
Inhibiting Myostatin Expression by the Antisense Oligonucleotides Improves Muscle Wasting in a Chronic Kidney Disease Mouse Model

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
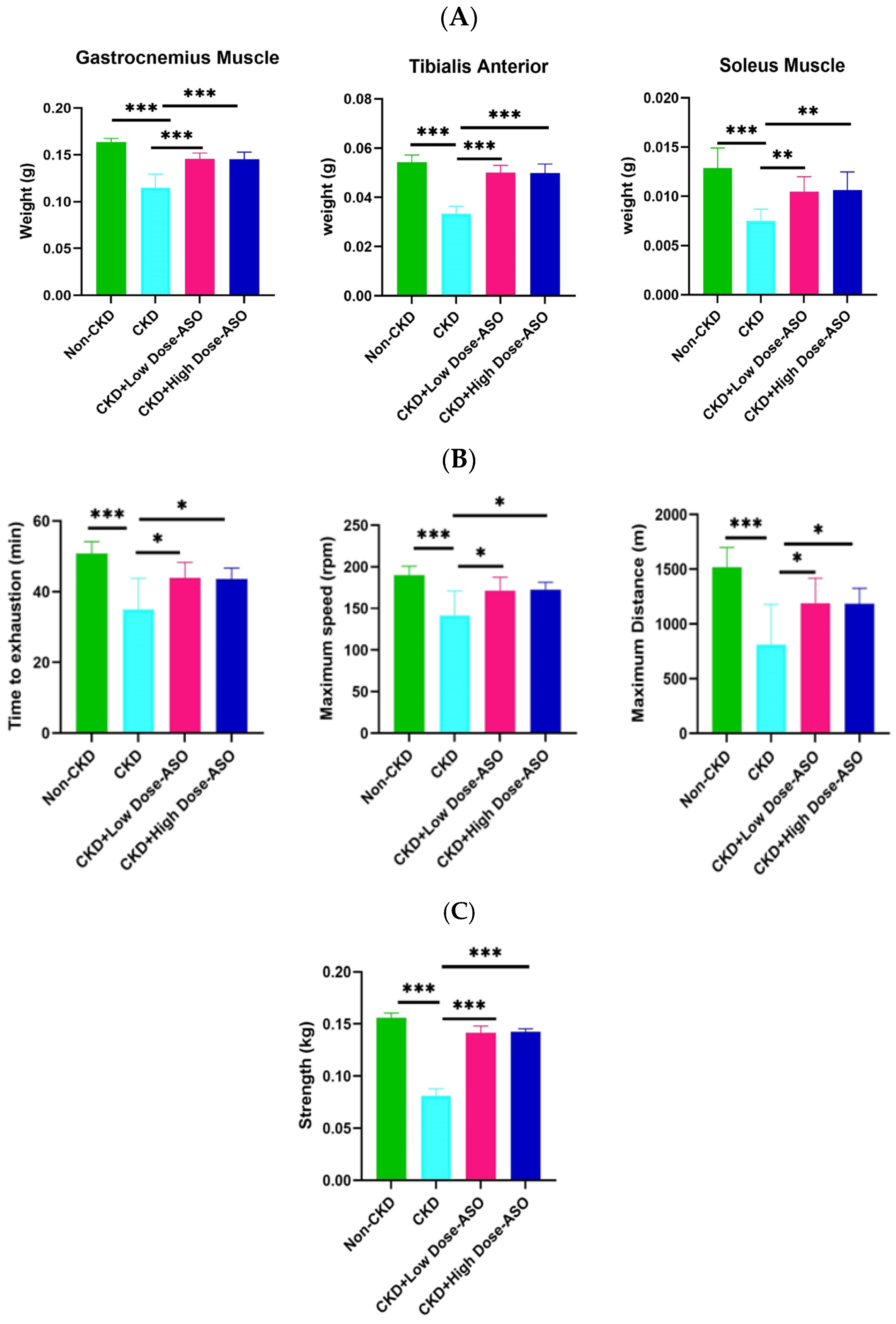
2.1. MSTN-ASO Treatment in Mice Increases Skeletal Muscle Mass, Strength, and Function
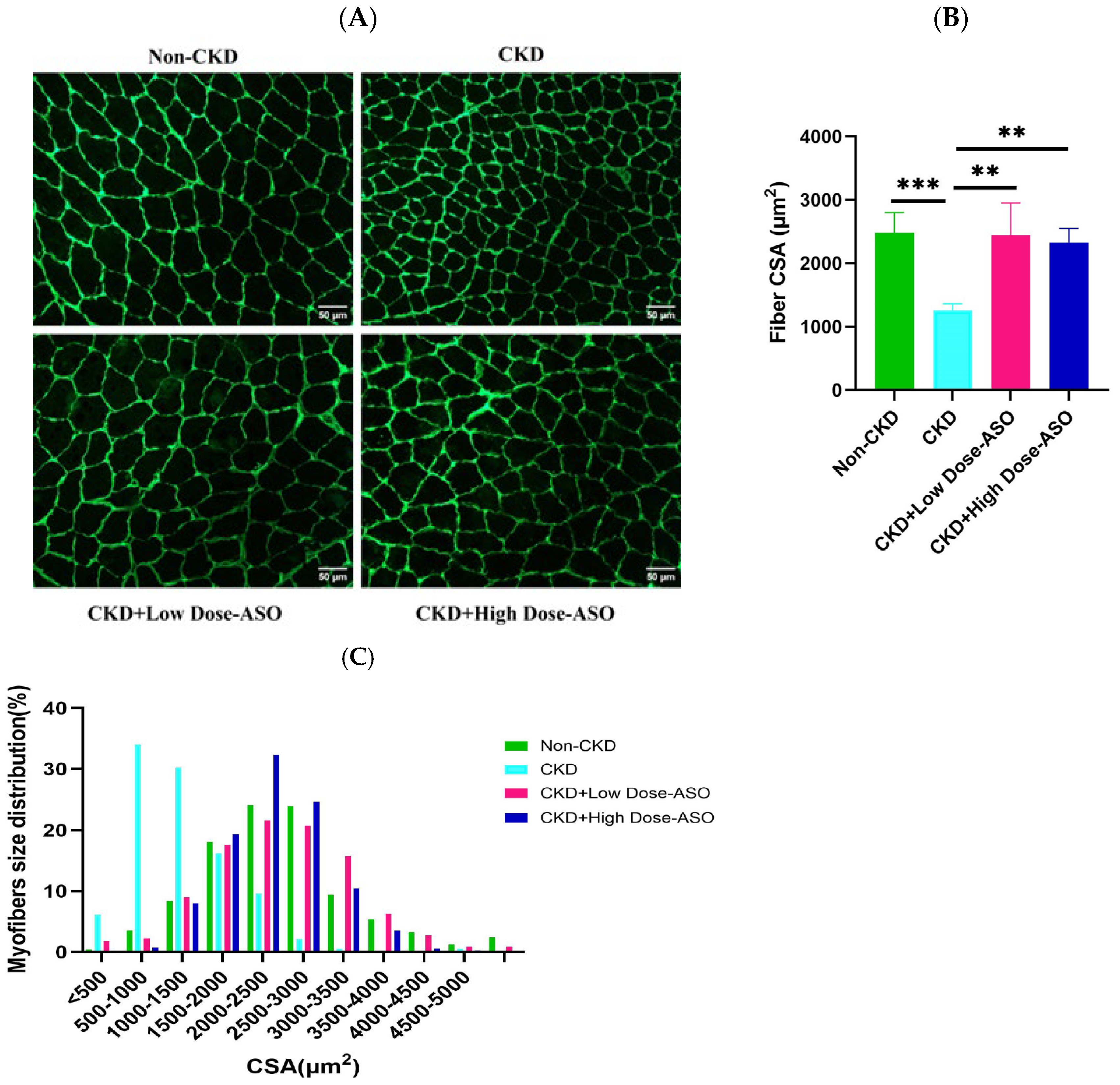
2.2. Mstn-Aso Treatment Improves Muscle Atrophy Induced by CKD
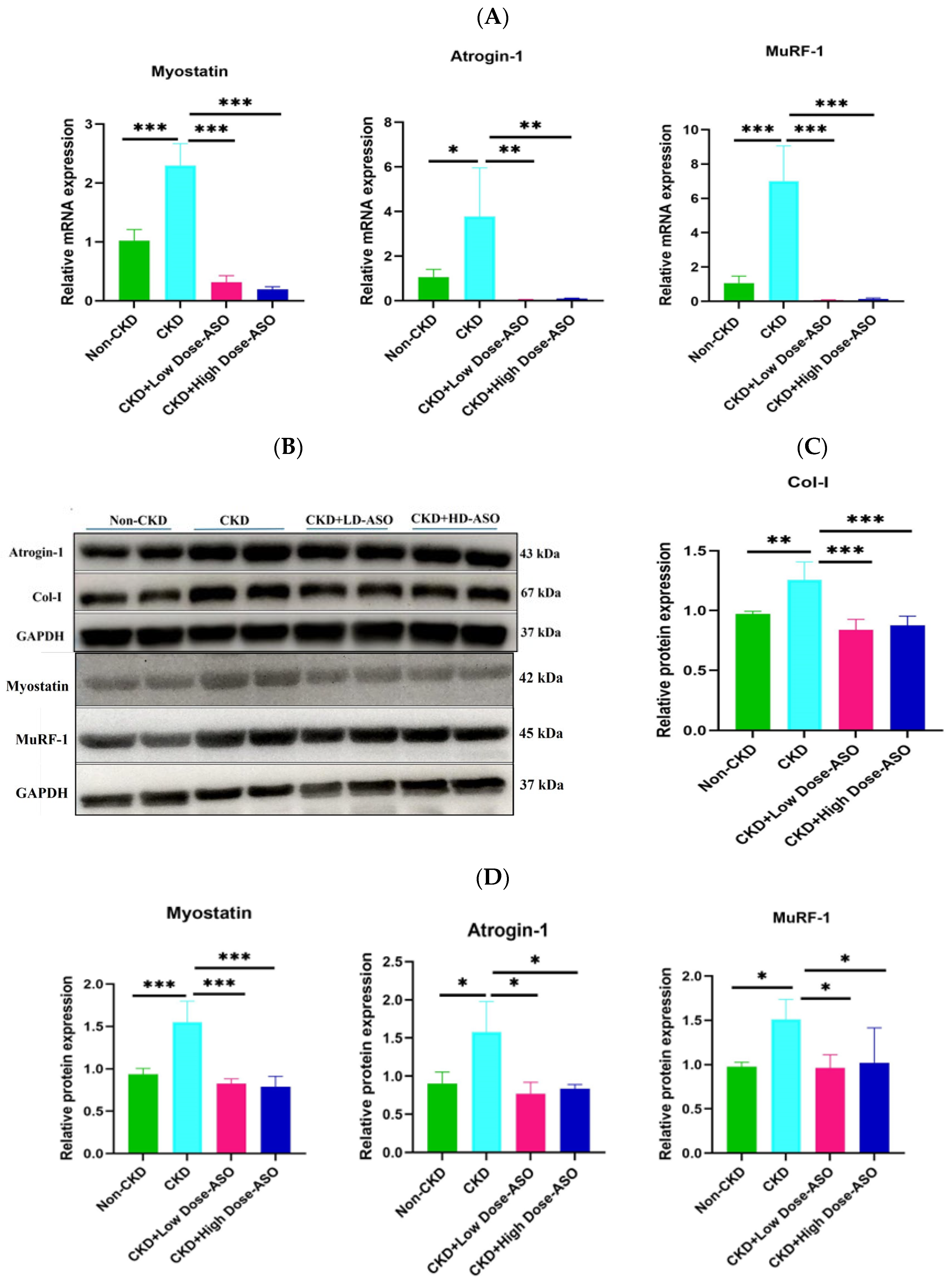
2.3. Atrophic Gene mRNA and Protein Expression Are Suppressed by MSTN-ASO Injections in Mice Skeletal Muscle
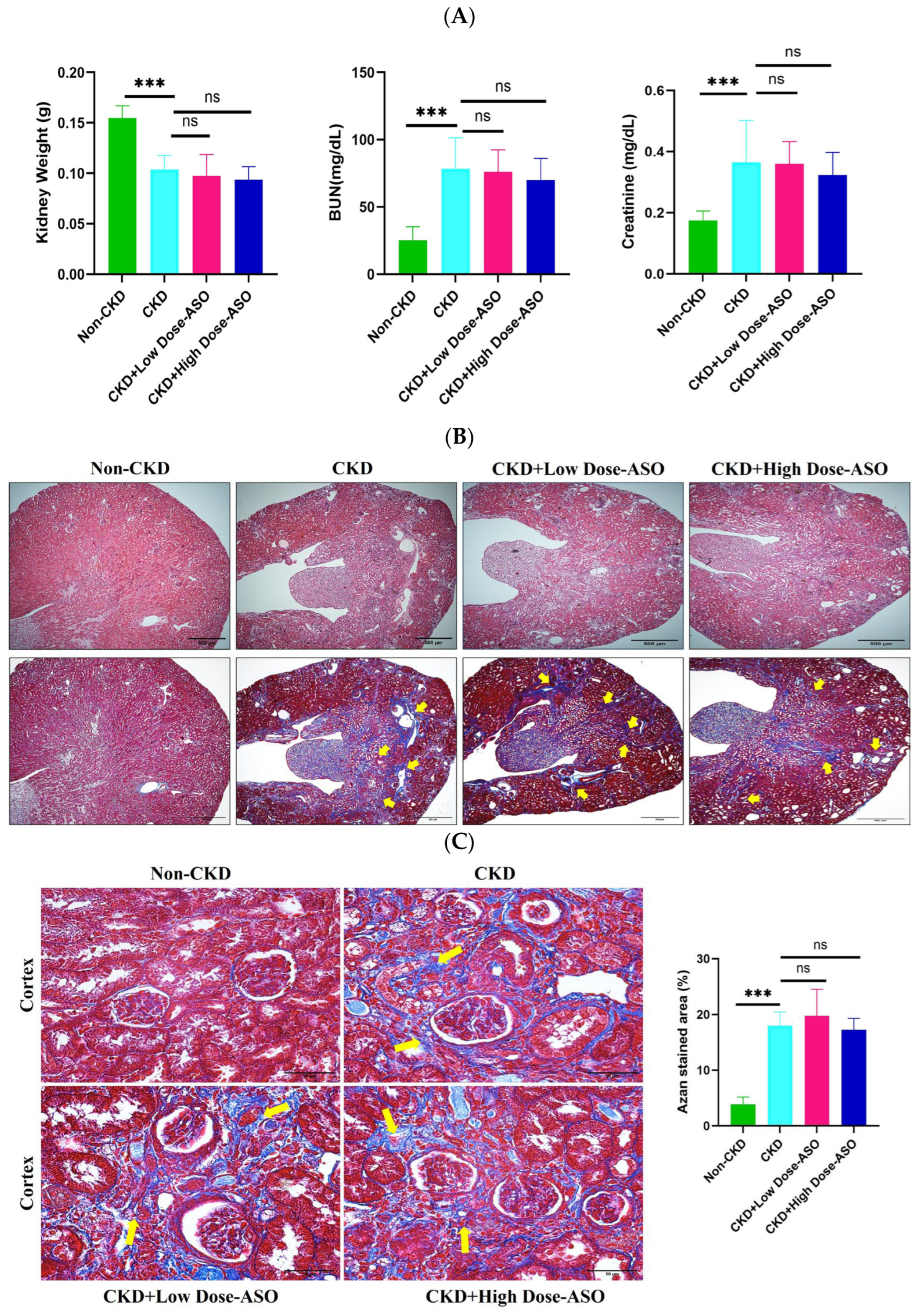
2.4. MSTN-ASO Treatment Does Not Affect Kidney Functions
3. Discussion
4. Materials and Methods
4.1. Animal Experiment
4.2. Treadmill Exhaustion Test
4.3. Forelimb Grip Strength Test
4.4. Immunohistochemistry Staining
4.5. Quantitative Real-Time PCR
4.6. Western Blotting
4.7. Measurement of BUN and Creatinine Concentrations
4.8. HE and Azan Staining
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | Chronic Kidney Disease |
| MSTN | Myostatin |
| GDF-8 | Growth Differentiation factor-8 |
| Atrogin-1 | Atrophy F-box gene 1 |
| MuRF-1 | Muscle RING-finger protein-1 |
| ASO | Antisense Oligonucleotide |
| PMOs | Phosphorodiamidate Morpholino Oligomers |
| SMA | Spinal Muscular Atrophy |
| DMD | Duchenne Muscular Dystrophy |
| GC | Gastrocnemius |
| TA | Tibialis Anterior |
| CSA | Cross-Sectional Area |
| Col-I | Collagen-1 |
| BUN | Blood Urea Nitrogen |
References
- Chatzipetrou, V.; Bégin, M.-J.; Hars, M.; Trombetti, A. Sarcopenia in Chronic Kidney Disease: A Scoping Review of Prevalence, Risk Factors, Association with Outcomes, and Treatment. Calcif. Tissue Int. 2022, 110, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Massini, G.; Caldiroli, L.; Molinari, P.; Carminati, F.M.I.; Castellano, G.; Vettoretti, S. Nutritional Strategies to Prevent Muscle Loss and Sarcopenia in Chronic Kidney Disease: What Do We Currently Know? Nutrients 2023, 15, 3107. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, R.; Corbella, X.; Mattace-Raso, F.; Tap, L.; Sieber, C.; Freiberger, E.; Kostka, T.; Guligowska, A.; Melzer, I.; Melzer, Y.; et al. Prevalence of Sarcopenia in Community-Dwelling Older Adults Using the Updated EWGSOP2 Definition According to Kidney Function and Albuminuria: The Screening for CKD among Older People across Europe (SCOPE) Study. BMC Geriatr. 2020, 20, 327. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wu, Y.; Cao, X.; Yang, K.; Tong, Y.; Zhang, F.; Wang, C.; Cui, R.; Ren, J.; Li, Q.; et al. Association between Sarcopenia and New-Onset Chronic Kidney Disease among Middle-Aged and Elder Adults: Findings from the China Health and Retirement Longitudinal Study. BMC Geriatr. 2024, 24, 134. [Google Scholar] [CrossRef]
