.; Wang, Z.; Yang, R.-M.; Yang, L.; et al. The isl2a transcription factor regulates pituitary development in zebrafish. Front. Endocrinol. 2023, 14, 920548. [Google Scholar] [CrossRef]
- Dozio, E.; Watanobe, H.; Ruscica, M.; Maggi, R.; Motta, M.; Magni, P. Expression of functional ciliary neurotrophic factor receptors in immortalized gonadotropin-releasing hormone-secreting neurons. J. Neuroendocrinol. 2005, 17, 286–291. [Google Scholar] [PubMed]
- Jachter, S.L. Kit Ligand Effect on Growth of Bovine Follicles in Culture and the Effect of Kit Ligand on the Function of Theca Cells. Bachelor’s Thesis, Oregon State University, Corvallis, OR, USA, 3 June 2020. [Google Scholar]
- Hsieh, M.; Boerboom, D.; Shimada, M.; Lo, Y.; Parlow, A.F.; Luhmann, U.F.; Berger, W.; Richards, J.S. Mice null for Frizzled4 (Fzd4−/−) are infertile and exhibit impaired corpora lutea formation and function. Biol. Reprod. 2005, 73, 1135–1146. [Google Scholar]
- Lapointe, E.; Boyer, A.; Rico, C.; Paquet, M.; Franco, H.L.; Gossen, J.; DeMayo, F.J.; Richards, J.S.; Boerboom, D. FZD1 regulates cumulus expansion genes and is required for normal female fertility in mice. Biol. Reprod. 2012, 87, 104. [Google Scholar]
- Zimmermann, R.C.; Jovanovic, V.P.; Sauer, C.M.; Shawber, C.; Gomez, R.; Wang, X.; Sauer, M.V.; Kitajewski, J.K. Intraovarian regulation of gonadotropin-dependent folliculogenesis depends on Notch receptor signaling pathways not involving Delta-like ligand 4 (Dll4). Reprod. Biol. Endocrinol. 2013, 11, 43. [Google Scholar]
- Albertini, D.F. Cytoplasmic microtubular dynamics and chromatin organization during mammalian oogenesis and oocyte maturation. Mutat. Res. Rev. Genet. Toxicol. 1992, 296, 57–68. [Google Scholar]
- Zampolla, T.; Spikings, E.; Rawson, D.; Zhang, T. Cytoskeleton proteins F-actin and tubulin distribution and interaction with mitochondria in the granulosa cells surrounding stage III zebrafish (Danio rerio) oocytes. Theriogenology 2011, 76, 1110–1119. [Google Scholar]
- Moenter, S.M.; Caraty, A.; Locatelli, A.; Karsch, F.J. Pattern of gonadotropin-releasing hormone (GnRH) secretion leading up to ovulation in the ewe: Existence of a preovulatory GnRH surge. Endocrinology 1991, 129, 1175–1182. [Google Scholar]
- Vieyra-Valdez, E.; Linares-Culebro, R.; Rosas-Gavilán, G.; Ramírez-Hernández, D.; Domínguez-Casalá, R.; Morales-Ledesma, L. Roles of the cholinergic system and vagal innervation in the regulation of GnRH secretion and ovulation: Experimental evidence. Brain Res. Bull. 2020, 165, 129–138. [Google Scholar] [PubMed]
- Wei, Q.; Fedail, J.S.; Kong, L.; Zheng, K.; Meng, C.; Fadlalla, M.B.; Shi, F. Thyroid hormones alter estrous cyclicity and antioxidative status in the ovaries of rats. Anim. Sci. J. 2018, 89, 513–526. [Google Scholar] [PubMed]
- Fedail, J.S.; Zheng, K.; Wei, Q.; Kong, L.; Shi, F. Roles of thyroid hormones in follicular development in the ovary of neonatal and immature rats. Endocrine 2014, 46, 594–604. [Google Scholar]
