
Basic Pathological Mechanisms in Peripheral Nerve Diseases

 , ,
, ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
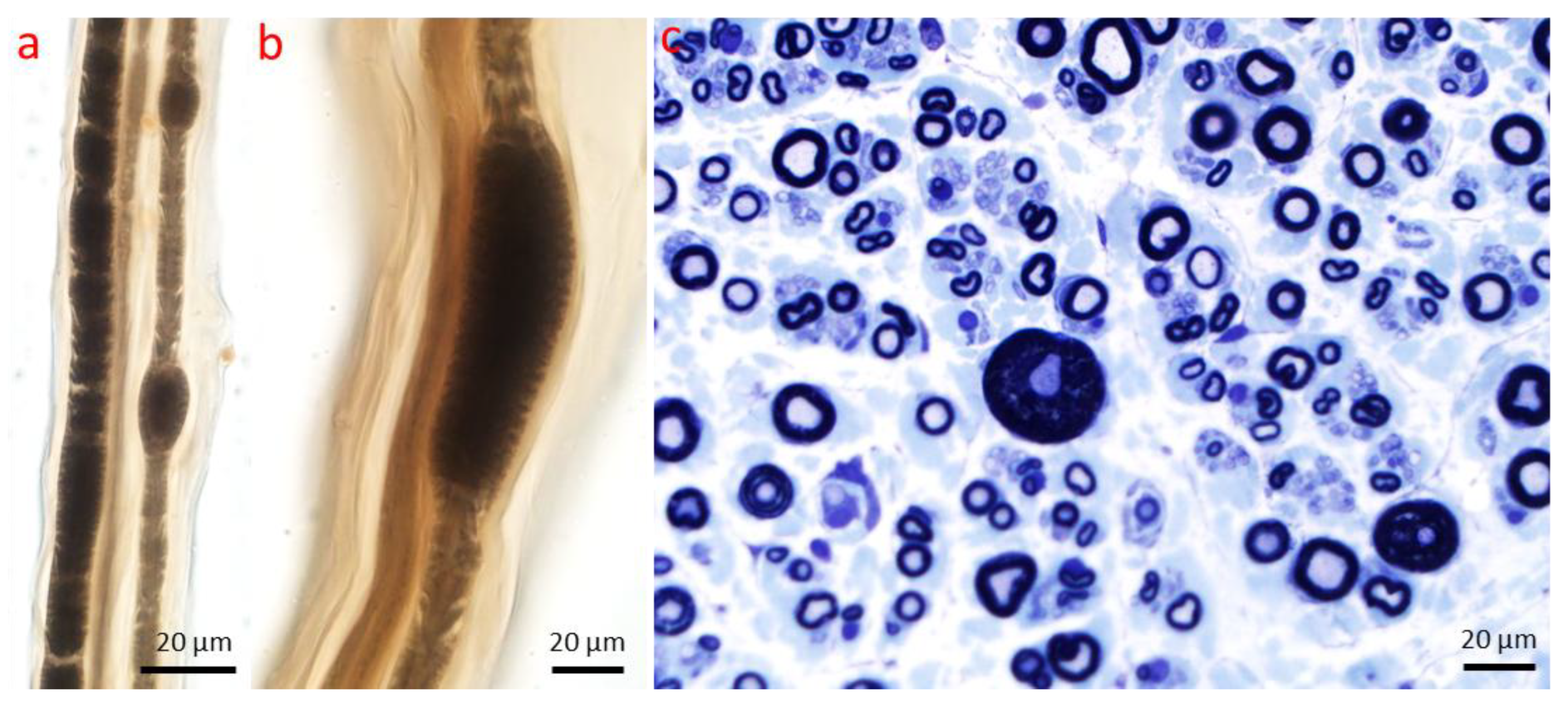
2. Axonopathy
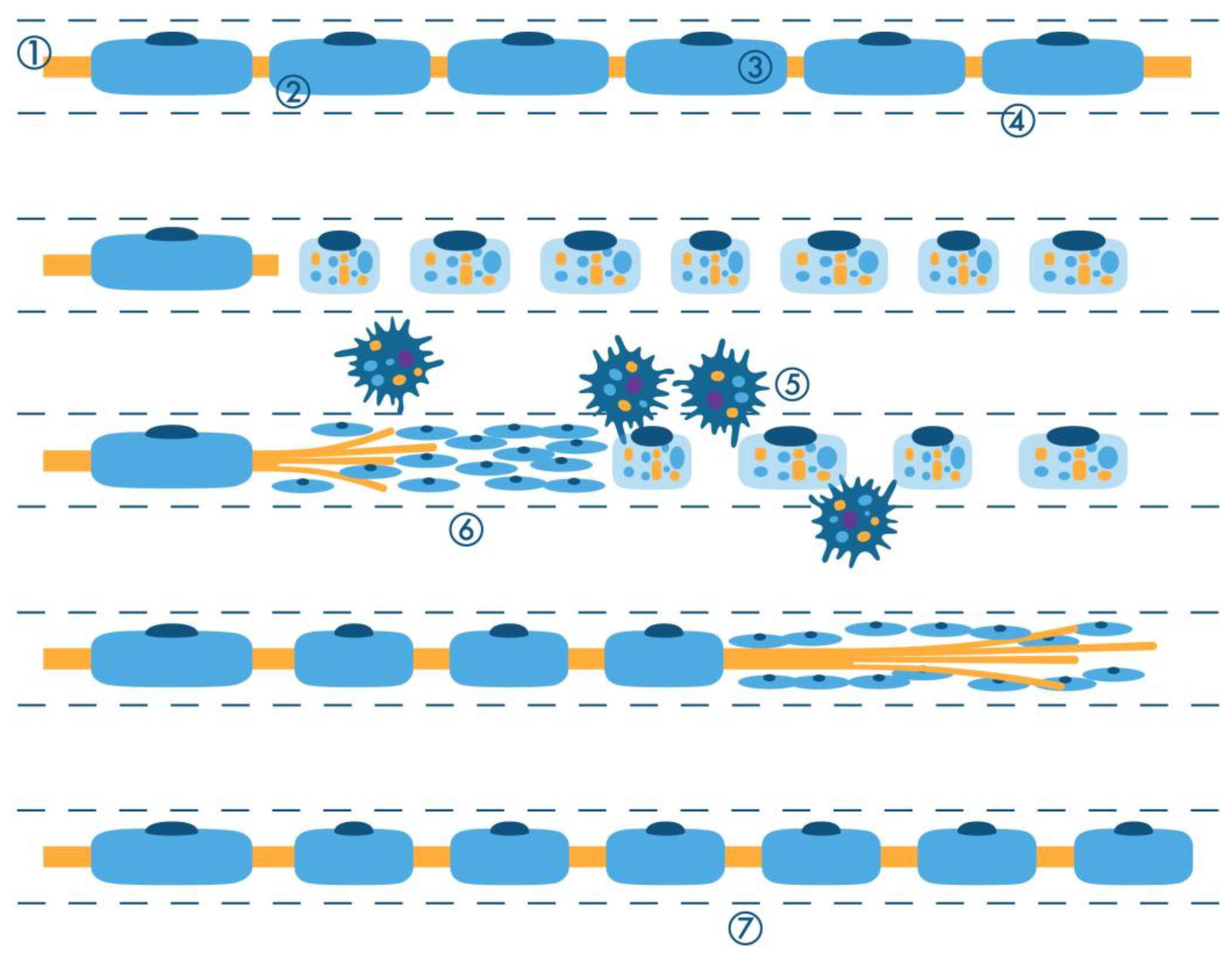
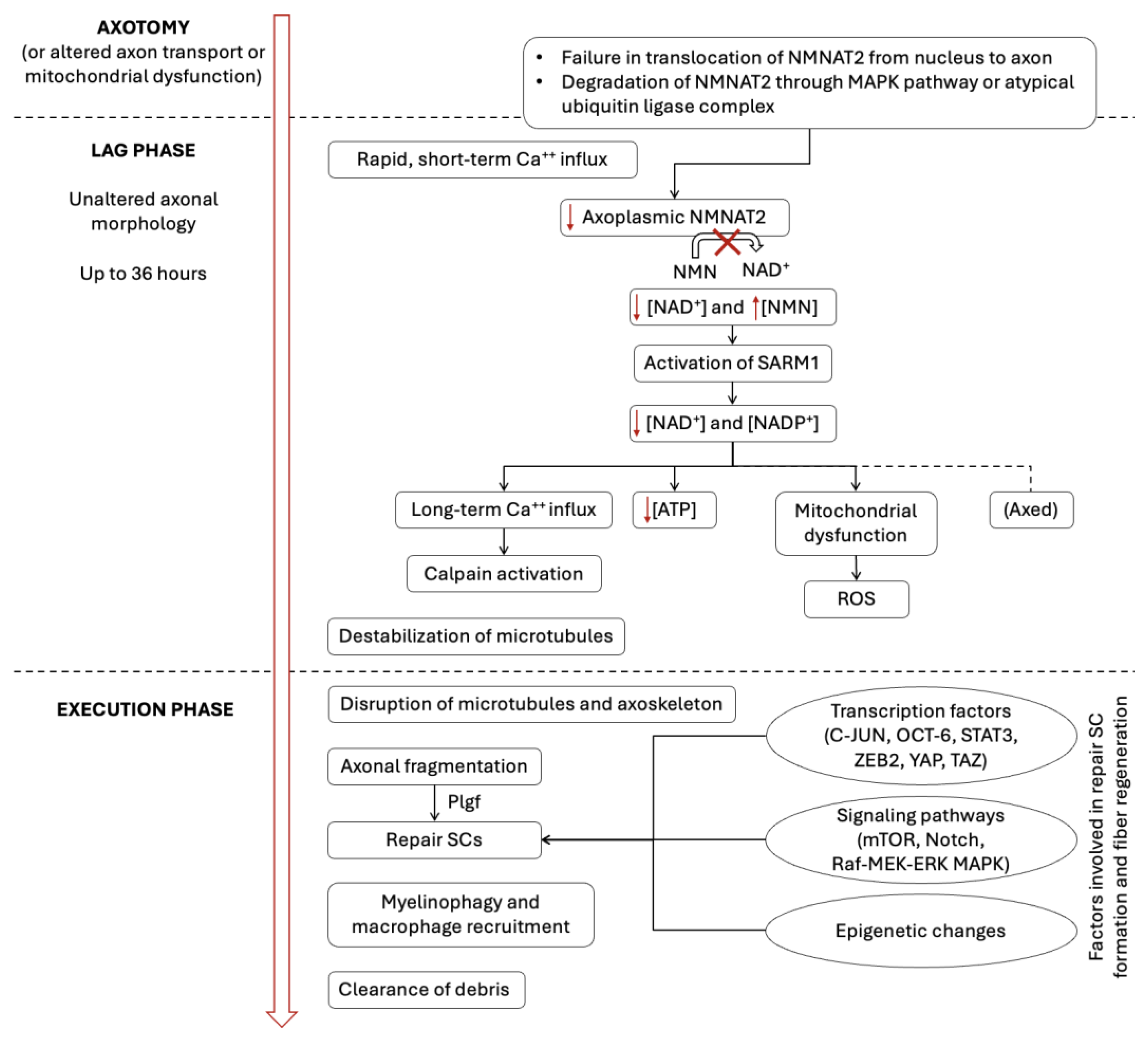
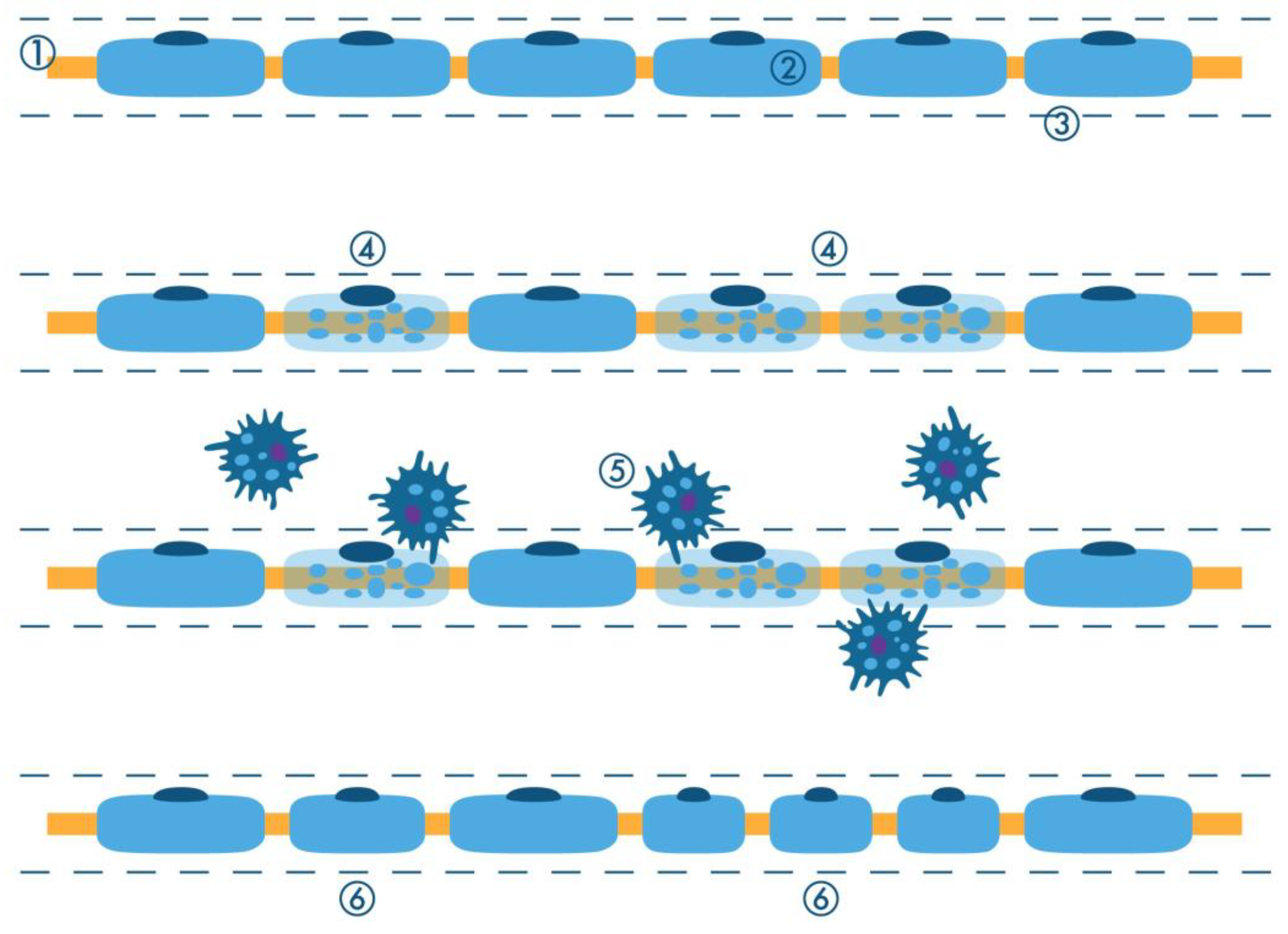
2.1. Active Axonal (Wallerian) Degeneration
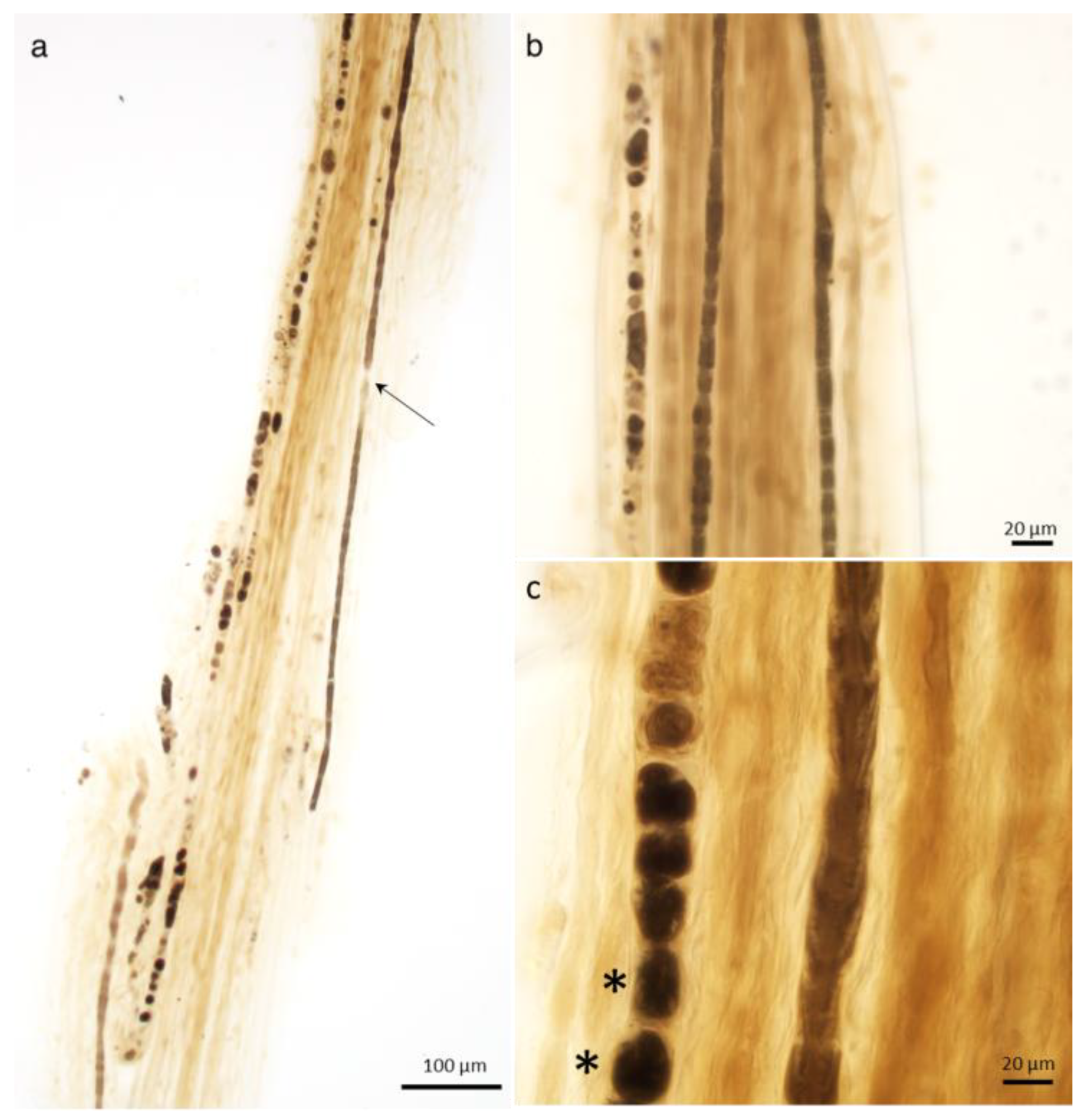
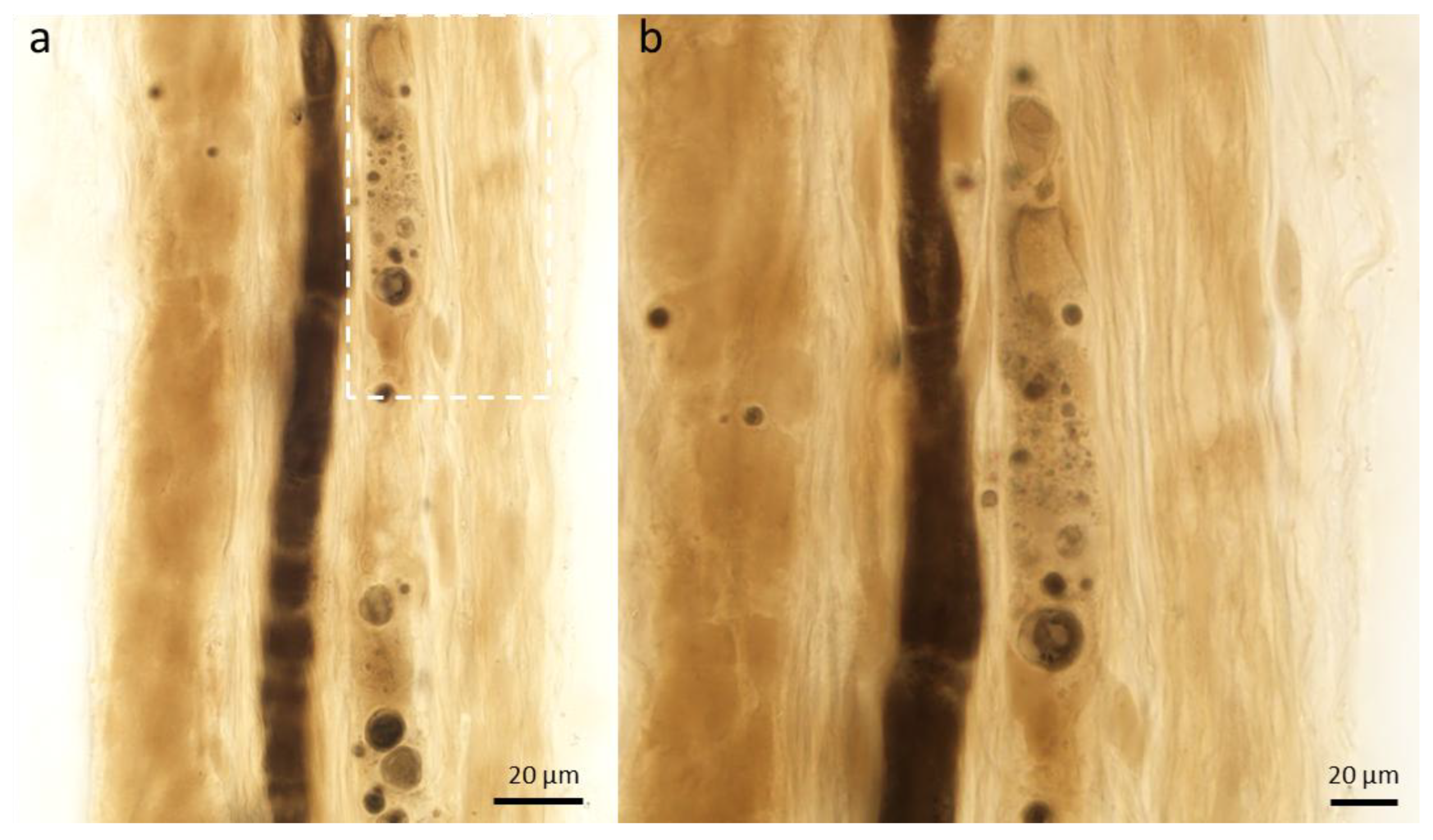
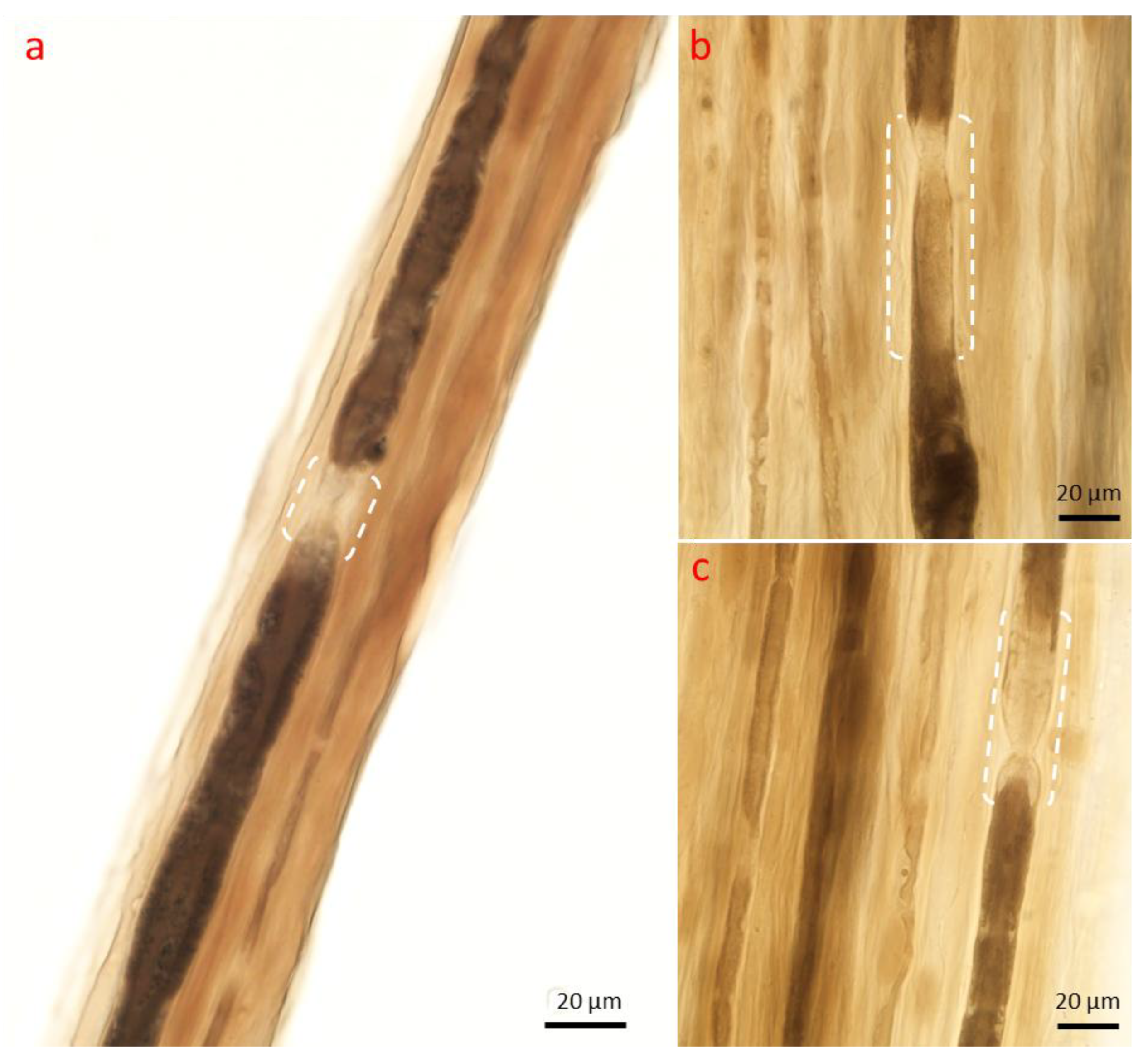
2.1.1. Changes in the Axon
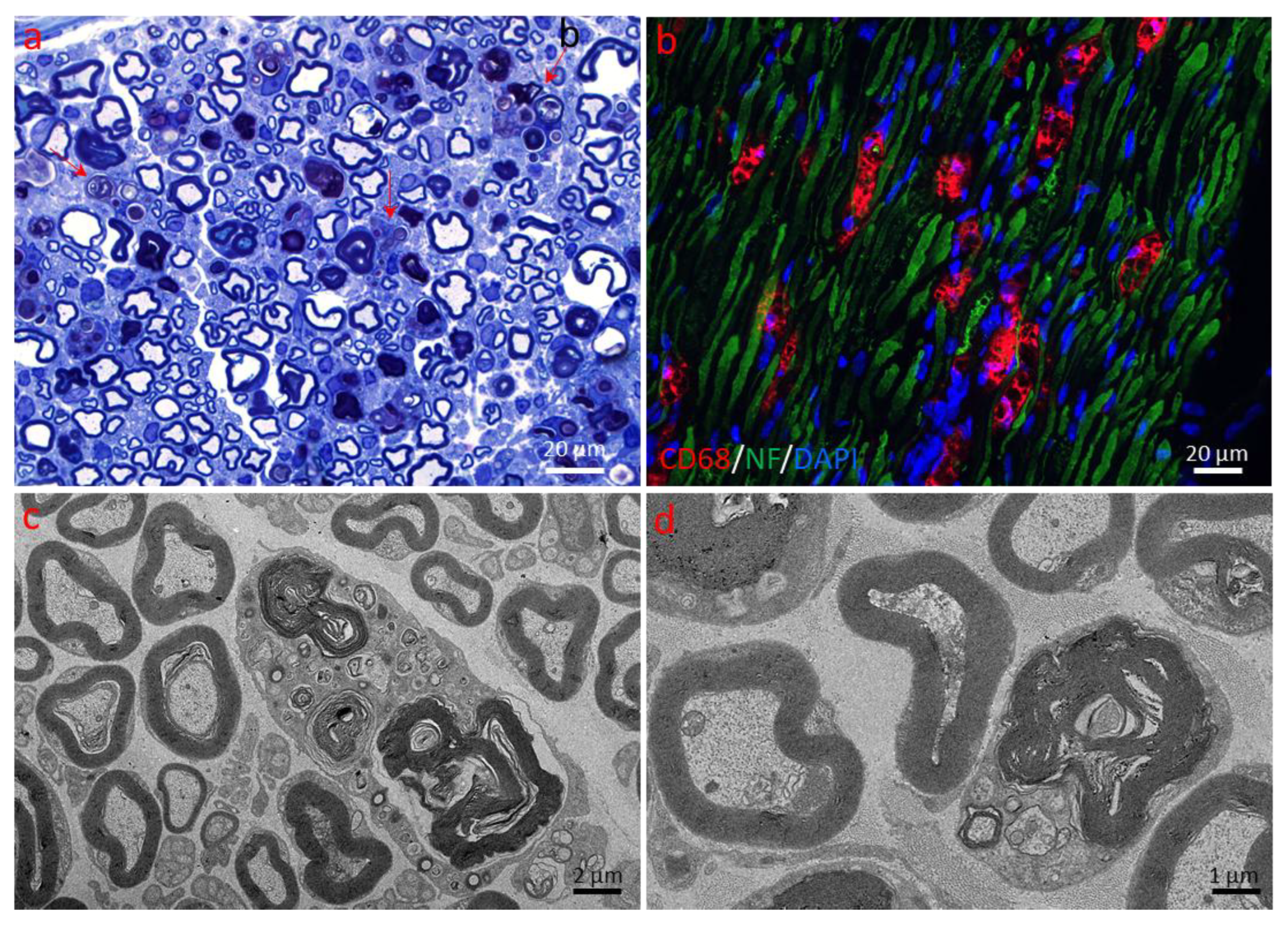
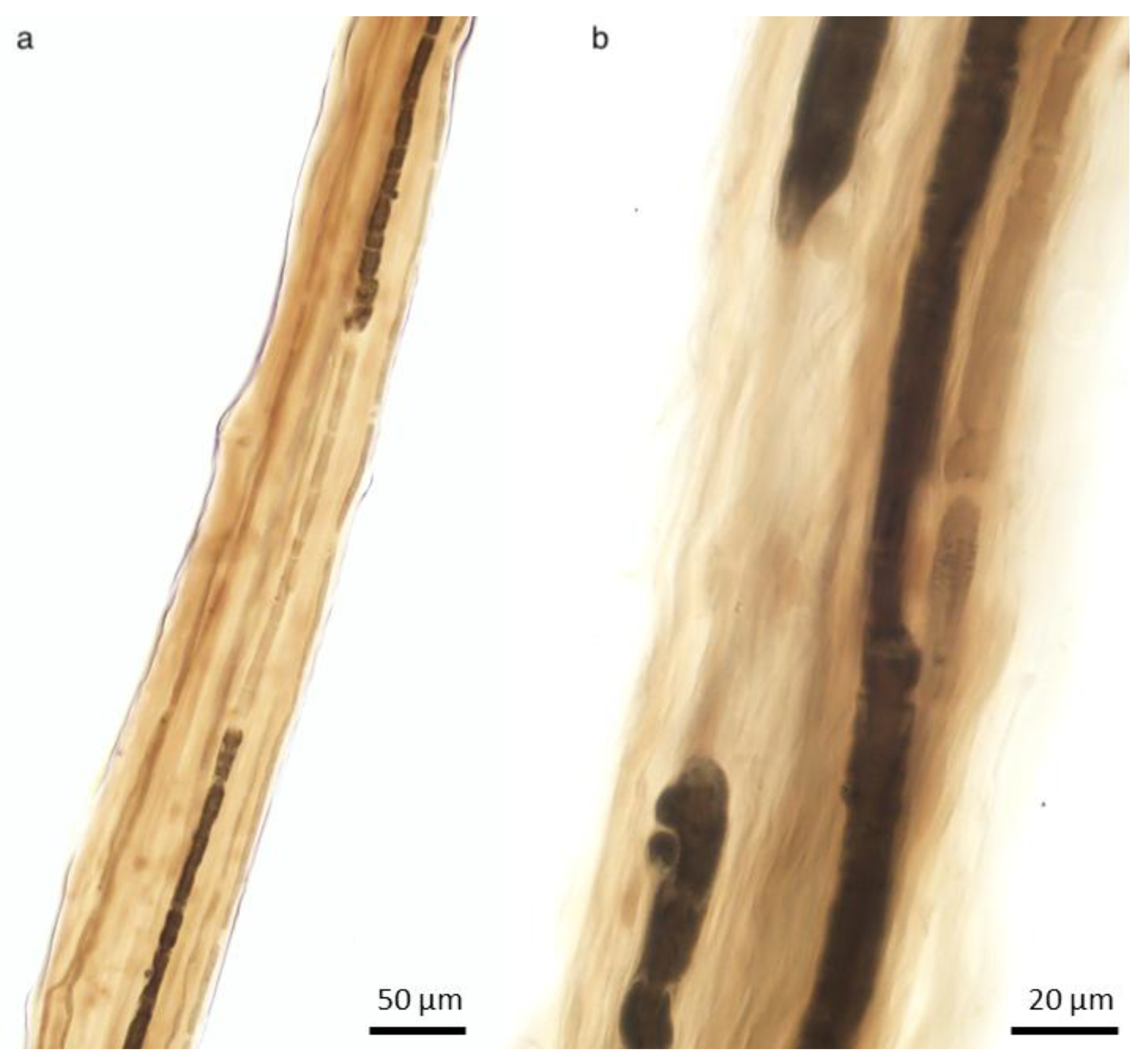
2.1.2. Schwann Cell and Macrophage Activity
2.1.3. Regeneration of the Nerve Fiber
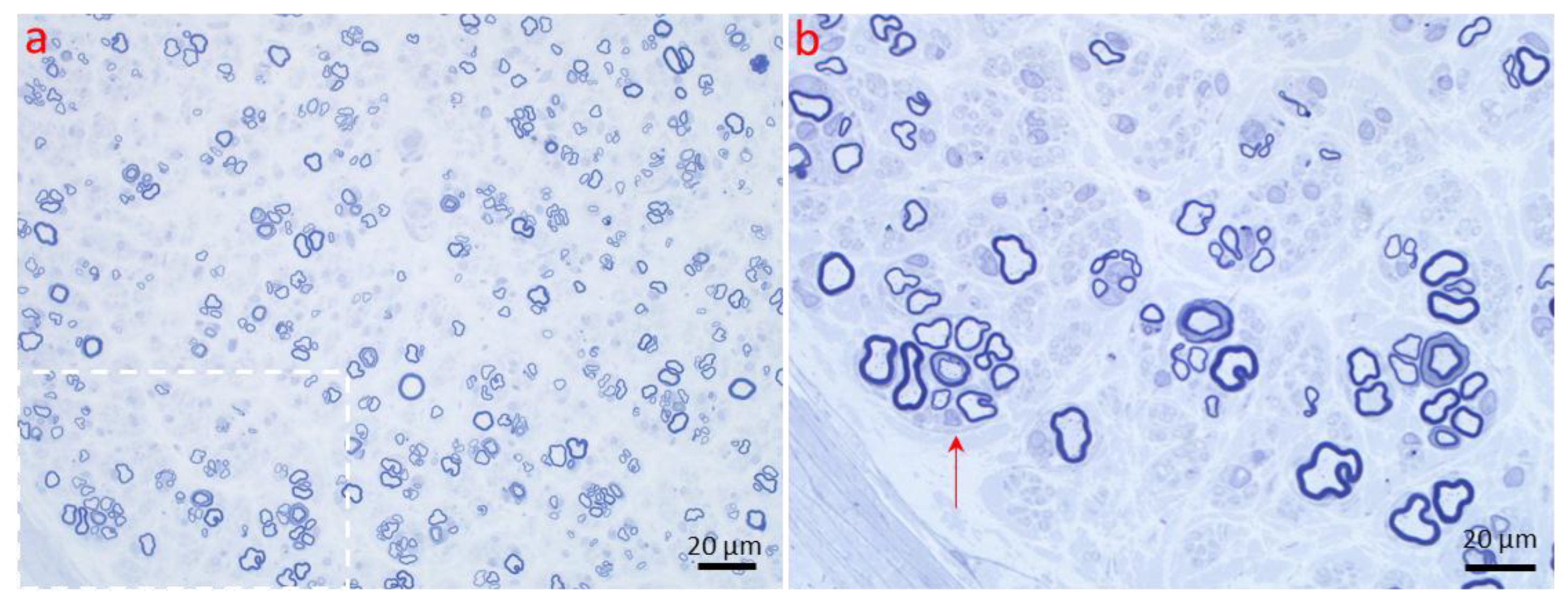
2.2. Axonal Atrophy
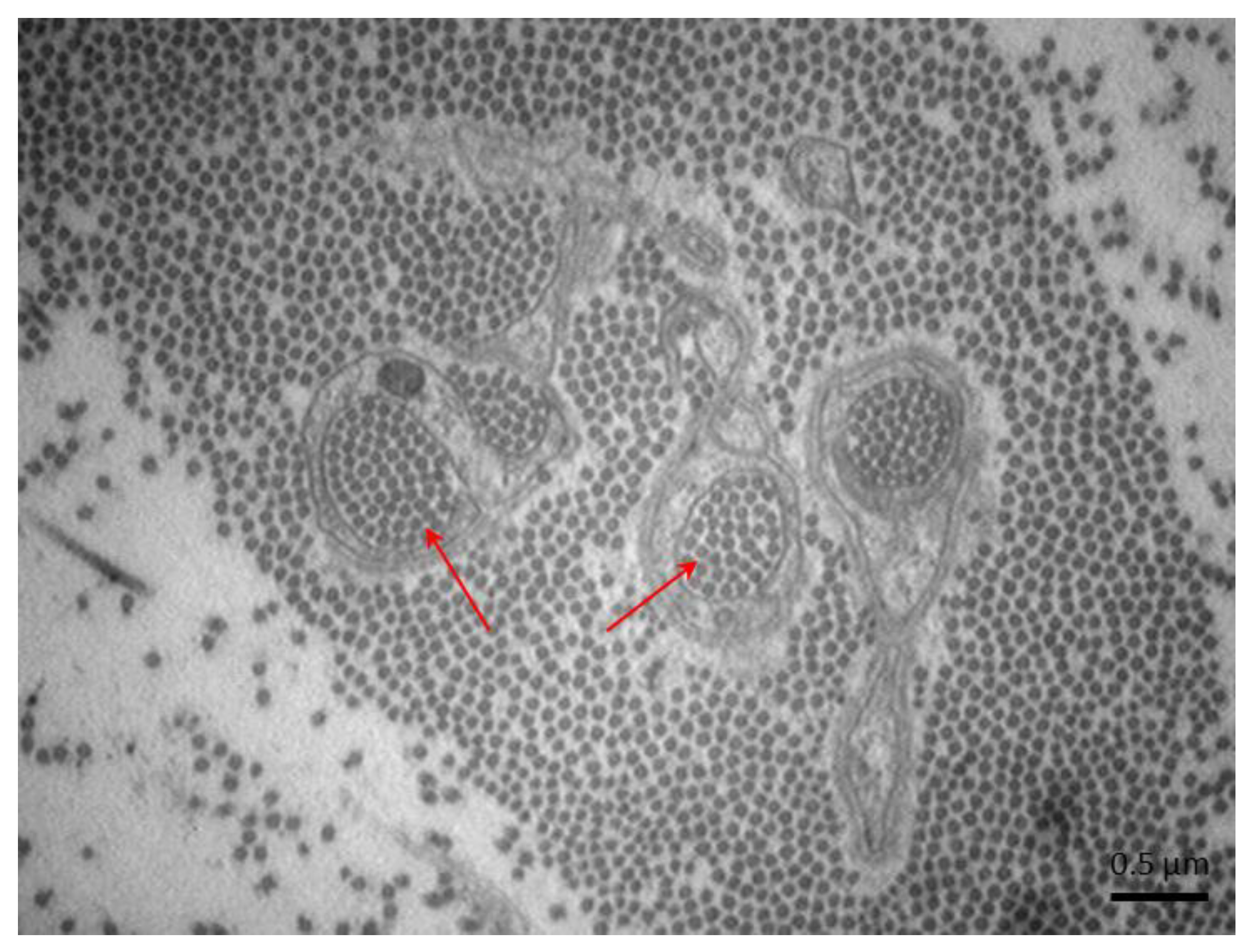
2.3. Axonal Dystrophy
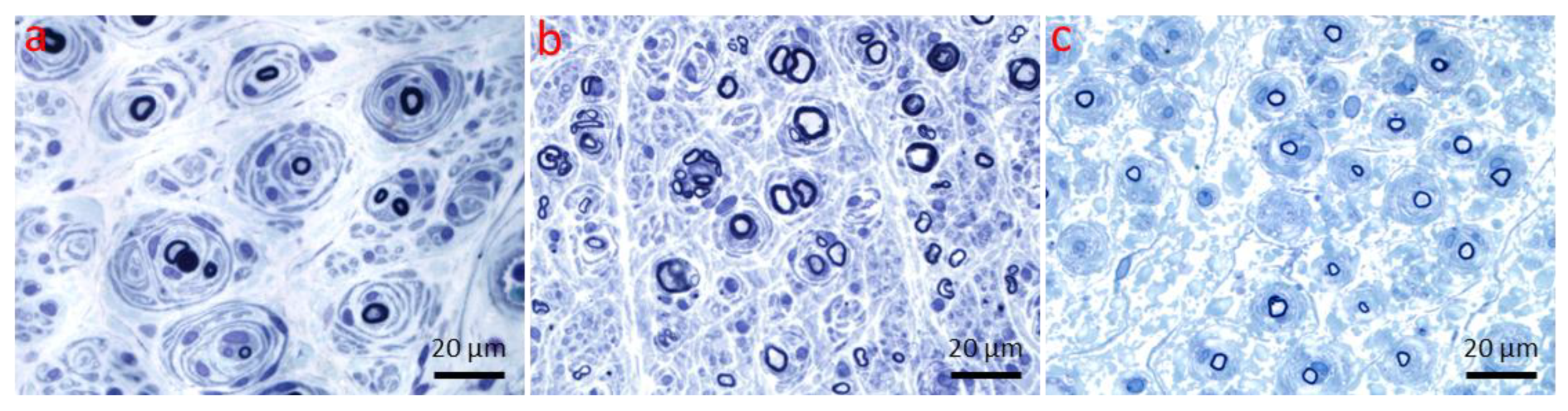
3. Myelinopathy
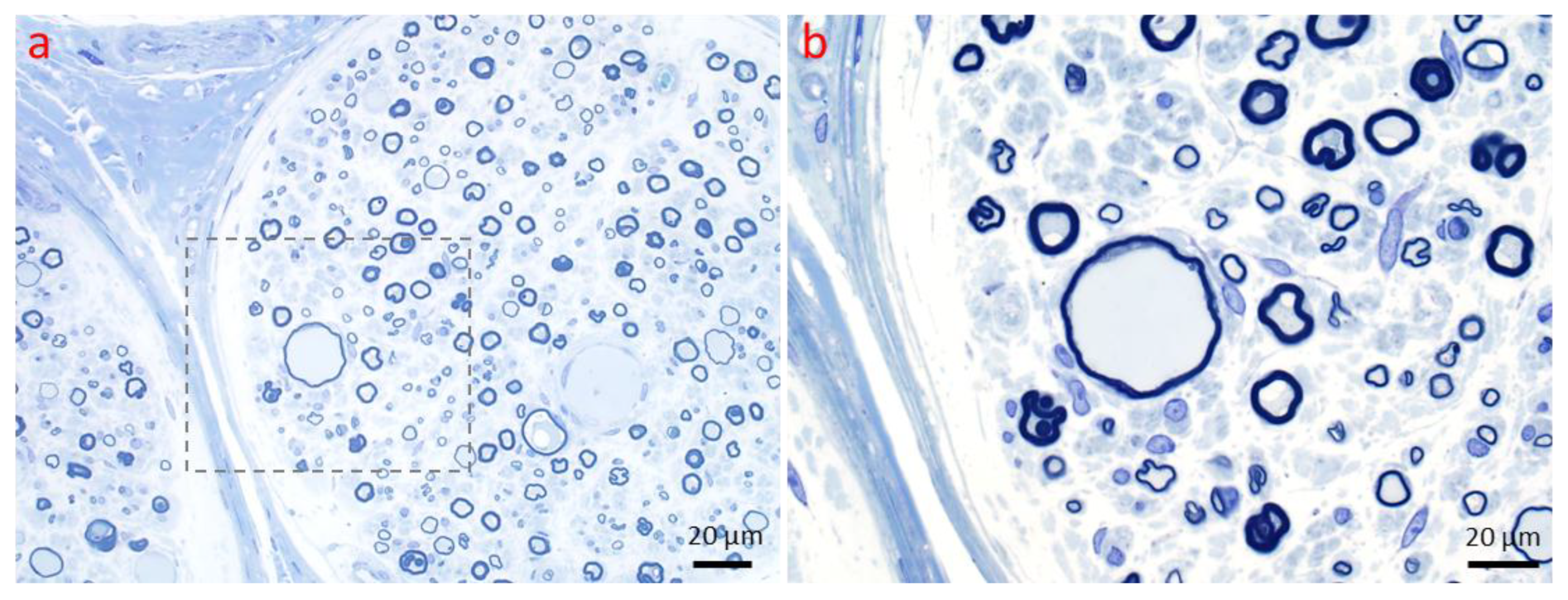
3.1. Primary Segmental Demyelination
3.2. Dysmyelination
3.3. Secondary Demyelination
3.4. Myelin Thickening and Changes in Myelin Folding
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watson, J.C.; Dyck, P.J.B. Peripheral Neuropathy: A Practical Approach to Diagnosis and Symptom Management. Mayo Clin. Proc. 2015, 90, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.M.; Mauermann, M.L. The Evaluation of Polyneuropathies. Neurology 2011, 76, S6–S13. [Google Scholar] [CrossRef]
- Prada, V.; Massucco, S.; Venturi, C.; Geroldi, A.; Bellone, E.; Mandich, P.; Minuto, M.; Varaldo, E.; Mancardi, G.; Grandis, M.; et al. Diagnostic Value of Sural Nerve Biopsy: Retrospective Analysis of Clinical Cases from 1981 to 2017. Front. Neurol. 2019, 10, 1218. [Google Scholar] [CrossRef]
- Sommer, C.L.; Brandner, S.; Dyck, P.J.; Harati, Y.; Lacroix, C.; Lammens, M.; Magy, L.; Mellgren, S.I.; Morbin, M.; Navarro, C.; et al. Peripheral Nerve Society Guideline on Processing and Evaluation of Nerve Biopsies. J. Peripher. Nerv. Syst. 2010, 15, 164–175. [Google Scholar] [CrossRef]
- Vallat, J.M.; Funalot, B.; Magy, L. Nerve Biopsy: Requirements for Diagnosis and Clinical Value. Acta Neuropathol. 2011, 121, 313–326. [Google Scholar] [CrossRef] [PubMed]
- England, J.D.; Asbury, A.K. Peripheral Neuropathy. Lancet 2004, 363, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romozzi, M.; Bisogni, G.; Cardellini, D.; Cavallaro, T.; Di Paolantonio, A.; Fabrizi, G.M.; Fenu, S.; Gentile, L.; Grandis, M.; et al. HATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sci. 2020, 10, 780. [Google Scholar] [CrossRef]
- Davidoff, L.M. Degeneration and Regeneration of the Nervous System. Am. J. Psychiatry 1929, 86, 212–218. [Google Scholar] [CrossRef]
- Waller, A. Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres. Philos. Trans. R. Soc. Lond. 1850, 140, 423–429. [Google Scholar]
- Kalinski, A.L.; Yoon, C.; Huffman, L.D.; Duncker, P.C.; Kohen, R.; Passino, R.; Hafner, H.; Johnson, C.; Kawaguchi, R.; Carbajal, K.S.; et al. Analysis of the Immune Response to Sciatic Nerve Injury Identifies Efferocytosis as a Key Mechanism of Nerve Debridement. eLlife 2020, 9, e60223. [Google Scholar] [CrossRef]
- Burnett, M.G.; Zager, E.L. Pathophysiology of Peripheral Nerve Injury: A Brief Review. Neurosurg. Focus. 2004, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, V.; Glass, J.D.; Griffin, J.W. Wallerian Degeneration in Peripheral Nerve Disease. Neurol. Clin. 1992, 10, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.L.; Neukomm, L.J. Axon Death Signalling in Wallerian Degeneration among Species and in Disease. Open Biol. 2019, 9, 190118. [Google Scholar] [CrossRef]
- Adalbert, R.; Morreale, G.; Paizs, M.; Conforti, L.; Walker, S.A.; Roderick, H.L.; Bootman, M.D.; Siklós, L.; Coleman, M.P. Intra-Axonal Calcium Changes after Axotomy in Wild-Type and Slow Wallerian Degeneration Axons. Neuroscience 2012, 225, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Villegas, R.; Martinez, N.W.; Lillo, J.; Pihan, P.; Hernandez, D.; Twiss, J.L.; Court, F.A. Calcium Release from Intra-Axonal Endoplasmic Reticulum Leads to Axon Degeneration through Mitochondrial Dysfunction. J. Neurosci. 2014, 34, 7179–7189. [Google Scholar] [CrossRef]
- George, E.; Glass, J.; Griffin, J. Axotomy-Induced Axonal Degeneration Is Mediated by Calcium Influx through Ion-Specific Channels. J. Neurosci. 1995, 15, 6445–6452. [Google Scholar] [CrossRef]