- Magagnoli, L.; Cozzolino, M.; Caskey, F.J.; Evans, M.; Torino, C.; Porto, G.; Szymczak, M.; Krajewska, M.; Drechsler, C.; Stenvinkel, P.; et al. Association between CKD-MBD and Mortality in Older Patients with Advanced CKD—Results from the EQUAL Study. Nephrol. Dial. Transplant. 2023, 38, 2562–2575. [Google Scholar] [CrossRef]
- Martins, P.; Marques, E.A.; Leal, D.V.; Ferreira, A.; Wilund, K.R.; Viana, J.L. Association between Physical Activity and Mortality in End-Stage Kidney Disease: A Systematic Review of Observational Studies. BMC Nephrol. 2021, 22, 227. [Google Scholar] [CrossRef]
- Jha, V.; Al-Ghamdi, S.M.G.; Li, G.; Wu, M.-S.; Stafylas, P.; Retat, L.; Card-Gowers, J.; Barone, S.; Cabrera, C.; Garcia Sanchez, J.J. Global Economic Burden Associated with Chronic Kidney Disease: A Pragmatic Review of Medical Costs for the Inside CKD Research Programme. Adv. Ther. 2023, 40, 4405–4420. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, X. Sarcopenia, the Economic Challenges in Healthcare and Individual Struggles Arise. Open Access Libr. J. 2024, 11, 1–7. [Google Scholar] [CrossRef]
- Bahat, G.; Ozkok, S. The Current Landscape of Pharmacotherapies for Sarcopenia. Drugs Aging 2024, 41, 83–112. [Google Scholar] [CrossRef]
- Oliveira, E.A.; Zheng, R.; Carter, C.E.; Mak, R.H. Cachexia/Protein Energy Wasting Syndrome in CKD: Causation and Treatment. Semin. Dial. 2019, 32, 493–499. [Google Scholar] [CrossRef]
- Noor, H.; Reid, J.; Slee, A. Resistance Exercise and Nutritional Interventions for Augmenting Sarcopenia Outcomes in Chronic Kidney Disease: A Narrative Review. J. Cachexia Sarcopenia Muscle 2021, 12, 1621–1640. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Q.; Tang, M.; Qi, G.; Qiu, C.; Huang, Y.; Yu, W.; Wang, W.; Sun, H.; Ni, X.; et al. Chronic Kidney Disease-Induced Muscle Atrophy: Molecular Mechanisms and Promising Therapies. Biochem. Pharmacol. 2023, 208, 115407. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Wang, P.-C.; Hsiung, T.; Fan, Y.-H.; Wu, J.-T.; Kan, W.-C.; Shiao, C.-C. Sarcopenia in Chronic Kidney Disease: A Narrative Review from Pathophysiology to Therapeutic Approaches. Biomedicines 2025, 13, 352. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Enoki, Y.; Maruyama, T. Sarcopenia in Chronic Kidney Disease: Factors, Mechanisms, and Therapeutic Interventions. Biol. Pharm. Bull. 2019, 42, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Klein, J.D.; Du, J.; Franch, H.A.; Zhang, L.; Hassounah, F.; Hudson, M.B.; Wang, X.H. Chronic Kidney Disease Induces Autophagy Leading to Dysfunction of Mitochondria in Skeletal Muscle. Am. J. Physiol. Renal Physiol. 2017, 312, F1128. [Google Scholar] [CrossRef]