- Li, S.; Yang, Q.; Li, M.; Lan, Y.; Song, Z. Integrated miRNA and mRNA sequencing reveals the sterility mechanism in hybrid yellow catfish resulting from Pelteobagrus fulvidraco (♀) × Pelteobagrus vachelli (♂). Animals 2024, 14, 1586. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Yu, W.; Yu, S.; Yin, M.; Wu, L.; Chen, Q.; Cai, R.; Suo, L.; Wang, L.; Lyu, Q.; et al. Progesterone affects clinic oocyte yields by coordinating with follicle stimulating hormone via PI3K/AKT and MAPK pathways. J. Adv. Res. 2021, 33, 189–199. [Google Scholar]
- Yin, H.; Hou, X.; Zhang, T.; Shi, L.; Su, Y.Q. Participation of EML6 in the regulation of oocyte meiotic progression in mice. J. Biomed. Res. 2019, 34, 44–53. [Google Scholar]
- Tian, Z.; Wang, Y.; Tang, J.; Chu, M. Polymorphism of SYNE2 gene and its association with litter size in Small Tail Han Sheep. Pak. J. Zool. 2021, 53, 537. [Google Scholar]
- Sell-Kubiak, E.; Duijvesteijn, N.; Lopes, M.S.; Janss, L.L.G.; Knol, E.F.; Bijma, P.; Mulder, H.A. Genome-wide association study reveals novel loci for litter size and its variability in a Large White pig population. BMC Genom. 2015, 16, 1049. [Google Scholar]
- Ozanne, J.; Lewis, M.; Schwenzer, A.; Kurian, D.; Brady, J.; Pritchard, D.; McLachlan, G.; Farquharson, C.; Midwood, K.S. Extracellular matrix complexity in biomarker studies: A novel assay detecting total serum tenascin-C reveals different distribution to isoform-specific assays. Front. Immunol. 2023, 14, 1275361. [Google Scholar]
- Tshilate, T.S.; Ishengoma, E.; Rhode, C. Construction of a high-density linkage map and QTL detection for growth traits in South African abalone (Haliotis midae). Anim. Genet. 2024, 55, 744–760. [Google Scholar] [PubMed]
- Philippe, F.X.; Mahmoudi, Y.; Cinq-Mars, D.; Lefrançois, M.; Moula, N.; Palacios, J.; Pelletier, F.; Godbout, S. Comparison of egg production, quality and composition in three production systems for laying hens. Livest. Sci. 2020, 232, 103917. [Google Scholar]
- Cunningham, J.B.; McCrum-Gardner, E. Power, effect and sample size using GPower: Practical issues for researchers and members of research ethics committees. Evid. Based Midwifery 2007, 5, 132–137. [Google Scholar]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA methylation profiles on sheep ovaries associated with prolificacy using whole-genome bisulfite sequencing. BMC Genom. 2017, 18, 759. [Google Scholar]
- Li, Y.; Gao, G.; Lin, Y.; Hu, S.; Luo, Y.; Wang, G.; Jin, L.; Wang, Q.; Wang, J.; Tang, Q.; et al. Pacific Biosciences assembly with Hi-C mapping generates an improved, chromosome-level goose genome. GigaScience 2020, 9, giaa114. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | HLRG | LLRG | p-Value |
|---|---|---|---|
| Sample | 17 | 17 | |
| TEP | 55.76 ± 1.61 | 31.29 ± 1.23 | <0.001 |
| MEP | 13.94 ± 0.40 | 7.82 ± 0.31 | <0.001 |
| LR/% | 46.86 ± 1.35 | 26.30 ± 1.03 | <0.001 |