- Avery, M.A.; Rooney, T.M.; Pandya, J.D.; Wishart, T.M.; Gillingwater, T.H.; Geddes, J.W.; Sullivan, P.G.; Freeman, M.R. WldS Prevents Axon Degeneration through Increased Mitochondrial Flux and Enhanced Mitochondrial Ca2+ Buffering. Curr. Biol. 2012, 22, 596–600. [Google Scholar] [CrossRef]
- Park, J.Y.; Jang, S.Y.; Shin, Y.K.; Koh, H.; Suh, D.J.; Shinji, T.; Araki, T.; Park, H.T. Mitochondrial Swelling and Microtubule Depolymerization Are Associated with Energy Depletion in Axon Degeneration. Neuroscience 2013, 238, 258–269. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, J.; Kim, A.; Liu, Q.; Watts, R.; Hoopfer, E.; Mitchison, T.; Luo, L.; He, Z. Involvement of the Ubiquitin-Proteasome System in the Early Stages of Wallerian Degeneration. Neuron 2003, 39, 217–225. [Google Scholar] [CrossRef]
- Shen, H.; Hyrc, K.L.; Goldberg, M.P. Maintaining Energy Homeostasis Is an Essential Component of WldS-Mediated Axon Protection. Neurobiol. Dis. 2013, 59, 69–79. [Google Scholar] [CrossRef]
- Coleman, M. Axon Degeneration Mechanisms: Commonality amid Diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.D.; Culver, D.G.; Levey, A.I.; Nash, N.R. Very Early Activation of M-Calpain in Peripheral Nerve during Wallerian Degeneration. J. Neurol. Sci. 2002, 196, 9–20. [Google Scholar] [CrossRef]
- Beirowski, B.; Adalbert, R.; Wagner, D.; Grumme, D.S.; Addicks, K.; Ribchester, R.R.; Coleman, M.P. The Progressive Nature of Wallerian Degeneration in Wild-Type and Slow Wallerian Degeneration (WldS) Nerves. BMC Neurosci. 2005, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.E.; Bilbao, J.M. Diseases of Peripheral Nerves. In Greenfield’s Neuropathology; CRC Press: Boca Raton, FL, USA, 2015; Volume 2, pp. 1413–1514. ISBN 9781315382715. [Google Scholar]
- Finn, J.T.; Weil, M.; Archer, F.; Siman, R.; Srinivasan, A.; Raff, M.C. Evidence That Wallerian Degeneration and Localized Axon Degeneration Induced by Local Neurotrophin Deprivation Do Not Involve Caspases. J. Neurosci. 2000, 20, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Lunn, E.R.; Perry, V.H.; Brown, M.C.; Rosen, H.; Gordon, S. Absence of Wallerian Degeneration Does Not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1989, 1, 27–33. [Google Scholar] [CrossRef]
- Coleman, M.P.; Conforti, L.; Buckmaster, E.A.; Tarlton, A.; Ewing, R.M.; Brown, M.C.; Lyon, M.F.; Perry, V.H. An 85-Kb Tandem Triplication in the Slow Wallerian Degeneration (Wlds) Mouse. Proc. Natl. Acad. Sci. USA 1998, 95, 9985–9990. [Google Scholar] [CrossRef]
- Lyon, M.F.; Ogunkolade, B.W.; Brown, M.C.; Atherton, D.J.; Perry, V.H. A Gene Affecting Wallerian Nerve Degeneration Maps Distally on Mouse Chromosome 4. Proc. Natl. Acad. Sci. USA 1993, 90, 9717–9720. [Google Scholar] [CrossRef]
- Mack, T.G.A.; Reiner, M.; Beirowski, B.; Mi, W.; Emanuelli, M.; Wagner, D.; Thomson, D.; Gillingwater, T.; Court, F.; Conforti, L.; et al. Wallerian Degeneration of Injured Axons and Synapses Is Delayed by a Ube4b/Nmnat Chimeric Gene. Nat. Neurosci. 2001, 4, 1199–1206. [Google Scholar] [CrossRef]
- Hoopfer, E.D.; McLaughlin, T.; Watts, R.J.; Schuldiner, O.; O’Leary, D.D.M.; Luo, L. Wlds Protection Distinguishes Axon Degeneration Following Injury from Naturally Occurring Developmental Pruning. Neuron 2006, 50, 883–895. [Google Scholar] [CrossRef]
- Schoenmann, Z.; Assa-Kunik, E.; Tiomny, S.; Minis, A.; Haklai-Topper, L.; Arama, E.; Yaron, A. Axonal Degeneration Is Regulated by the Apoptotic Machinery or a NAD + -Sensitive Pathway in Insects and Mammals. J. Neurosci. 2010, 30, 6375–6386. [Google Scholar] [CrossRef]
- Tao, J.; Rolls, M.M. Dendrites Have a Rapid Program of Injury-Induced Degeneration That Is Molecularly Distinct from Developmental Pruning. J. Neurosci. 2011, 31, 5398–5405. [Google Scholar] [CrossRef] [PubMed]
- van Lier, M.; Smit-Rigter, L.; Krimpenfort, R.; Saiepour, M.H.; Ruimschotel, E.; Kamphuis, W.; Heime, J.A.; Levelt, C.N. NMNAT Proteins That Limit Wallerian Degeneration Also Regulate Critical Period Plasticity in the Visual Cortex. eNeuro 2019, 6, ENEURO.0277-18.2018. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, M.; Araki, T. Mechanism of Initiation and Regulation of Axonal Degeneration with Special Reference to NMNATs and Sarm1. Neurosci. Res. 2021, 197, 3–8. [Google Scholar] [CrossRef]