- Bataille, S.; Chauveau, P.; Fouque, D.; Aparicio, M.; Koppe, L. Myostatin and Muscle Atrophy during Chronic Kidney Disease. Nephrol. Dial. Transplant. 2021, 36, 1986–1993. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Moon, J.S.; Park, S.-Y.; Ho Lim, J.; Chun, H.J.; Qadri, A.F.; Hwang, Y.C.; Jan, A.T.; Ahmad, S.S.; et al. Myostatin and Its Regulation: A Comprehensive Review of Myostatin Inhibiting Strategies. Front. Physiol. 2022, 13, 876078. [Google Scholar] [CrossRef]
- Wang, D.-T.; Yang, Y.-J.; Huang, R.-H.; Zhang, Z.-H.; Lin, X. Myostatin Activates the Ubiquitin-Proteasome and Autophagy-Lysosome Systems Contributing to Muscle Wasting in Chronic Kidney Disease. Oxid. Med. Cell Longev. 2015, 2015, 684965. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal Muscle Atrophy and the E3 Ubiquitin Ligases MuRF1 and MAFbx/Atrogin-1. Am. J. Physiol-Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef]
- Degens, H.; Patel, K.; Matsakas, A. Myostatin Knockout Mice Have Larger Muscle Fibers with Normal Function and Morphology. Muscle Nerve 2025. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.; Nunan, E.; Koilpillai, J.; Harris, T.; Wright, C.; Gore, J.; Butcher, J. Deletion of Myostatin Significantly and Preferentially Improves Muscle Function in a Sex-Dependent Manner in Both an Obese and Aging Mouse Model. Physiology 2024, 39, 1888. [Google Scholar] [CrossRef]
- Csürhés, T.; Szabó, F.; Holló, G.; Mikó, E.; Török, M.; Bene, S. Relationship between Some Myostatin Variants and Meat Production Related Calving, Weaning and Muscularity Traits in Charolais Cattle. Animals 2023, 13, 1895. [Google Scholar] [CrossRef]
- Chen, M.-M.; Zhao, Y.; Yu, K.; Xu, X.-L.; Zhang, X.-S.; Zhang, J.-L.; Wu, S.-J.; Liu, Z.-M.; Yuan, Y.-M.; Guo, X.-F.; et al. A MSTNDel73C Mutation with FGF5 Knockout Sheep by CRISPR/Cas9 Promotes Skeletal Muscle Myofiber Hyperplasia. eLife 2024, 12, RP86827. [Google Scholar] [CrossRef]
- Grochowska, E.; Borys, B.; Mroczkowski, S. Effects of Intronic SNPs in the Myostatin Gene on Growth and Carcass Traits in Colored Polish Merino Sheep. Genes 2019, 11, 2. [Google Scholar] [CrossRef]
- Ceccobelli, S.; Perini, F.; Trombetta, M.F.; Tavoletti, S.; Lasagna, E.; Pasquini, M. Effect of Myostatin Gene Mutation on Slaughtering Performance and Meat Quality in Marchigiana Bulls. Animals 2022, 12, 518. [Google Scholar] [CrossRef]
- Kornegay, J.N.; Bogan, D.J.; Bogan, J.R.; Dow, J.L.; Wang, J.; Fan, Z.; Liu, N.; Warsing, L.C.; Grange, R.W.; Ahn, M.; et al. Dystrophin-Deficient Dogs with Reduced Myostatin Have Unequal Muscle Growth and Greater Joint Contractures. Skelet. Muscle 2016, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Xu, K.; Xiao, W.; Shu, C.; Li, N.; Li, K.; Gu, H.; Zhu, Z.; Zhang, L.; Ren, H.; et al. Dual Single Guide RNAs-Mediating Deletion of Mature Myostatin Peptide Results in Concomitant Muscle Fibre Hyperplasia and Adipocyte Hypotrophy in Pigs. Biochem. Biophys. Res. Commun. 2023, 673, 145–152. [Google Scholar] [CrossRef]