| Groups | Sample | Raw Reads | Clean Reads | RB (G) | CB (G) | ER (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|---|
| HLRG | 1HPO | 21,006,508 | 19,754,481 | 6.30 | 5.93 | 94.04 | 97.38 | 93.12 |
| 2HPO | 21,321,613 | 20,127,809 | 6.40 | 6.04 | 94.40 | 97.72 | 94.24 | |
| 3HPO | 25,219,828 | 23,791,538 | 7.57 | 7.14 | 94.34 | 97.24 | 92.77 | |
| LLRG | 1LPO | 33,732,858 | 32,942,426 | 10.12 | 9.88 | 97.66 | 97.19 | 92.75 |
| 2LPO | 21,102,732 | 20,168,463 | 6.33 | 6.05 | 95.57 | 97.50 | 93.23 | |
| 3LPO | 26,338,862 | 24,998,942 | 7.90 | 7.50 | 94.91 | 97.59 | 93.53 |
| Groups | Sample | RR (Gb) | CB (Gb) | Clean Ratio (%) | Mapping rate (%) | BCR (%) |
|---|---|---|---|---|---|---|
| HLRG | 1HPO | 20.18 | 15.45 | 76.57 | 74.60 | 99.90 |
| 2HPO | 18.16 | 15.16 | 83.44 | 81.40 | 99.90 | |
| 3HPO | 20.30 | 16.21 | 79.85 | 77.80 | 99.90 | |
| LLRG | 1LPO | 18.25 | 14.70 | 80.56 | 78.50 | 99.90 |
| 2LPO | 18.97 | 15.53 | 81.85 | 79.90 | 99.90 | |
| 3LPO | 18.66 | 15.87 | 85.08 | 82.40 | 99.89 |
| Groups | HLRG | LLRG |
|---|---|---|
| mCG (%) | 145,307,448 (63.90%) | 153,422,600 (63.86%) |
| mCHG (%) | 706,246,411 (0.92%) | 750,555,445 (0.99%) |
| mCHH (%) | 2,233,876,832 (0.97%) | 2,393,932,911 (1.06%) |
| Gene | MDL | FC | p-Value | Padj | LR |
|---|---|---|---|---|---|
| CUL9 | −0.31 | 1.95 | 1.93 × 10−5 | 1.75 × 10−3 | 21.78 |
| MEGF6 | −0.31 | 1.65 | 3.59 × 10−4 | 1.57 × 10−2 | 18.81 |
| EMAL6 | −0.27 | 1.07 | 8.84 × 10−4 | 3.61 × 10−2 | 11.06 |
| SYNE2 | −0.44 | 1.51 | 6.69 × 10−4 | 2.91 × 10−2 | 11.57 |
| AK1BA | −0.28 | 1.59 | 7.46 × 10−6 | 1.06 × 10−3 | 20.07 |
| MYCT1 | 0.38 | 1.72 | 4.37 × 10−5 | 3.28 × 10−3 | 22.21 |
| F16B1 | 0.34 | 1.49 | 3.97 × 10−5 | 3.27 × 10−3 | 18.21 |
| TENA | 0.45 | 1.73 | 1.77 × 10−7 | 5.74 × 10−5 | 27.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, L.; Zhao, X.; Wang, H.; Chen, Z.; Zhang, K.; Xue, J.; Luo, Y.; Liu, H.; Jiang, X.; Wang, J.; et al. DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese. Int. J. Mol. Sci. 2025, 26, 3408. https://doi.org/10.3390/ijms26073408
Ma L, Zhao X, Wang H, Chen Z, Zhang K, Xue J, Luo Y, Liu H, Jiang X, Wang J, et al. DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese. International Journal of Molecular Sciences. 2025; 26(7):3408. https://doi.org/10.3390/ijms26073408
Chicago/Turabian StyleMa, Lin, Xianzhi Zhao, Haiwei Wang, Zhuping Chen, Keshan Zhang, Jiajia Xue, Yi Luo, Hanyu Liu, Xinshuai Jiang, Jiayue Wang, and et al. 2025. "DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese" International Journal of Molecular Sciences 26, no. 7: 3408. https://doi.org/10.3390/ijms26073408
APA StyleMa, L., Zhao, X., Wang, H., Chen, Z., Zhang, K., Xue, J., Luo, Y., Liu, H., Jiang, X., Wang, J., Ma, X., Mao, F., Zhong, Y., Liu, Y., Deng, R., Zhou, Y., Wang, C., Xie, Y., Chen, Y., ... Gao, G. (2025). DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese. International Journal of Molecular Sciences, 26(7), 3408. https://doi.org/10.3390/ijms26073408