- McGuinness, H.Y.; Gu, W.; Shi, Y.; Kobe, B.; Ve, T. SARM1-Dependent Axon Degeneration: Nucleotide Signaling, Neurodegenerative Disorders, Toxicity, and Therapeutic Opportunities. Neuroscientist 2024, 30, 473–492. [Google Scholar]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. DSarm/Sarm1 Is Required for Activation of an Injury-Induced Axon Death Pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef]
- Wakatsuki, S.; Furuno, A.; Ohshima, M.; Araki, T. Oxidative Stress–Dependent Phosphorylation Activates ZNRF1 to Induce Neuronal/Axonal Degeneration. J. Cell Biol. 2015, 211, 881–896. [Google Scholar] [CrossRef]
- Miller, B.R.; Press, C.; Daniels, R.W.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. A Dual Leucine Kinase–Dependent Axon Self-Destruction Program Promotes Wallerian Degeneration. Nat. Neurosci. 2009, 12, 387–389. [Google Scholar] [CrossRef]
- Walker, L.J.; Summers, D.W.; Sasaki, Y.; Brace, E.J.; Milbrandt, J.; DiAntonio, A. MAPK Signaling Promotes Axonal Degeneration by Speeding the Turnover of the Axonal Maintenance Factor NMNAT2. eLife 2017, 6, e22540. [Google Scholar] [CrossRef]
- Neukomm, L.J.; Burdett, T.C.; Seeds, A.M.; Hampel, S.; Coutinho-Budd, J.C.; Farley, J.E.; Wong, J.; Karadeniz, Y.B.; Osterloh, J.M.; Sheehan, A.E.; et al. Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron 2017, 95, 78–91.e5. [Google Scholar] [CrossRef]
- Spencer, P.S.; Schaumburg, H.H. Central Peripheral Distal Axonopathy: The Pathology of Dying Back Poly Neuropathies. In Progress in Neuropathology; Grune & Stratton: New York, NY, USA, 1977; pp. 253–295. [Google Scholar]
- Cavanagh, J.B. The “dying Back” Process. A Common Denominator in Many Naturally Occurring and Toxic Neuropathies. Arch. Pathol. Lab. Med. 1979, 103, 659–664. [Google Scholar] [PubMed]
- Raff, M.C.; Whitmore, A.V.; Finn, J.T. Axonal Self-Destruction and Neurodegeneration. Science 2002, 296, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Cavaletti, G.; Lauria, G.; Meregalli, C. Ubiquitin Proteasome System and Microtubules Are Master Regulators of Central and Peripheral Nervous System Axon Degeneration. Cells 2022, 11, 1358. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, J.J.; Ghiretti, A.E.; Holzbaur, E.L.F. The Impact of Cytoskeletal Organization on the Local Regulation of Neuronal Transport. Nat. Rev. Neurosci. 2017, 18, 585–597. [Google Scholar] [CrossRef]
- Midroni, G.; Bilbao, J. Biopsy Diagnosis of Peripheral Neuropathy; Butterworth-Heinemann: Boston, MA, USA, 1995. [Google Scholar]
- Catenaccio, A.; Hurtado, M.L.; Diaz, P.; Lamont, D.J.; Wishart, T.M.; Court, F.A. Molecular Analysis of Axonal-Intrinsic and Glial-Associated Co-Regulation of Axon Degeneration. Cell Death Dis. 2017, 8, e3166. [Google Scholar] [CrossRef]
- Jablonka-Shariff, A.; Lu, C.; Campbell, K.; Monk, K.R.; Snyder-Warwick, A.K. Gpr126/Adgrg6 Contributes to the Terminal Schwann Cell Response at the Neuromuscular Junction Following Peripheral Nerve Injury. Glia 2020, 68, 1182–1200. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.; Coleman, M.P. Lessons from Injury: How Nerve Injury Studies Reveal Basic Biological Mechanisms and Therapeutic Opportunities for Peripheral Nerve Diseases. Neurotherapeutics 2021, 18, 2200–2221. [Google Scholar] [CrossRef]
- Hirata, K.; Kawabuchi, M. Myelin Phagocytosis by Macrophages and Nonmacrophages during Wallerian Degeneration. Microsc. Res. Tech. 2002, 57, 541–547. [Google Scholar] [CrossRef]
- LeBlanc, A.C.; Poduslo, J.F. Axonal Modulation of Myelin Gene Expression in the Peripheral Nerve. J. Neurosci. Res. 1990, 26, 317–326. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann Cell Autophagy, Myelinophagy, Initiates Myelin Clearance from Injured Nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.M.; Taniuchi, M.; DiStefano, P.S. Expression and Possible Function of Nerve Growth Factor Receptors on Schwann Cells. Trends Neurosci. 1988, 11, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Kioussi, C.; Gruss, P. Making of a Schwann. Trends Genet. 1996, 12, 84–86. [Google Scholar] [CrossRef]
- Zorick, T.S.; Syroid, D.E.; Arroyo, E.; Scherer, S.S.; Lemke, G. The Transcription Factors SCIP and Krox-20 Mark Distinct Stages and Cell Fates in Schwann Cell Differentiation. Mol. Cell. Neurosci. 1996, 8, 129–145. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Pilch, K.S.; van der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. C-Jun Reprograms Schwann Cells of Injured Nerves to Generate a Repair Cell Essential for Regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef]