- Liu, L.; Hu, R.; You, H.; Li, J.; Liu, Y.; Li, Q.; Wu, X.; Huang, J.; Cai, X.; Wang, M.; et al. Formononetin Ameliorates Muscle Atrophy by Regulating Myostatin-Mediated PI3K/Akt/FoxO3a Pathway and Satellite Cell Function in Chronic Kidney Disease. J. Cell Mol. Med. 2021, 25, 1493–1506. [Google Scholar] [CrossRef]
- Yu, R.; Chen, J.-A.; Xu, J.; Cao, J.; Wang, Y.; Thomas, S.S.; Hu, Z. Suppression of Muscle Wasting by the Plant-Derived Compound Ursolic Acid in a Model of Chronic Kidney Disease. J. Cachexia Sarcopenia Muscle 2017, 8, 327–341. [Google Scholar] [CrossRef]
- Heitman, K.; Alexander, M.S.; Faul, C. Skeletal Muscle Injury in Chronic Kidney Disease-From Histologic Changes to Molecular Mechanisms and to Novel Therapies. Int. J. Mol. Sci. 2024, 25, 5117. [Google Scholar] [CrossRef] [PubMed]
- Eilers, W.; Cleasby, M.; Foster, K. Development of Antisense-Mediated Myostatin Knockdown for the Treatment of Insulin Resistance. Sci. Rep. 2021, 11, 1604. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin Inhibition in Combination with Antisense Oligonucleotide Therapy Improves Outcomes in Spinal Muscular Atrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid. Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef]
- Kemaladewi, D.U.; Hoogaars, W.M.; van Heiningen, S.H.; Terlouw, S.; de Gorter, D.J.; den Dunnen, J.T.; van Ommen, G.J.B.; Aartsma-Rus, A.; ten Dijke, P.; ’t Hoen, P.A. Dual Exon Skipping in Myostatin and Dystrophin for Duchenne Muscular Dystrophy. BMC Med. Genomics. 2011, 4, 36. [Google Scholar] [CrossRef]
- Li, Y.; Tan, Y.; Zhang, R.; Wang, T.; Na, N.; Zheng, T.; Veedu, R.N.; Chen, S. Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease. Kidney Dial. 2022, 2, 16–37. [Google Scholar] [CrossRef]
- Maeta, K.; Farea, M.; Nishio, H.; Matsuo, M. An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor. Int. J. Mol. Sci. 2022, 23, 5016. [Google Scholar] [CrossRef]
- Wang, M.; You, L.; He, X.; Peng, Y.; Wang, R.; Zhang, Z.; Shu, J.; Zhang, P.; Sun, X.; Jia, L.; et al. Multiomics Analysis Reveals Therapeutic Targets for Chronic Kidney Disease with Sarcopenia. J. Cachexia Sarcopenia Muscle. 2025, 16, e13696. [Google Scholar] [CrossRef]
- Heitman, K.; Bollenbecker, S.; Bradley, J.; Czaya, B.; Fajol, A.; Thomas, S.M.; Li, Q.; Komarova, S.; Krick, S.; Rowe, G.C.; et al. Hyperphosphatemia Contributes to Skeletal Muscle Atrophy in Mice. Int. J. Mol. Sci. 2024, 25, 9308. [Google Scholar] [CrossRef]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal Myopathy in CKD: A Comparison of Adenine-Induced Nephropathy and 5/6 Nephrectomy Models in Mice. Am. J. PhysiolRen. Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef]
