
The Yeast HMGB Protein Hmo1 Is a Multifaceted Regulator of DNA Damage Tolerance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
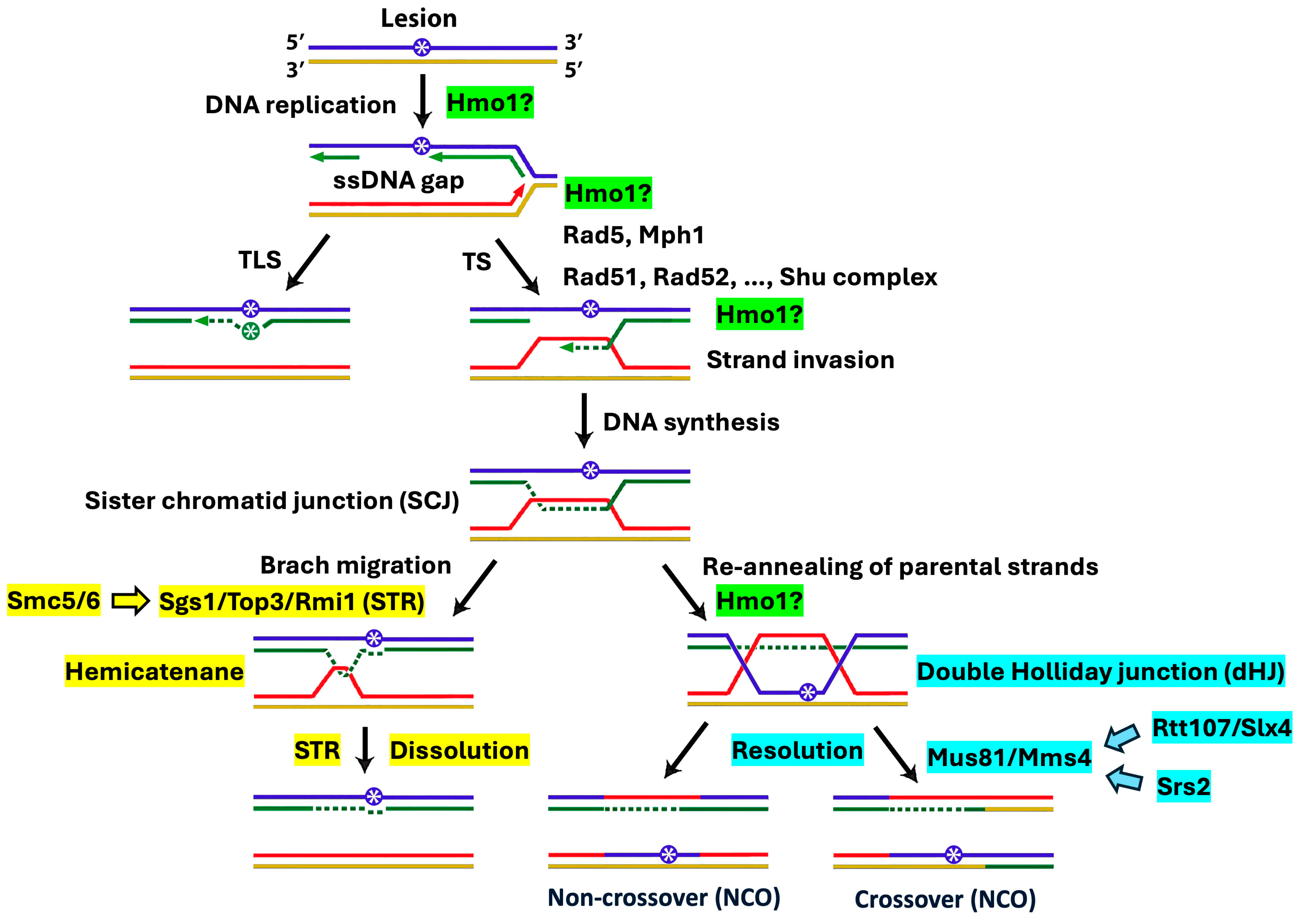
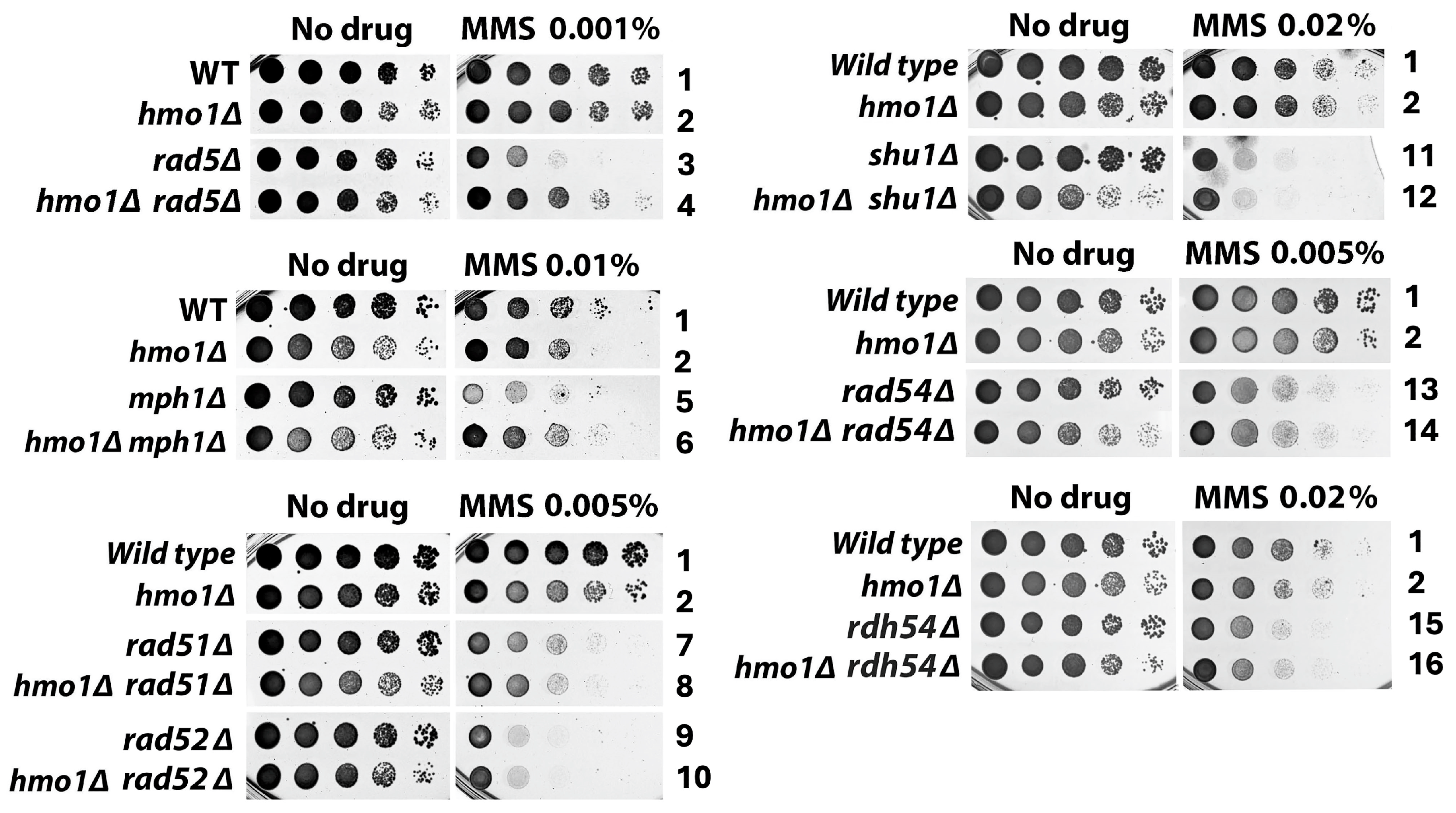
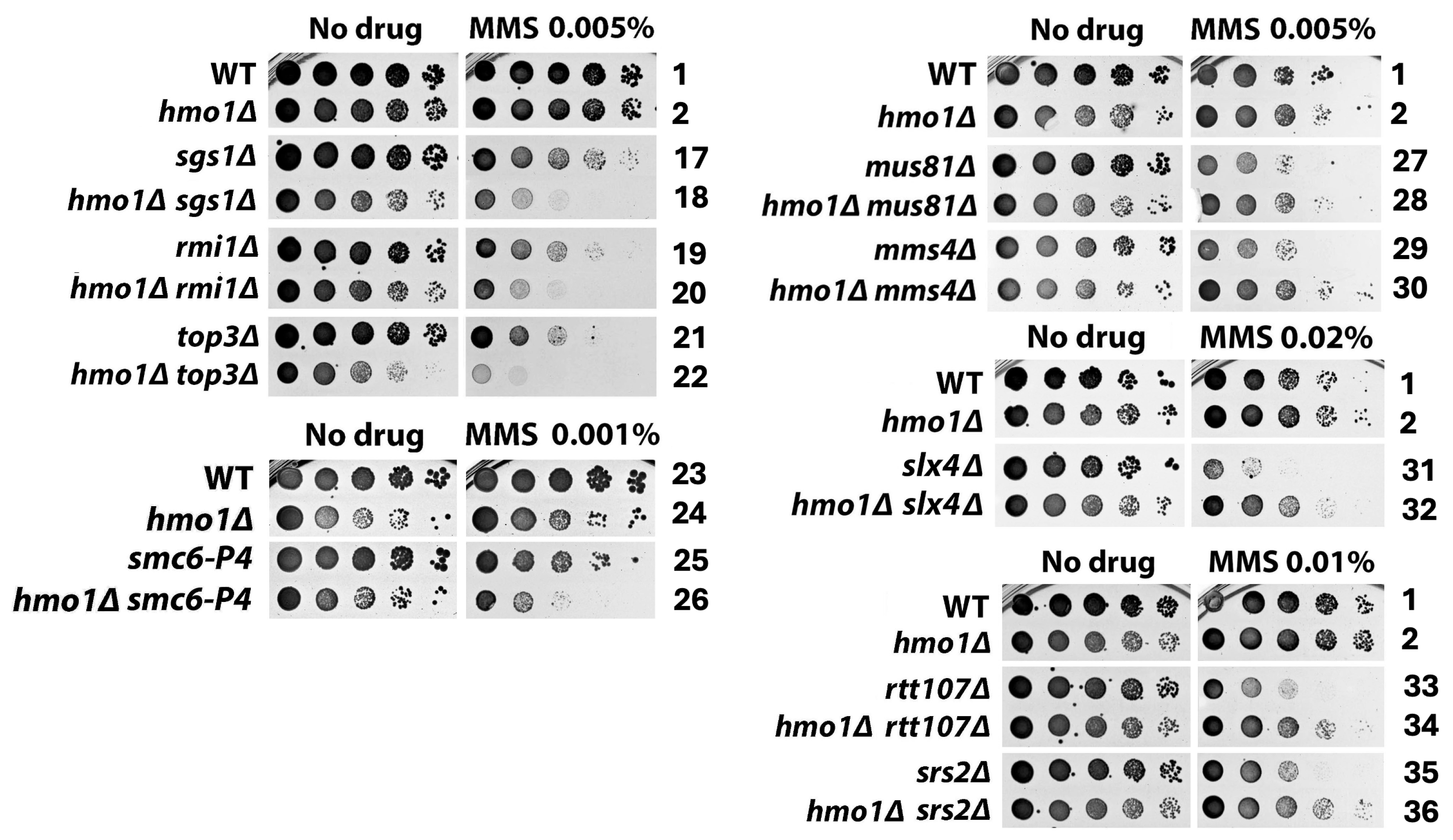
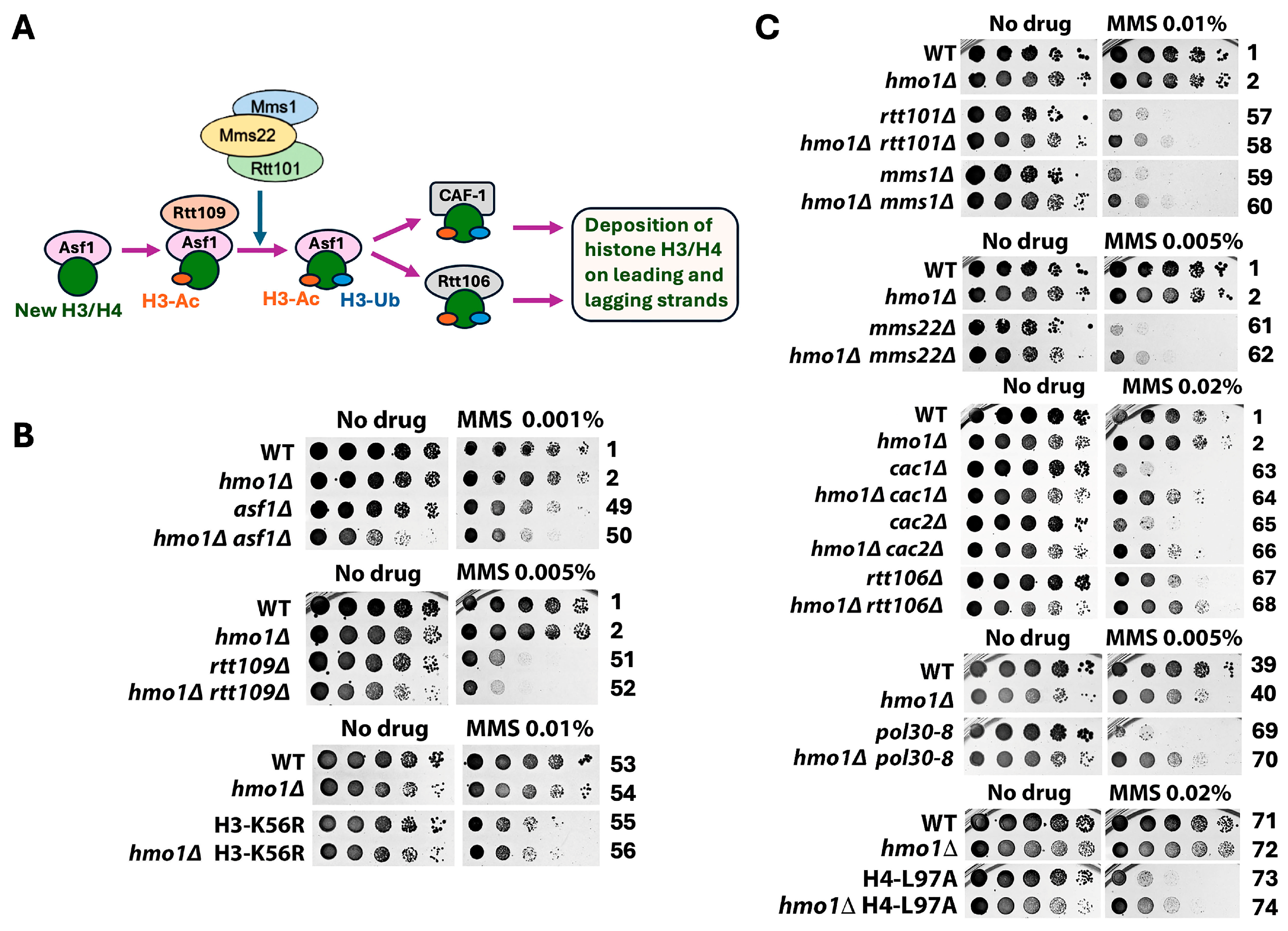
2.1. Distinct Functional Interactions of Hmo1 with DDT Factors Involved in the Formation and Processing/Removal of the SCJ Intermediate of Template Switching
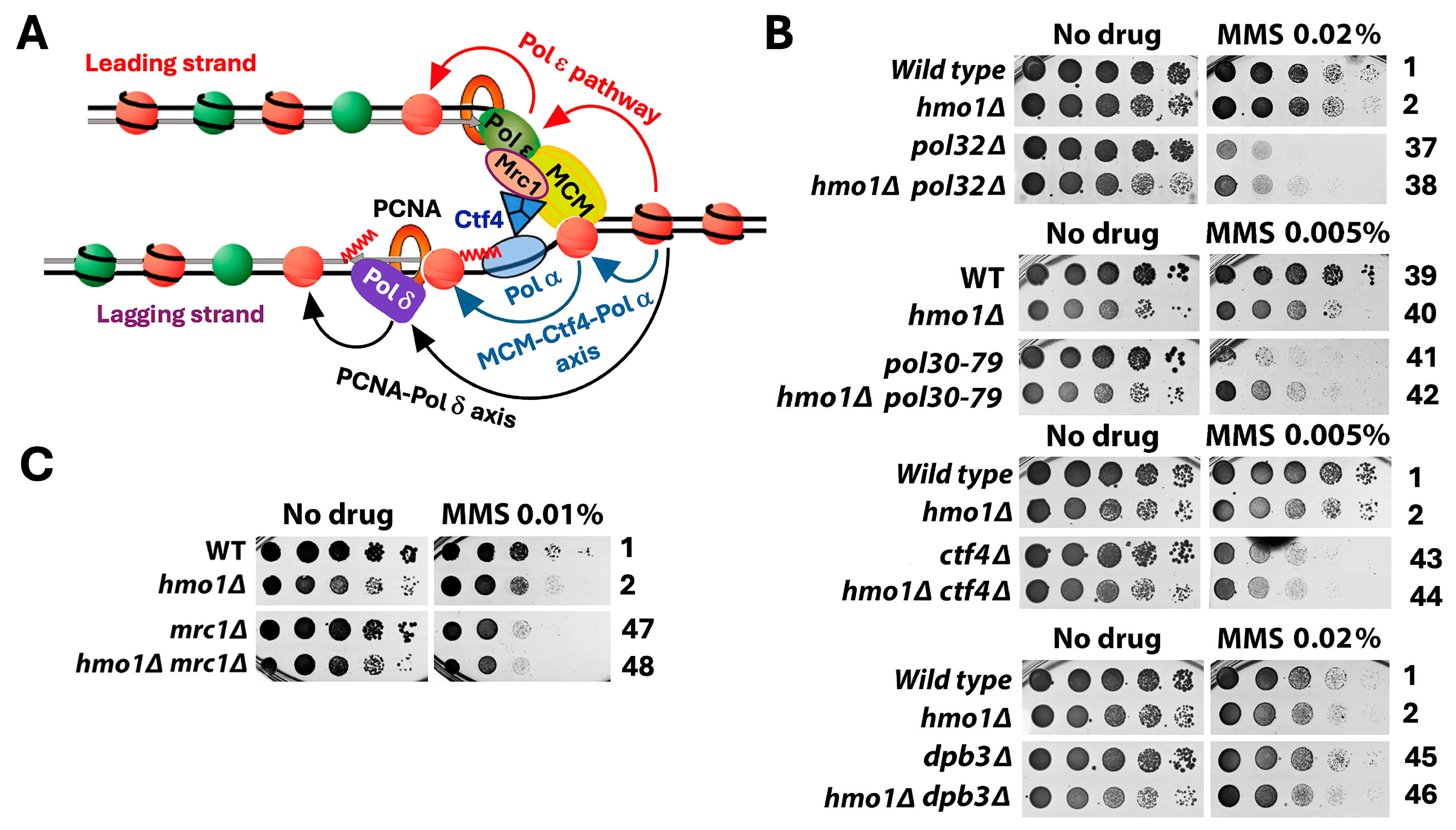
2.2. Hmo1 May Contribute to DDT by Functioning in DNA Replication-Coupled Chromatin Assembly
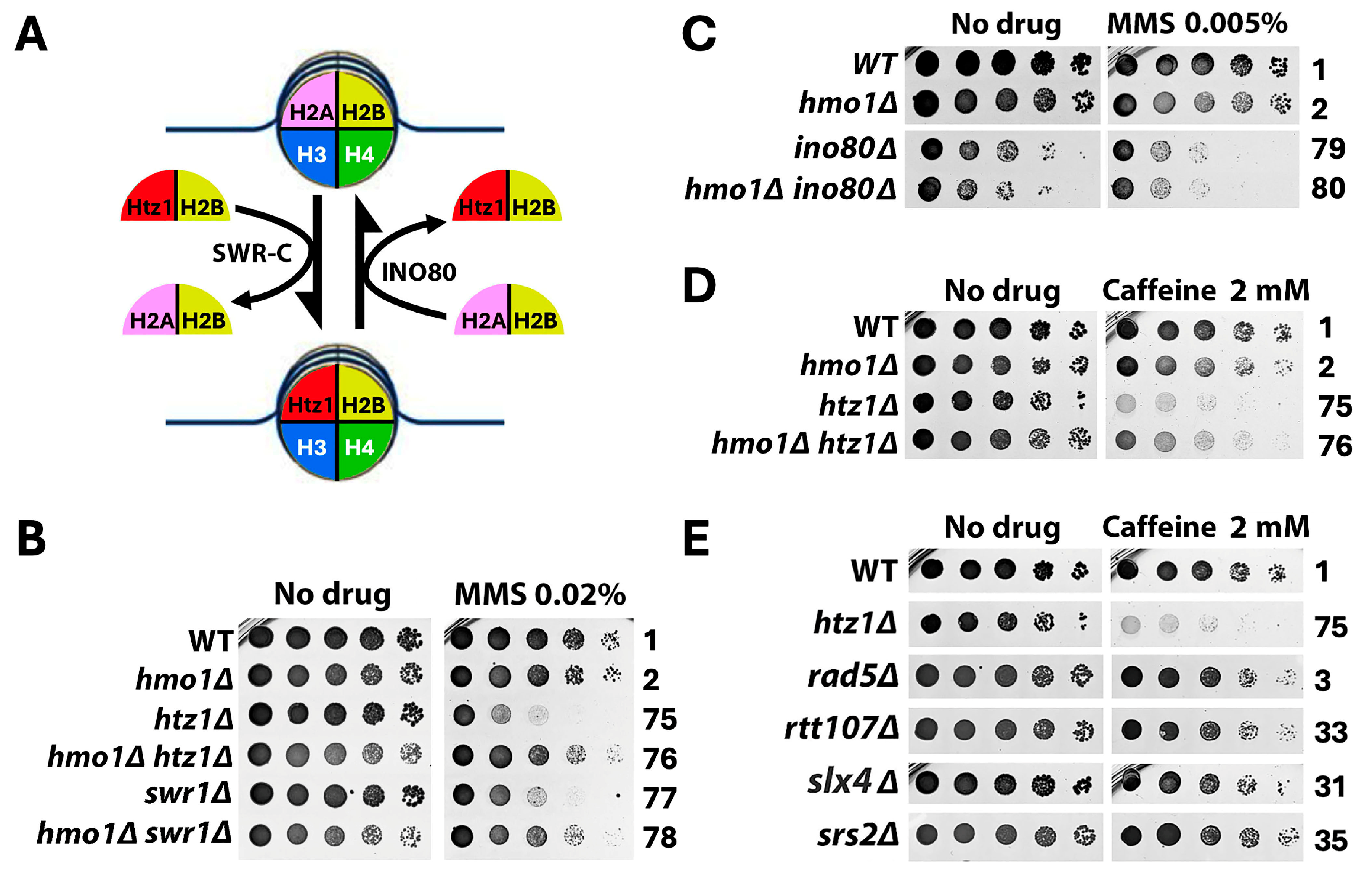
2.3. Hmo1 Antagonizes the Function of the Histone H2A Variant H2A.Z (Htz1 in Yeast) in DDT
2.4. Genetic Background-Dependent Differential Requirement of the CTD of Hmo1 for Its Function in DDT
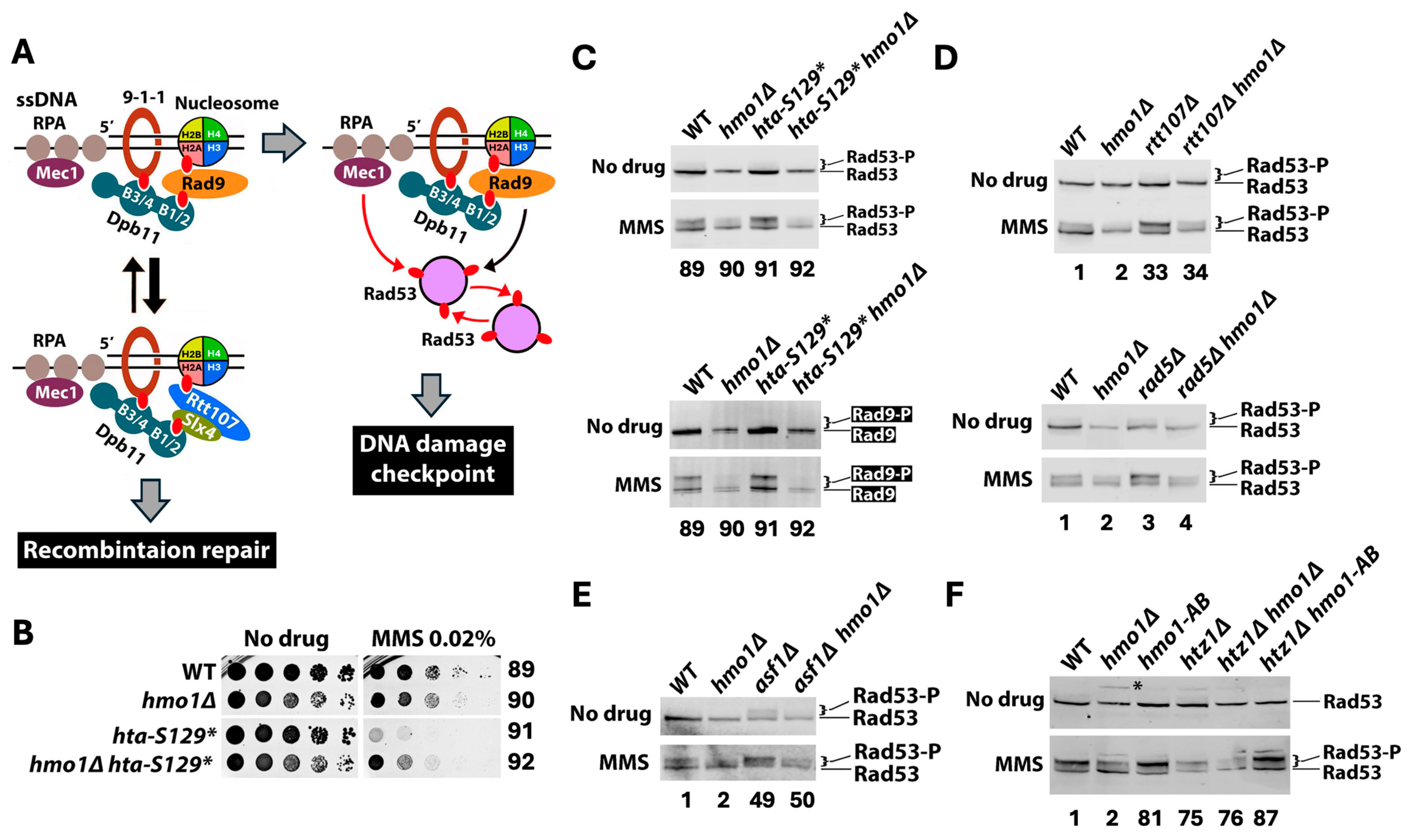
2.5. Hmo1 Promotes DNA Damage Checkpoint Signaling Induced by MMS
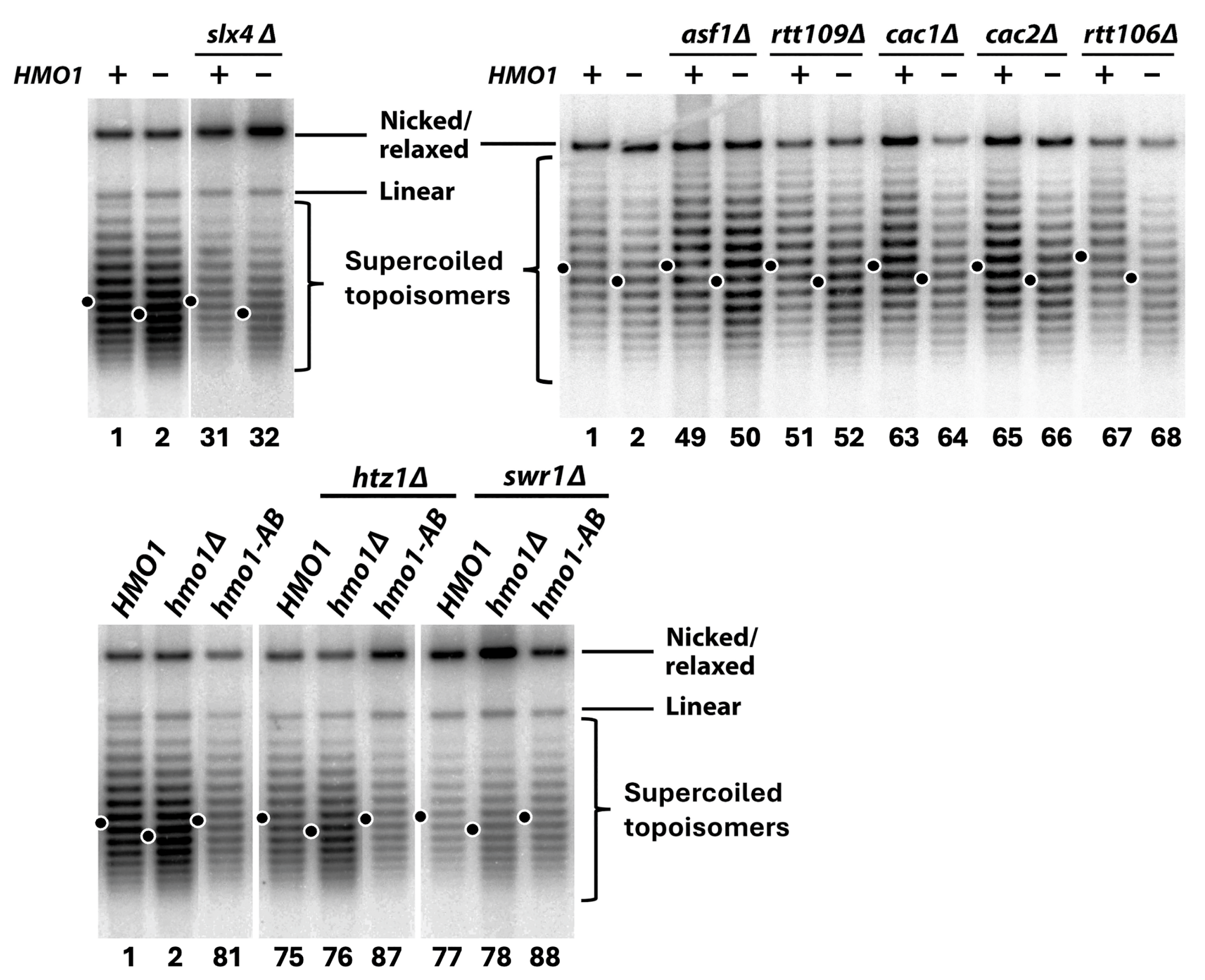
2.6. Hmo1 Regulates DDT Independently of Its Promotion of Negative DNA Supercoiling as a Proxy of the Chromatin Structure
3. Discussion
3.1. Hmo1 May Direct Persistent SCJs to the Mus81/Mms4 Nuclease-Mediated Resolution Pathway in Template Switching
3.2. Hmo1 May Regulate DDT by Modulating Replication-Coupled Chromatin Assembly
3.3. Hmo1 Counteracts the Function of Histone H2A Variant H2A.Z (Htz1) in DDT
3.4. The Contribution of the CTD of Hmo1 to DDT Is Dependent on the Genetic Background of the Host
3.5. Hmo1 Promotes DNA Damage Checkpoint Signaling
4. Materials and Methods
4.1. Yeast Strains
4.2. Examination of the Yeast Growth Phenotype
4.3. SDS-PAGE and Western Blotting
4.4. Analysis of the Topology of the Yeast 2-Micron (2μ) Plasmid
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosom core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Stros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Postnikov, Y.V.; Bustin, M. Functional interplay between histone H1 and HMG proteins in chromatin. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 462–467. [Google Scholar] [CrossRef]