- Atanasoski, S.; Scherer, S.S.; Sirkowski, E.; Leone, D.; Garratt, A.N.; Birchmeier, C.; Suter, U. ErbB2 Signaling in Schwann Cells Is Mostly Dispensable for Maintenance of Myelinated Peripheral Nerves and Proliferation of Adult Schwann Cells after Injury. J. Neurosci. 2006, 26, 2124–2131. [Google Scholar] [CrossRef]
- Fricker, F.R.; Antunes-Martins, A.; Galino, J.; Paramsothy, R.; La Russa, F.; Perkins, J.; Goldberg, R.; Brelstaff, J.; Zhu, N.; McMahon, S.B.; et al. Axonal Neuregulin 1 Is a Rate Limiting but Not Essential Factor for Nerve Remyelination. Brain 2013, 136, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Fricker, F.R.; Lago, N.; Balarajah, S.; Tsantoulas, C.; Tanna, S.; Zhu, N.; Fageiry, S.K.; Jenkins, M.; Garratt, A.N.; Birchmeier, C.; et al. Axonally Derived Neuregulin-1 Is Required for Remyelination and Regeneration after Nerve Injury in Adulthood. J. Neurosci. 2011, 31, 3225–3233. [Google Scholar] [CrossRef]
- Stassart, R.M.; Fledrich, R.; Velanac, V.; Brinkmann, B.G.; Schwab, M.H.; Meijer, D.; Sereda, M.W.; Nave, K.-A. A Role for Schwann Cell–Derived Neuregulin-1 in Remyelination. Nat. Neurosci. 2013, 16, 48–54. [Google Scholar] [CrossRef]
- Han, S.B.; Kim, H.; Lee, H.; Grove, M.; Smith, G.M.; Son, Y.-J. Postinjury Induction of Activated ErbB2 Selectively Hyperactivates Denervated Schwann Cells and Promotes Robust Dorsal Root Axon Regeneration. J. Neurosci. 2017, 37, 10955–10970. [Google Scholar] [CrossRef]
- Michailov, G.V.; Sereda, M.W.; Brinkmann, B.G.; Fischer, T.M.; Haug, B.; Birchmeier, C.; Role, L.; Lai, C.; Schwab, M.H.; Nave, K.-A. Axonal Neuregulin-1 Regulates Myelin Sheath Thickness. Science 2004, 304, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Stassart, R.M.; Möbius, W.; Nave, K.A.; Edgar, J.M. The Axon-Myelin Unit in Development and Degenerative Disease. Front. Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef]
- Brück, W. The Role of Macrophages in Wallerian Degeneration. Brain Pathol. 1997, 7, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Msheik, Z.; El Massry, M.; Rovini, A.; Billet, F.; Desmoulière, A. The Macrophage: A Key Player in the Pathophysiology of Peripheral Neuropathies. J. Neuroinflammation 2022, 19, 97. [Google Scholar] [CrossRef]
- Brück, W.; Brück, Y.; Maruschak, B.; Friede, R.L. Mechanisms of Macrophage Recruitment in Wallerian Degeneration. Acta Neuropathol. 1995, 89, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Perrin, F.E. Involvement of Monocyte Chemoattractant Protein-1, Macrophage Inflammatory Protein-1 and Interleukin-1 in Wallerian Degeneration. Brain 2005, 128, 854–866. [Google Scholar] [CrossRef]
- Fenrich, K.; Gordon, T. Canadian Association of Neuroscience Review: Axonal Regeneration in the Peripheral and Central Nervous Systems—Current Issues and Advances. Can. J. Neurol. Sci./J. Can. Des Sci. Neurol. 2004, 31, 142–156. [Google Scholar] [CrossRef]
- Shamash, S.; Reichert, F.; Rotshenker, S. The Cytokine Network of Wallerian Degeneration: Tumor Necrosis Factor-α, Interleukin-1α, and Interleukin-1β. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [CrossRef]
- Chen, P.; Piao, X.; Bonaldo, P. Role of Macrophages in Wallerian Degeneration and Axonal Regeneration after Peripheral Nerve Injury. Acta Neuropathol. 2015, 130, 605–618. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian Degeneration: Gaining Perspective on Inflammatory Events after Peripheral Nerve Injury. J. Neuroinflammation 2011, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, W.; Yamasaki, R.; Hashimoto, Y.; Ko, S.; Kobayakawa, Y.; Isobe, N.; Matsushita, T.; Kira, J. ichi Clearance of Peripheral Nerve Misfolded Mutant Protein by Infiltrated Macrophages Correlates with Motor Neuron Disease Progression. Sci. Rep. 2021, 11, 16438. [Google Scholar] [CrossRef]
- Contreras, E.; Bolívar, S.; Navarro, X.; Udina, E. New Insights into Peripheral Nerve Regeneration: The Role of Secretomes. Exp. Neurol. 2022, 354, 114069. [Google Scholar] [CrossRef]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann Cell Update: Adaptive Reprogramming, EMT, and Stemness in Regenerating Nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Akram, R.; Anwar, H.; Javed, M.S.; Rasul, A.; Imran, A.; Malik, S.A.; Raza, C.; Khan, I.U.; Sajid, F.; Iman, T.; et al. Axonal Regeneration: Underlying Molecular Mechanisms and Potential Therapeutic Targets. Biomedicines 2022, 10, 3186. [Google Scholar] [CrossRef] [PubMed]