- Tinklenberg, J.A.; Siebers, E.M.; Beatka, M.J.; Meng, H.; Yang, L.; Zhang, Z.; Ross, J.A.; Ochala, J.; Morris, C.; Owens, J.M.; et al. Myostatin Inhibition Using mRK35 Produces Skeletal Muscle Growth and Tubular Aggregate Formation in Wild Type and TgACTA1D286G Nemaline Myopathy Mice. Hum. Mol. Genet. 2018, 27, 638–648. [Google Scholar] [CrossRef] [PubMed]
- St. Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A Mouse Anti-Myostatin Antibody Increases Muscle Mass and Improves Muscle Strength and Contractility in the Mdx Mouse Model of Duchenne Muscular Dystrophy and Its Humanized Equivalent, Domagrozumab (PF-06252616), Increases Muscle Volume in Cynomolgus Monkeys. Skeletal Muscle 2017, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kasukawa, Y.; Nozaka, K.; Tsuchie, H.; Kudo, D.; Kinoshita, H.; Ono, Y.; Igarashi, S.; Kasama, F.; Harata, S.; et al. Changes in Skeletal Muscle Atrophy over Time in a Rat Model of Adenine-Induced Chronic Kidney Disease. Appl. Sci. 2024, 14, 9106. [Google Scholar] [CrossRef]
- Lindqvist, J.; Granzier, H. Pharmacological Inhibition of Myostatin in a Mouse Model of Typical Nemaline Myopathy Increases Muscle Size and Force. Int. J. Mol. Sci. 2023, 24, 15124. [Google Scholar] [CrossRef] [PubMed]
- Bunn, R.C.; Adatorwovor, R.; Smith, R.R.; Ray, P.D.; Fields, S.E.; Keeble, A.R.; Fry, C.S.; Uppuganti, S.; Nyman, J.S.; Fowlkes, J.L.; et al. Pharmacologic Inhibition of Myostatin with a Myostatin Antibody Improves the Skeletal Muscle and Bone Phenotype of Male Insulin-Deficient Diabetic Mice. JBMR Plus. 2023, 7, e10833. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription Factor FoxO1, the Dominant Mediator of Muscle Wasting in Chronic Kidney Disease, Is Inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J.; et al. Myostatin Blockade with a Fully Human Monoclonal Antibody Induces Muscle Hypertrophy and Reverses Muscle Atrophy in Young and Aged Mice. Skeletal Muscle 2015, 5, 34. [Google Scholar] [CrossRef]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/Myostatin Inhibitor, GDF11 Propeptide-Fc, Increases Skeletal Muscle Mass and Improves Muscle Strength in Dystrophic Mdx Mice. Skeletal Muscle 2019, 9, 16. [Google Scholar] [CrossRef]
- Avin, K.G.; Chen, N.X.; Organ, J.M.; Zarse, C.; O’Neill, K.; Conway, R.G.; Konrad, R.J.; Bacallao, R.L.; Allen, M.R.; Moe, S.M. Skeletal Muscle Regeneration and Oxidative Stress Are Altered in Chronic Kidney Disease. PLoS ONE 2016, 11, e0159411. [Google Scholar] [CrossRef]
- Cruz, A.; Ferian, A.; Alves, P.K.N.; Silva, W.J.; Bento, M.R.; Gasch, A.; Labeit, S.; Moriscot, A.S. Skeletal Muscle Anti-Atrophic Effects of Leucine Involve Myostatin Inhibition. DNA Cell Biol. 2020, 39, 2289–2299. [Google Scholar] [CrossRef]