- Lu, J.; Kobayashi, R.; Brill, S.J. Characterization of a high mobility group 1/2 homolog in yeast. J. Biol. Chem. 1996, 271, 33678–33685. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Grove, A. Yeast HMO1: Linker Histone Reinvented. Microbiol. Mol. Biol. Rev. 2017, 81, e00037-16. [Google Scholar] [CrossRef] [PubMed]
- Bi, X. Hmo1: A versatile member of the high mobility group box family of chromosomal architecture proteins. World J. Biol. Chem. 2024, 15, 97938. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.J.; Huo, R.; Becker, N.; Holte, M.N.; Muthurajan, U.M.; Rouzina, I.; Luger, K.; Maher, L.J., 3rd; Israeloff, N.E.; Williams, M.C. Single and double box HMGB proteins differentially destabilize nucleosomes. Nucleic Acids Res. 2019, 47, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Malinina, D.K.; Sivkina, A.L.; Korovina, A.N.; McCullough, L.L.; Formosa, T.; Kirpichnikov, M.P.; Studitsky, V.M.; Feofanov, A.V. Hmo1 Protein Affects the Nucleosome Structure and Supports the Nucleosome Reorganization Activity of Yeast FACT. Cells 2022, 11, 2931. [Google Scholar] [CrossRef]
- Hepp, M.I.; Smolle, M.; Gidi, C.; Amigo, R.; Valenzuela, N.; Arriagada, A.; Maureira, A.; Gogol, M.M.; Torrejón, M.; Workman, J.L.; et al. Role of Nhp6 and Hmo1 in SWI/SNF occupancy and nucleosome landscape at gene regulatory regions. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Amigo, R.; Farkas, C.; Gidi, C.; Hepp, M.I.; Cartes, N.; Tarifeño, E.; Workman, J.L.; Gutiérrez, J.L. The linker histone Hho1 modulates the activity of ATP-dependent chromatin remodeling complexes. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194781. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Grove, A. The high mobility group protein HMO1 functions as a linker histone in yeast. Epigenetics Chromatin 2016, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Adhil, M.; Choudhary, R.; Gilbert, N.; Foiani, M. Negative supercoil at gene boundaries modulates gene topology. Nature 2020, 577, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Xiao, L.; Grove, A. Yeast high mobility group protein HMO1 stabilizes chromatin and is evicted during repair of DNA double strand breaks. Nucleic Acids Res. 2015, 43, 5759–5770. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Xiao, L.; Gupta, A.; Grove, A. Control of DNA end resection by yeast Hmo1p affects efficiency of DNA end-joining. DNA Repair 2017, 53, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Huici, V.; Szakal, B.; Urulangodi, M.; Psakhye, I.; Castellucci, F.; Menolfi, D.; Rajakumara, E.; Fumasoni, M.; Bermejo, R.; Jentsch, S.; et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014, 33, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [PubMed]
- Bi, X. Mechanism of DNA damage tolerance. World J. Biol. Chem. 2015, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Psakhye, I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016, 40, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhao, X. Replication fork regression and its regulation. FEMS Yeast Res. 2017, 17, fow110. [Google Scholar] [CrossRef] [PubMed]
- Kamau, E.; Bauerle, K.T.; Grove, A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains. J. Biol. Chem. 2004, 279, 55234–55240. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.N.; Zhao, X. Replication-Associated Recombinational Repair: Lessons from Budding Yeast. Genes 2016, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Martino, J.; Bernstein, K.A. The Shu complex is a conserved regulator of homologous recombination. FEMS Yeast Res. 2016, 16, fow073. [Google Scholar] [CrossRef] [PubMed]
- Gaines, W.A.; Godin, S.K.; Kabbinavar, F.F.; Rao, T.; VanDemark, A.P.; Sung, P.; Bernstein, K.A. Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat. Commun. 2015, 6, 7834. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Zhang, Z.; Herken, B.W.; Westmoreland, J.W.; Lee, A.G.; Mihalevic, M.J.; Yu, Z.; Sobol, R.W.; Resnick, M.A.; Bernstein, K.A. The Shu complex promotes error-free tolerance of alkylation-induced base excision repair products. Nucleic Acids Res. 2016, 44, 8199–8215. [Google Scholar] [CrossRef] [PubMed]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP Hydrolysis Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394.e18. [Google Scholar] [CrossRef] [PubMed]