- Fontana, X.; Hristova, M.; Da Costa, C.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.R.; et al. C-Jun in Schwann Cells Promotes Axonal Regeneration and Motoneuron Survival via Paracrine Signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef]
- Ito, Y.; Yamamoto, M.; Li, M.; Doyu, M.; Tanaka, F.; Mutch, T.; Mitsuma, T.; Sobue, G. Differential Temporal Expression of MRNAs for Ciliary Neurotrophic Factor (CNTF), Leukemia Inhibitory Factor (LIF), Interleukin-6 (IL-6), and Their Receptors (CNTFRα, LIFRβ, IL-6Rα and Gp130) in Injured Peripheral Nerves. Brain Res. 1998, 793, 321–327. [Google Scholar] [CrossRef]
- Cheng, H.; Randolph, A.; Yee, D.; Delafontaine, P.; Tennekoon, G.; Feldman, E.L. Characterization of Insulin-Like Growth Factor-I and Its Receptor and Binding Proteins in Transected Nerves and Cultured Schwann Cells. J. Neurochem. 1996, 66, 525–536. [Google Scholar] [CrossRef]
- Araki, T.; Milbrandt, J. Ninjurin, a Novel Adhesion Molecule, Is Induced by Nerve Injury and Promotes Axonal Growth. Neuron 1996, 17, 353–361. [Google Scholar] [CrossRef]
- Dyck, P.; Dick, J.; Engelstad, J. Pathologic Alterations of Nerves. In Peripheral Neuropathy; Elsevier-Saunders: Philadelphia, PA, USA, 2005; pp. 733–829. [Google Scholar]
- Martini, R. Expression and Functional Roles of Neural Cell Surface Molecules and Extracellular Matrix Components during Development and Regeneration of Peripheral Nerves. J. Neurocytol. 1994, 23, 1–28. [Google Scholar] [CrossRef]
- Brügger, V.; Duman, M.; Bochud, M.; Münger, E.; Heller, M.; Ruff, S.; Jacob, C. Delaying Histone Deacetylase Response to Injury Accelerates Conversion into Repair Schwann Cells and Nerve Regeneration. Nat. Commun. 2017, 8, 14272. [Google Scholar] [CrossRef]
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmaine, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef]
- Ma, K.H.; Svaren, J. Epigenetic Control of Schwann Cells. Neuroscientist 2018, 24, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Fugleholm, K.; Schmalbruch, H.; Krarup, C. Early Peripheral Nerve Regeneration after Crushing, Sectioning, and Freeze Studied by Implanted Electrodes in the Cat. J. Neurosci. 1994, 14, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Müller, H.W. Nerve Injury, Axonal Degeneration and Neural Regeneration: Basic Insights. Brain Pathol. 1999, 9, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Martini, R.; Schachner, M.; Brushart, T. The L2/HNK-1 Carbohydrate Is Preferentially Expressed by Previously Motor Axon-Associated Schwann Cells in Reinnervated Peripheral Nerves. J. Neurosci. 1994, 14, 7180–7191. [Google Scholar] [CrossRef]
- Capodivento, G.; Camera, M.; Liessi, N.; Trada, A.; Debellis, D.; Schenone, A.; Armirotti, A.; Visigalli, D.; Nobbio, L. Monitoring Myelin Lipid Composition and the Structure of Myelinated Fibers Reveals a Maturation Delay in CMT1A. Int. J. Mol. Sci. 2024, 25, 11244. [Google Scholar] [CrossRef]
- Yuan, A.; Nixon, R.A. Neurofilament Proteins as Biomarkers to Monitor Neurological Diseases and the Efficacy of Therapies. Front. Neurosci. 2021, 15, 689938. [Google Scholar] [CrossRef]
- Petzold, A. The 2022 Lady Estelle Wolfson Lectureship on Neurofilaments. J. Neurochem. 2022, 163, 179–219. [Google Scholar] [CrossRef]
- Nardocci, N.; Zorzi, G. Axonal Dystrophies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 1919–1924. [Google Scholar]
- Friede, R.L. Axon Dystrophies. In Developmental Neuropathology; Springer: Berlin/Heidelberg, Germany, 1989; pp. 552–560. ISBN 978-3-642-73699-5. [Google Scholar]
- Vuia, O. Neuroaxonal Dystrophy, a Juvenile-Adult Form. Clin. Neurol. Neurosurg. 1976, 79, 307–315. [Google Scholar] [CrossRef]
- Williamson, K.; Sima, A.A.F.; Curry, B.; Ludwin, S.K. Neuroaxonal Dystrophy in Young Adults: A Clinicopathological Study of Two Unrelated Cases. Ann. Neurol. 1982, 11, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Sasaki, Y.; Warlo, I.; Goebel, H.H. Axonal Pathology of the Skin in Infantile Neuroaxonal Dystrophy. Acta Neuropathol. 1987, 75, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.D.; Litt, M.; Kramer, P.; Pandolfo, M.; Angelini, L.; Nardocci, N.; Davis, S.; Pineda, M.; Hattori, H.; Flett, P.J.; et al. Homozygosity Mapping of Hallervorden–Spatz Syndrome to Chromosome 20p12.3–P13. Nat. Genet. 1996, 14, 479–481. [Google Scholar] [CrossRef]