- Dong, J.; Dong, Y.; Chen, Z.; Mitch, W.E.; Zhang, L. The Pathway to Muscle Fibrosis Depends on Myostatin Stimulating the Differentiation of Fibro/Adipogenic Progenitor Cells in Chronic Kidney Disease. Kidney Int. 2017, 91, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Miyakoshi, N.; Kasukawa, Y.; Nozaka, K.; Tsuchie, H.; Sato, C.; Abe, K.; Shoji, R.; Shimada, Y. Analysis of Bone in Adenine-Induced Chronic Kidney Disease Model Rats. Osteoporos. Sarcopenia. 2021, 7, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Yamazaki, D.; Sufiun, A.; Kitada, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. A Novel Approach to Adenine-Induced Chronic Kidney Disease Associated Anemia in Rodents. PLoS ONE 2018, 13, e0192531. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Huang, J.; Hu, R.; You, H.; Liu, L.; Wang, D.; Wei, L. Paeoniflorin Ameliorates Skeletal Muscle Atrophy in Chronic Kidney Disease via AMPK/SIRT1/PGC-1α-Mediated Oxidative Stress and Mitochondrial Dysfunction. Front. Pharmacol. 2022, 13, 859723. [Google Scholar] [CrossRef]
- Atay, J.C.L.; Elsborg, S.H.; Palmfeldt, J.; Nejsum, L.N.; Nørregaard, R. Recovery of Water Homeostasis in Adenine-Induced Kidney Disease Is Mediated by Increased AQP2 Membrane Targeting. Int. J. Mol. Sci. 2024, 25, 3447. [Google Scholar] [CrossRef]
- Li, Z.B.; Zhang, J.; Wagner, K.R. Inhibiting Myostatin Reverses Muscle Fibrosis through Apoptosis. J. Cell Sci. 2012, 125, 3957–3965. [Google Scholar] [CrossRef]
- Ju, L.; Diao, J.; Zhang, J.; Dai, F.; Zhou, H.; Han, Z.; Hu, R.; Pei, T.; Wang, F.; He, Z.; et al. Shenshuai Yingyang Jiaonang Ameliorates Chronic Kidney Disease-Associated Muscle Atrophy in Rats by Inhibiting Ferroptosis Mediated by the HIF-1α/SLC7A11 Pathway. Heliyon 2024, 10, e29093. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.-P. Role of Activin A and Myostatin in Human Cancer Cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef]
- Hanada, K.; Fukasawa, K.; Hinata, H.; Imai, S.; Takayama, K.; Hirai, H.; Ohfusa, R.; Hayashi, Y.; Itoh, F. Combination Therapy with Anamorelin and a Myostatin Inhibitor Is Advantageous for Cancer Cachexia in a Mouse Model. Cancer Sci. 2022, 113, 3547–3557. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Coleman, L.A.; Miller, R.; Rooks, D.S.; Laurent, D.; Petricoul, O.; Praestgaard, J.; Swan, T.; Wade, T.; Perry, R.G.; et al. Effect of Bimagrumab vs Placebo on Body Fat Mass Among Adults with Type 2 Diabetes and Obesity: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open. 2021, 4, e2033457. [Google Scholar] [CrossRef]