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.N.; Choi, K.; Xue, X.; Torres, N.P.; Szakal, B.; Wei, L.; Wan, B.; Arter, M.; Matos, J.; Sung, P.; et al. Smc5/6 Mediated Sumoylation of the Sgs1-Top3-Rmi1 Complex Promotes Removal of Recombination Intermediates. Cell Rep. 2016, 16, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Chavdarova, M.; Marini, V.; Sisakova, A.; Sedlackova, H.; Vigasova, D.; Brill, S.J.; Lisby, M.; Krejci, L. Srs2 promotes Mus81-Mms4-mediated resolution of recombination intermediates. Nucleic Acids Res. 2015, 43, 3626–3642. [Google Scholar] [CrossRef] [PubMed]
- Hammond-Martel, I.; Verreault, A.; Wurtele, H. Chromatin dynamics and DNA replication roadblocks. DNA Repair 2021, 104, 103140. [Google Scholar] [CrossRef] [PubMed]
- Serra-Cardona, A.; Zhang, Z. Replication-Coupled Nucleosome Assembly in the Passage of Epigenetic Information and Cell Identity. Trends Biochem. Sci. 2018, 43, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gan, H.; Serra-Cardona, A.; Zhang, L.; Gan, S.; Sharma, S.; Johansson, E.; Chabes, A.; Xu, R.M.; Zhang, Z. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 2018, 361, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Borel, V.; Logan, C.; Svendsen, J.; Cox, D.E.; Nye, E.; Metcalfe, K.; O’Connell, S.M.; Stamp, G.; Flynn, H.R.; et al. Polε Instability Drives Replication Stress, Abnormal Development, and Tumorigenesis. Mol. Cell 2018, 70, 707–721.e7. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Serra-Cardona, A.; Hua, X.; Zhou, H.; Labib, K.; Yu, C.; Zhang, Z. The Mcm2-Ctf4-Polα Axis Facilitates Parental Histone H3-H4 Transfer to Lagging Strands. Mol. Cell 2018, 72, 140–151.e3. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhang, Q.; Jia, J.; Zhou, J.; Zhang, Z.; Karri, S.; Jiang, J.; Dickinson, Q.; Yao, Y.; Tang, X.; et al. DNA polymerase delta governs parental histone transfer to DNA replication lagging strand. Proc. Natl. Acad. Sci. USA 2024, 121, e2400610121. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Yang, C.; Wu, J.; Lei, Y.; Hu, J.; Feng, J.; Li, Q. DNA polymerase δ subunit Pol32 binds histone H3-H4 and couples nucleosome assembly with Okazaki fragment processing. Sci. Adv. 2024, 10, eado1739. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, Y.; Fang, Y.; Paulo, J.A.; Yaghoubi, D.; Hua, X.; Shipkovenska, G.; Toda, T.; Zhang, Z.; Gygi, S.P.; et al. A replisome-associated histone H3-H4 chaperone required for epigenetic inheritance. Cell 2024, 187, 5010–5028.e24. [Google Scholar] [CrossRef] [PubMed]