- Cork, L.C.; Troncoso, J.C.; Price, D.L.; Stanley, E.F.; Griffin, J.W. Canine Neuroaxonal Dystrophy. J. Neuropathol. Exp. Neurol. 1983, 42, 286–296. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Plurad, S.B.; Modert, C.W. Neuroaxonal Dystrophy in the Autonomic Ganglia of Aged Rats. J. Neuropathol. Exp. Neurol. 1983, 42, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Chrisman, C.L.; Cork, L.C.; Gamble, D.A. Neuroaxonal Dystrophy of Rottweiler Dogs. J. Am. Vet. Med. Assoc. 1984, 184, 464–467. [Google Scholar] [CrossRef]
- Carmichael, K.P.; Howerth, E.W.; Oliver, J.E.; Klappenbach, K. Neuroaxonal Dystrophy in a Group of Related Cats. J. Vet. Diagn. Investig. 1993, 5, 585–590. [Google Scholar] [CrossRef]
- Sacre, B.J.; Cummings, J.F.; De Lahunta, A. Neuroaxonal Dystrophy in a Jack Russell Terrier Pup Resembling Human Infantile Neuroaxonal Dystrophy. Cornell Vet. 1993, 83, 133–142. [Google Scholar]
- Franklin, R.J.M.; Jeffery, N.D.; Ramsey, I.K. Neuroaxonal Dystrophy in a Litter of Papillon Pups. J. Small Anim. Pract. 1995, 36, 441–444. [Google Scholar] [CrossRef]
- Adams, A.P.; Collatos, C.; Fuentealba, C.; Illanes, O.; Blanchard, R. Neuroaxonal Dystrophy in a Two-Year-Old Quarter Horse Filly. Can. Vet. J. 1996, 37, 43–44. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Gurney, M.E. Development of Central Nervous System Pathology in a Murine Transgenic Model of Human Amyotrophic Lateral Sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar] [PubMed]
- Schmidt, R.E.; Dorsey, D.A.; Beaudet, L.N.; Plurad, S.B.; Parvin, C.A.; Bruch, L.A. Vacuolar Neuritic Dystrophy in Aged Mouse Superior Cervical Sympathetic Ganglia Is Strain-Specific. Brain Res. 1998, 806, 141–151. [Google Scholar] [CrossRef]
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the Brain and Testis Results in Progressive Neurodegeneration and Sterility in Mice. Nat. Genet. 2000, 26, 300–306. [Google Scholar] [CrossRef]
- Liedtke, W.; Leman, E.E.; Fyffe, R.E.W.; Raine, C.S.; Schubart, U.K. Stathmin-Deficient Mice Develop an Age-Dependent Axonopathy of the Central and Peripheral Nervous Systems. Am. J. Pathol. 2002, 160, 469–480. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kikuchi, T.; Kikuchi, H.; Ichihara, N.; Ishikawa, A.; Ishijima, Y.; Tachibana, M. A New Mouse Model for Infantile Neuroaxonal Dystrophy, Inad Mouse, Maps to Mouse Chromosome 1. Mamm. Genome 2005, 16, 73–78. [Google Scholar] [CrossRef]
- Schmidt, R.E. Synaptic Dysplasia in Sympathetic Autonomic Ganglia. J. Neurocytol. 1996, 25, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Groves, M.; Miedzybrodzka, Z.; Roper, H.; Willis, T.; Winer, J.; Cole, G.; Reilly, M.M. New Mutations, Genotype Phenotype Studies and Manifesting Carriers in Giant Axonal Neuropathy. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Lampert, P.W. A Comparative Electron Microscopic Study of Reactive, Degenerating, Regenerating, and Dystrophic Axons. J. Neuropathol. Exp. Neurol. 1967, 26, 345–368. [Google Scholar] [CrossRef]
- Koestner, A.; Norton, S. Nervous System. In A Handbook of Toxicologic Pathology; Academic Press: San Diego, CA, USA, 1991; pp. 625–674. [Google Scholar]
- Bouley, D.M.; McIntire, J.J.; Harris, B.T.; Tolwani, R.J.; Otto, G.M.; DeKruyff, R.H.; Hayflick, S.J. Spontaneous Murine Neuroaxonal Dystrophy: A Model of Infantile Neuroaxonal Dystrophy. J. Comp. Pathol. 2006, 134, 161–170. [Google Scholar] [CrossRef]
- Korhonen, L.; Lindholm, D. The Ubiquitin Proteasome System in Synaptic and Axonal Degeneration. J. Cell Biol. 2004, 165, 27–30. [Google Scholar] [CrossRef]
- Edgar, J.M.; Garbern, J. The Myelinated Axon Is Dependent on the Myelinating Cell for Support and Maintenance: Molecules Involved. J. Neurosci. Res. 2004, 76, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, E.J.; Xu, Y.T.; Zhou, L.; Messing, A.; Peles, E.; Chiu, S.Y.; Scherer, S.S. Myelinating Schwann Cells Determine the Internodal Localization of Kv1.1, Kv1.2, Kvbeta2, and Caspr. J. Neurocytol. 1999, 28, 333–347. [Google Scholar] [CrossRef]