- Eilers, W.; Chambers, D.; Cleasby, M.; Foster, K. Local Myostatin Inhibition Improves Skeletal Muscle Glucose Uptake in Insulin-Resistant High-Fat Diet-Fed Mice. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E163–E174. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Sun, L.; Liu, C.; Tien, P. Inhibition of Atrogin-1/MAFbx Expression by Adenovirus-Delivered Small Hairpin RNAs Attenuates Muscle Atrophy in Fasting Mice. Hum. Gene Ther. 2011, 22, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle Sparing in Muscle RING Finger 1 Null Mice: Response to Synthetic Glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pan, Y.; Pang, Q.; Zhang, A. Irisin Ameliorates Muscle Atrophy by Inhibiting the Upregulation of the Ubiquitin–Proteasome System in Chronic Kidney Disease. Calcif. Tissue Int. 2024, 115, 712–724. [Google Scholar] [CrossRef]
- Chen, M.-M.; Zhao, Y.-P.; Zhao, Y.; Deng, S.-L.; Yu, K. Regulation of Myostatin on the Growth and Development of Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 785712. [Google Scholar] [CrossRef]
- Wilhelmsen, A.; Stephens, F.B.; Bennett, A.J.; Karagounis, L.G.; Jones, S.W.; Tsintzas, K. Skeletal Muscle Myostatin mRNA Expression Is Upregulated in Aged Human Adults with Excess Adiposity but Is Not Associated with Insulin Resistance and Ageing. GeroScience 2023, 46, 2033–2049. [Google Scholar] [CrossRef]
- Li, N.; Yang, Q.; Walker, R.G.; Thompson, T.B.; Du, M.; Rodgers, B.D. Myostatin Attenuation In Vivo Reduces Adiposity, but Activates Adipogenesis. Endocrinology 2016, 157, 282–291. [Google Scholar] [CrossRef]
- Verzola, D.; Milanesi, S.; Viazzi, F.; Ansaldo, F.; Saio, M.; Garibaldi, S.; Carta, A.; Costigliolo, F.; Salvidio, G.; Barisione, C.; et al. Enhanced Myostatin Expression and Signalling Promote Tubulo-interstitial Inflammation in Diabetic Nephropathy. Sci. Rep. 2020, 10, 6343. [Google Scholar] [CrossRef]
- Verzola, D.; Barisione, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging Role of Myostatin and Its Inhibition in the Setting of Chronic Kidney Disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef]
- Marchildon, F.; Chi, J.; O’Connor, S.; Bediako, H.; Cohen, P. Beige Fat Is Dispensable for the Metabolic Benefits Associated with Myostatin Deletion. Mol. Metab. 2021, 43, 101120. [Google Scholar] [CrossRef]
- Rodrigues, G.G.C.; Dellê, H.; Brito, R.B.O.; Cardoso, V.O.; Fernandes, K.P.S.; Mesquita-Ferrari, R.A.; Cunha, R.S.; Stinghen, A.E.M.; Dalboni, M.A.; Barreto, F.C. Indoxyl Sulfate Contributes to Uremic Sarcopenia by Inducing Apoptosis in Myoblasts. Arch. Med. Res. 2020, 51, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Takemura, K.; Higashihara, T.; Inagi, R. Uremic Sarcopenia: Clinical Evidence and Basic Experimental Approach. Nutrients 2020, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Bataille, S.; Dou, L.; Bartoli, M.; Sallée, M.; Aniort, J.; Ferkak, B.; Chermiti, R.; McKay, N.; Da Silva, N.; Burtey, S.; et al. Mechanisms of Myostatin and Activin A Accumulation in Chronic Kidney Disease. Nephrol. Dial. Transpl. 