- Charlton, S.J.; Flury, V.; Kanoh, Y.; Genzor, A.V.; Kollenstart, L.; Ao, W.; Brøgger, P.; Weisser, M.B.; Adamus, M.; Alcaraz, N.; et al. The fork protection complex promotes parental histone recycling and epigenetic memory. Cell 2024, 187, 5029–5047.e21. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Fang, Y.; Shan, C.M.; Hua, X.; Kim, J.K.; Tang, L.C.; Jovanovic, M.; Tong, L.; Qiao, F.; Zhang, Z.; et al. Mrc1 regulates parental histone segregation and heterochromatin inheritance. Mol. Cell 2024, 84, 3223–3236.e4. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Miller, K.M.; Maas, N.L.; Roguev, A.; Fillingham, J.; Chu, C.S.; Schuldiner, M.; Gebbia, M.; Recht, J.; Shales, M.; et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 2007, 446, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, H.; Zhang, H.; Wang, Z.; Zhou, H.; Zhang, Z. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 2013, 155, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Ayyagari, R.; Gomes, X.V.; Burgers, P.M. Mutations in yeast proliferating cell nuclear antigen define distinct sites for interaction with DNA polymerase delta and DNA polymerase epsilon. Mol. Cell. Biol. 1997, 17, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Serra-Cardona, A.; Hua, X.; McNutt, S.W.; Zhou, H.; Toda, T.; Jia, S.; Chu, F.; Zhang, Z. The PCNA-Pol δ complex couples lagging strand DNA synthesis to parental histone transfer for epigenetic inheritance. Sci. Adv. 2024, 10, eadn5175. [Google Scholar] [CrossRef] [PubMed]
- Dolce, V.; Dusi, S.; Giannattasio, M.; Joseph, C.R.; Fumasoni, M.; Branzei, D. Parental histone deposition on the replicated strands promotes error-free DNA damage tolerance and regulates drug resistance. Genes Dev. 2022, 36, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shibahara, K.; Stillman, B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000, 408, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Srinivasan, M.; Nakanishi, S.; Leatherwood, J.; Shilatifard, A.; Sternglanz, R. A conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol. Cell. Biol. 2011, 31, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef] [PubMed]