- de Waegh, S.M.; Lee, V.M.-Y.; Brady, S.T. Local Modulation of Neurofilament Phosphorylation, Axonal Caliber, and Slow Axonal Transport by Myelinating Schwann Cells. Cell 1992, 68, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Angelides, K. Clustering of Voltage-Dependent Sodium Channels on Axons Depends on Schwann Cell Contact. Nature 1992, 356, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Thomas, P.K. Peripheral Neuropathy; Elsevier-Saunders: Philadelphia, PA, USA, 2005. [Google Scholar]
- Li, J. Molecular Regulators of Nerve Conduction—Lessons from Inherited Neuropathies and Rodent Genetic Models. Exp. Neurol. 2015, 267, 209–218. [Google Scholar] [CrossRef]
- Uncini, A.; Cavallaro, T.; Fabrizi, G.M.; Manganelli, F.; Vallat, J.M. Conduction Slowing, Conduction Block and Temporal Dispersion in Demyelinating, Dysmyelinating and Axonal Neuropathies: Electrophysiology Meets Pathology. J. Peripher. Nerv. Syst. 2024, 29, 135–160. [Google Scholar] [CrossRef]
- Klein, D.; Martini, R. Myelin and Macrophages in the PNS: An Intimate Relationship in Trauma and Disease. Brain Res. 2016, 1641, 130–138. [Google Scholar] [CrossRef]
- Nobbio, L.; Mancardi, G.; Grandis, M.; Levi, G.; Suter, U.; Nave, K.; Windebank, A.J.; Abbruzzese, M.; Schenone, A. PMP22 Transgenic Dorsal Root Ganglia Cultures Show Myelin Abnormalities Similar to Those of Human CMT1A. Ann. Neurol. 2001, 50, 47–55. [Google Scholar] [CrossRef]
- Hanemann, C.O.; Gabreëls-Festen, A.A.W.M.; Stoll, G.; Müller, H.W. Schwann Cell Differentiation in Charcot-Marie-Tooth Disease Type 1A (CMT1A): Normal Number of Myelinating Schwann Cells in Young CMT1A Patients and Neural Cell Adhesion Molecule Expression in Onion Bulbs. Acta Neuropathol. 1997, 94, 310–315. [Google Scholar] [CrossRef]
- Uncini, A.; Kuwabara, S. Nodopathies of the Peripheral Nerve: An Emerging Concept. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1186–1195. [Google Scholar] [CrossRef]
- Dolma, S.; Joshi, A. The Node of Ranvier as an Interface for Axo-Glial Interactions: Perturbation of Axo-Glial Interactions in Various Neurological Disorders. J. Neuroimmune Pharmacol. 2023, 18, 215–234. [Google Scholar] [PubMed]
- Uncini, A. Autoimmune Nodo-paranodopathies 10 Years Later: Clinical Features, Pathophysiology and Treatment. J. Peripher. Nerv. Syst. 2023, 28, S23–S35. [Google Scholar] [CrossRef]
- Uncini, A.; Susuki, K.; Yuki, N. Nodo-Paranodopathy: Beyond the Demyelinating and Axonal Classification in Anti-Ganglioside Antibody-Mediated Neuropathies. Clin. Neurophysiol. 2013, 124, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- McGonigal, R.; Campbell, C.I.; Barrie, J.A.; Yao, D.; Cunningham, M.E.; Crawford, C.L.; Rinaldi, S.; Rowan, E.G.; Willison, H.J. Schwann Cell Nodal Membrane Disruption Triggers Bystander Axonal Degeneration in a Guillain-Barré Syndrome Mouse Model. J. Clin. Investig. 2022, 132, e158524. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, P.Y.K.; van Doorn, P.A.; Hadden, R.D.M.; Avau, B.; Vankrunkelsven, P.; Allen, J.A.; Attarian, S.; Blomkwist-Markens, P.H.; Cornblath, D.R.; Eftimov, F.; et al. European Academy of Neurology/Peripheral Nerve Society Guideline on Diagnosis and Treatment of Chronic Inflammatory Demyelinating Polyradiculoneuropathy: Report of a Joint Task Force—Second Revision. Eur. J. Neurol. 2021, 28, 3556–3583. [Google Scholar] [CrossRef]
- Querol, L.; Dalakas, M.C. The Discovery of Autoimmune Nodopathies and the Impact of IgG4 Antibodies in Autoimmune Neurology. Neurol. Neuroimmunol. Neuroinflammation 2025, 12, e200365. [Google Scholar] [CrossRef]
- Garcia, A.; Combarros, O.; Calleja, J.; Berciano, J. Charcot-Marie-Tooth Disease Type 1A with 17p Duplication in Infancy and Early Childhood A Longitudinal Clinical and Electrophysiologic Study. Neurology 1998, 50, 1061–1067. [Google Scholar]
- Yiu, E.M.; Burns, J.; Ryan, M.M.; Ouvrier, R.A. Neurophysiologic Abnormalities in Children with Charcot-Marie-Tooth Disease Type 1A. J. Peripher. Nerv. Syst. 2008, 13, 236–241. [Google Scholar] [CrossRef]
- Manganelli, F.; Pisciotta, C.; Reilly, M.M.; Tozza, S.; Schenone, A.; Fabrizi, G.M.; Cavallaro, T.; Vita, G.; Padua, L.; Gemignani, F.; et al. Nerve Conduction Velocity in CMT1A: What Else Can We Tell? Eur. J. Neurol. 2016, 23, 1566–1571. [Google Scholar] [CrossRef]