2022, 37, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Yasar, E.; Tek, N.A.; Tekbudak, M.Y.; Yurtdaş, G.; Gülbahar, Ö.; Uyar, G.Ö.; Ural, Z.; Çelik, Ö.M.; Erten, Y. The Relationship Between Myostatin, Inflammatory Markers, and Sarcopenia in Patients With Chronic Kidney Disease. J. Ren. Nutr. 2022, 32, 677–684. [Google Scholar] [CrossRef]
- Lee, T.; Awano, H.; Yagi, M.; Matsumoto, M.; Watanabe, N.; Goda, R.; Koizumi, M.; Takeshima, Y.; Matsuo, M. 2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping. Genes 2017, 8, 67. [Google Scholar] [CrossRef]
- Ito, K.; Takakusa, H.; Kakuta, M.; Kanda, A.; Takagi, N.; Nagase, H.; Watanabe, N.; Asano, D.; Goda, R.; Masuda, T.; et al. Renadirsen, a Novel 2′OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo. Curr. Issues Mol. Biol. 2021, 43, 1267–1281. [Google Scholar] [CrossRef]
- Arounleut, P.; Bialek, P.; Liang, L.-F.; Upadhyay, S.; Fulzele, S.; Johnson, M.; Elsalanty, M.; Isales, C.M.; Hamrick, M.W. A Myostatin Inhibitor (Propeptide-Fc) Increases Muscle Mass and Muscle Fiber Size in Aged Mice but Does Not Increase Bone Density or Bone Strength. Exp. Gerontol. 2013, 48, 898–904. [Google Scholar] [CrossRef]
- Sheikh, A.M.; Yano, S.; Mitaki, S.; Haque, M.A.; Yamaguchi, S.; Nagai, A. A Mesenchymal Stem Cell Line (B10) Increases Angiogenesis in a Rat MCAO Model. Exp. Neurol. 2019, 311, 182–193. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, A.; Md. Sheikh, A.; Yoshino, J.; Kanda, T.; Nagai, A.; Matsuo, M.; Yano, S. Inhibiting Myostatin Expression by the Antisense Oligonucleotides Improves Muscle Wasting in a Chronic Kidney Disease Mouse Model. Int. J. Mol. Sci. 2025, 26, 3098. https://doi.org/10.3390/ijms26073098
Akhter A, Md. Sheikh A, Yoshino J, Kanda T, Nagai A, Matsuo M, Yano S. Inhibiting Myostatin Expression by the Antisense Oligonucleotides Improves Muscle Wasting in a Chronic Kidney Disease Mouse Model. International Journal of Molecular Sciences. 2025; 26(7):3098. https://doi.org/10.3390/ijms26073098
Chicago/Turabian StyleAkhter, Arju, Abdullah Md. Sheikh, Jun Yoshino, Takeshi Kanda, Atsushi Nagai, Masafumi Matsuo, and Shozo Yano. 2025. "Inhibiting Myostatin Expression by the Antisense Oligonucleotides Improves Muscle Wasting in a Chronic Kidney Disease Mouse Model" International Journal of Molecular Sciences 26, no. 7: 3098. https://doi.org/10.3390/ijms26073098
APA StyleAkhter, A., Md. Sheikh, A., Yoshino, J., Kanda, T., Nagai, A., Matsuo, M., & Yano, S. (2025). Inhibiting Myostatin Expression by the Antisense Oligonucleotides Improves Muscle Wasting in a Chronic Kidney Disease Mouse Model. International Journal of Molecular Sciences, 26(7), 3098. https://doi.org/10.3390/ijms26073098