- Ferrand, J.; Rondinelli, B.; Polo, S.E. Histone Variants: Guardians of Genome Integrity. Cells 2020, 9, 2424. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Kobor, M.S.; Venkatasubrahmanyam, S.; Meneghini, M.D.; Gin, J.W.; Jennings, J.L.; Link, A.J.; Madhani, H.D.; Rine, J. A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol. 2004, 2, E131. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Li, B.Z.; Szakal, B.; Branzei, D.; Putnam, C.D.; Kolodner, R.D. The Swr1 chromatin remodeling complex prevents genome instability induced by replication fork progression defects. Nat. Commun. 2018, 9, 3680. [Google Scholar] [CrossRef] [PubMed]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Tsabar, M.; Eapen, V.V.; Mason, J.M.; Memisoglu, G.; Waterman, D.P.; Long, M.J.; Bishop, D.K.; Haber, J.E. Caffeine impairs resection during DNA break repair by reducing the levels of nucleases Sae2 and Dna2. Nucleic Acids Res. 2015, 43, 6889–6901. [Google Scholar] [CrossRef] [PubMed]
- Bauerle, K.T.; Kamau, E.; Grove, A. Interactions between N- and C-terminal domains of the Saccharomyces cerevisiae high-mobility group protein HMO1 are required for DNA bending. Biochemistry 2006, 45, 3635–3645. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Williams, A.M.; Grove, A. The C-terminal domain of yeast high mobility group protein HMO1 mediates lateral protein accretion and in-phase DNA bending. Biochemistry 2010, 49, 4051–4059. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Kamau, E.; Donze, D.; Grove, A. Expression of yeast high mobility group protein HMO1 is regulated by TOR signaling. Gene 2011, 489, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Morawska, M.; Wong, R.P.; Daigaku, Y.; Ulrich, H.D. Spatial separation between replisome- and template-induced replication stress signaling. EMBO J. 2018, 37, e98369. [Google Scholar] [CrossRef] [PubMed]