- Shy, M.E.; Chen, L.; Swan, M.E.R.; Taube, B.R.; Krajewski, K.M.; Herrmann, D.; Lewis, R.A.; Mcdermott, M.P. Neuropathy Progression in Charcot-Marie-Tooth Disease Type 1A. Neurology 2008, 70, 378–383. [Google Scholar]
- Verhamme, C.; Van Schaik, I.N.; Koelman, J.H.T.M.; De Haan, R.J.; De Visser, M. The Natural History of Charcot-Marie-Tooth Type 1A in Adults: A 5-Year Follow-up Study. Brain 2009, 132, 3252–3262. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, F.; Nolano, M.; Pisciotta, C.; Provitera, V.; Fabrizi, G.M.; Cavallaro, T.; Stancanelli, A.; Caporaso, G.; Shy, M.E.; Santoro, L. Charcot-Marie-Tooth Disease: New Insights from Skin Biopsy. Neurology 2015, 85, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Saporta, M.A.; Katona, I.; Lewis, R.A.; Masse, S.; Shy, M.E.; Li, J. Shortened Internodal Length of Dermal Myelinated Nerve Fibres in Charcot-Marie-Tooth Disease Type 1A. Brain 2009, 132, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, K.M.; Lewis, R.A.; Fuerst, D.R.; Turansky, C.; Hinderer, S.R.; Garbern, J.; Kamholz, J.; Shy, M.E. Neurological Dysfunction and Axonal Degeneration in Charcot-Marie-Tooth Disease Type 1A. Brain 2000, 123, 1516–1527. [Google Scholar] [CrossRef]
- Li, J. Caveats in the Established Understanding of CMT1A. Ann. Clin. Transl. Neurol. 2017, 4, 601–607. [Google Scholar] [CrossRef]
- Oka, N.; Kawasaki, T.; Unuma, T.; Shigematsu, K.; Sugiyama, H. Different Profiles of Onion Bulb in CIDP and CMT1A in Relation to Extracellular Matrix. Clin. Neuropathol. 2013, 32, 406–412. [Google Scholar] [CrossRef]
- Fledrich, R.; Kungl, T.; Nave, K.A.; Stassart, R.M. Axo-Glial Interdependence in Peripheral Nerve Development. Development 2019, 146, dev151704. [Google Scholar] [CrossRef]
- Fledrich, R.; Abdelaal, T.; Rasch, L.; Bansal, V.; Schütza, V.; Brügger, B.; Lüchtenborg, C.; Prukop, T.; Stenzel, J.; Rahman, R.U.; et al. Targeting Myelin Lipid Metabolism as a Potential Therapeutic Strategy in a Model of CMT1A Neuropathy. Nat. Commun. 2018, 9, 3025. [Google Scholar] [CrossRef]
- Visigalli, D.; Capodivento, G.; Basit, A.; Fernández, R.; Hamid, Z.; Pencová, B.; Gemelli, C.; Marubbi, D.; Pastorino, C.; Luoma, A.M.; et al. Exploiting Sphingo- and Glycerophospholipid Impairment to Select Effective Drugs and Biomarkers for CMT1A. Front. Neurol. 2020, 11, 903. [Google Scholar] [CrossRef]
- Dyck, P.J.; Johnson, W.J.; Lambert, E.H.; O’Brien, P.C. Segmental Demyelination Secondary to Axonal Degeneration in Uremic Neuropathy. Mayo Clin. Proc. 1971, 46, 400–431. [Google Scholar]
- Gabriel, J.-M.; Erne, B.; Pareyson, D.; Sghirlanzoni, A.; Taroni, F.; Steck, A.J. Gene Dosage Effects in Hereditary Peripheral Neuropathy. Neurology 1997, 49, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Veneri, F.A.; Prada, V.; Mastrangelo, R.; Ferri, C.; Nobbio, L.; Passalacqua, M.; Milanesi, M.; Bianchi, F.; Del Carro, U.; Vallat, J.-M.; et al. A Novel Mouse Model of CMT1B Identifies Hyperglycosylation as a New Pathogenetic Mechanism. Hum. Mol. Genet. 2022, 31, 4255–4274. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schenone, A.; Massucco, S.; Schenone, C.; Venturi, C.B.; Nozza, P.; Prada, V.; Pomili, T.; Di Patrizi, I.; Capodivento, G.; Nobbio, L.; et al. Basic Pathological Mechanisms in Peripheral Nerve Diseases. Int. J. Mol. Sci. 2025, 26, 3377. https://doi.org/10.3390/ijms26073377
Schenone A, Massucco S, Schenone C, Venturi CB, Nozza P, Prada V, Pomili T, Di Patrizi I, Capodivento G, Nobbio L, et al. Basic Pathological Mechanisms in Peripheral Nerve Diseases. International Journal of Molecular Sciences. 2025; 26(7):3377. https://doi.org/10.3390/ijms26073377
Chicago/Turabian StyleSchenone, Angelo, Sara Massucco, Cristina Schenone, Consuelo Barbara Venturi, Paolo Nozza, Valeria Prada, Tania Pomili, Irene Di Patrizi, Giovanna Capodivento, Lucilla Nobbio, and et al. 2025. "Basic Pathological Mechanisms in Peripheral Nerve Diseases" International Journal of Molecular Sciences 26, no. 7: 3377. https://doi.org/10.3390/ijms26073377
APA StyleSchenone, A., Massucco, S., Schenone, C., Venturi, C. B., Nozza, P., Prada, V., Pomili, T., Di Patrizi, I., Capodivento, G., Nobbio, L., & Grandis, M. (2025). Basic Pathological Mechanisms in Peripheral Nerve Diseases. International Journal of Molecular Sciences, 26(7), 3377. https://doi.org/10.3390/ijms26073377