- Bacal, J.; Moriel-Carretero, M.; Pardo, B.; Barthe, A.; Sharma, S.; Chabes, A.; Lengronne, A.; Pasero, P. Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J. 2018, 37, e99319. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Crabbé, L.; Pasero, P. Signaling pathways of replication stress in yeast. FEMS Yeast Res. 2017, 17, fow101. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Williams, R.S.; Dovey, C.L.; Guenther, G.; Tainer, J.A.; Russell, P. γH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010, 29, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107–Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar]
- Mejia-Ramirez, E.; Limbo, O.; Langerak, P.; Russell, P. Critical function of gH2A in S-phase. PLoS Genet. 2015, 11, e1005517. [Google Scholar] [CrossRef]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Liu, Y.; Ma, C.J.; Smolka, M.B. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 2013, 493, 120–124. [Google Scholar] [CrossRef]
- Siler, J.; Guo, N.; Liu, Z.; Qin, Y.; Bi, X. γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2024, 25, 2462. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.T.; Thoma, F.; Brubaker, J.M. Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: A model system for study of higher order structure. Cell 1985, 42, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, Y.; Goulet, I.; Revet, B.; Le Bret, M.; Prunell, A. Chromatin reconstitution on small DNA rings. II. DNA supercoiling on the nucleosome. J. Mol. Biol. 1988, 200, 267–290. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-López, M.; Villoria, M.T.; Esteras, M.; Jarmuz, A.; Torres-Rosell, J.; Clemente-Blanco, A.; Aragon, L. Sgs1’s roles in DNA end resection, HJ dissolution, and crossover suppression require a two-step SUMO regulation dependent on Smc5/6. Genes Dev. 2016, 30, 1339–1356. [Google Scholar] [CrossRef] [PubMed]
- Gallo-Fernández, M.; Saugar, I.; Ortiz-Bazán, M.Á.; Vázquez, M.V.; Tercero, J.A. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 2012, 40, 8325–8335. [Google Scholar] [CrossRef] [PubMed]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, J.; Li, Q. The replisome guides nucleosome assembly during DNA replication. Cell Biosci. 2020, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.G.; Hanna, M.D.; Lambrecht, A.D.; Mitchell, B.A.; Ziola, B.; Cobb, J.A.; Xiao, W. The Mre11-Rad50-Xrs2 complex is required for yeast DNA post replication repair. PLoS ONE 2014, 9, e109292. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.; Dhoonmoon, A.; Straka, J.; Nicolae, C.M.; Moldovan, G.L. Multi-step processing of replication stress-derived nascent strand DNA gaps by MRE11 and EXO1 nucleases. Nat. Commun. 2023, 14, 6265. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Domínguez-García, I.; Domínguez-Pérez, M.D.C.; Huertas, P. EXO1 and DNA2-mediated ssDNA gap expansion is essential for ATR activation and to maintain viability in BRCA1-deficient cells. Nucleic Acids Res. 2024, 52, 6376–6391. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Yu, Q.; Siler, J.; Li, C.; Khan, A. Functions of Fun30 chromatin remodeler in regulating cellular resistance to genotoxic stress. PLoS ONE 2015, 10, e0121341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Choi, K.; Szakal, B.; Arenz, J.; Duan, X.; Ye, H.; Branzei, D.; Zhao, X. Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc. Natl. Acad. Sci. USA 2009, 106, 21252–21257. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, A.; Wysocki, R.; Jobin-Robitaille, O.; Altaf, M.; Côté, J.; Kron, S.J. Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 13771–13776. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, J.; Wei, A.; Guo, N.; Wang, R.; Bi, X. The Yeast HMGB Protein Hmo1 Is a Multifaceted Regulator of DNA Damage Tolerance. Int. J. Mol. Sci. 2025, 26, 3255. https://doi.org/10.3390/ijms26073255
Huo J, Wei A, Guo N, Wang R, Bi X. The Yeast HMGB Protein Hmo1 Is a Multifaceted Regulator of DNA Damage Tolerance. International Journal of Molecular Sciences. 2025; 26(7):3255. https://doi.org/10.3390/ijms26073255
Chicago/Turabian StyleHuo, Jinlong, Anhui Wei, Na Guo, Ruotong Wang, and Xin Bi. 2025. "The Yeast HMGB Protein Hmo1 Is a Multifaceted Regulator of DNA Damage Tolerance" International Journal of Molecular Sciences 26, no. 7: 3255. https://doi.org/10.3390/ijms26073255
APA StyleHuo, J., Wei, A., Guo, N., Wang, R., & Bi, X. (2025). The Yeast HMGB Protein Hmo1 Is a Multifaceted Regulator of DNA Damage Tolerance. International Journal of Molecular Sciences, 26(7), 3255. https://doi.org/10.3390/ijms26073255