
Novel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature

 , , , and
, , , and
Abstract
1. Introduction
2. Case Presentation
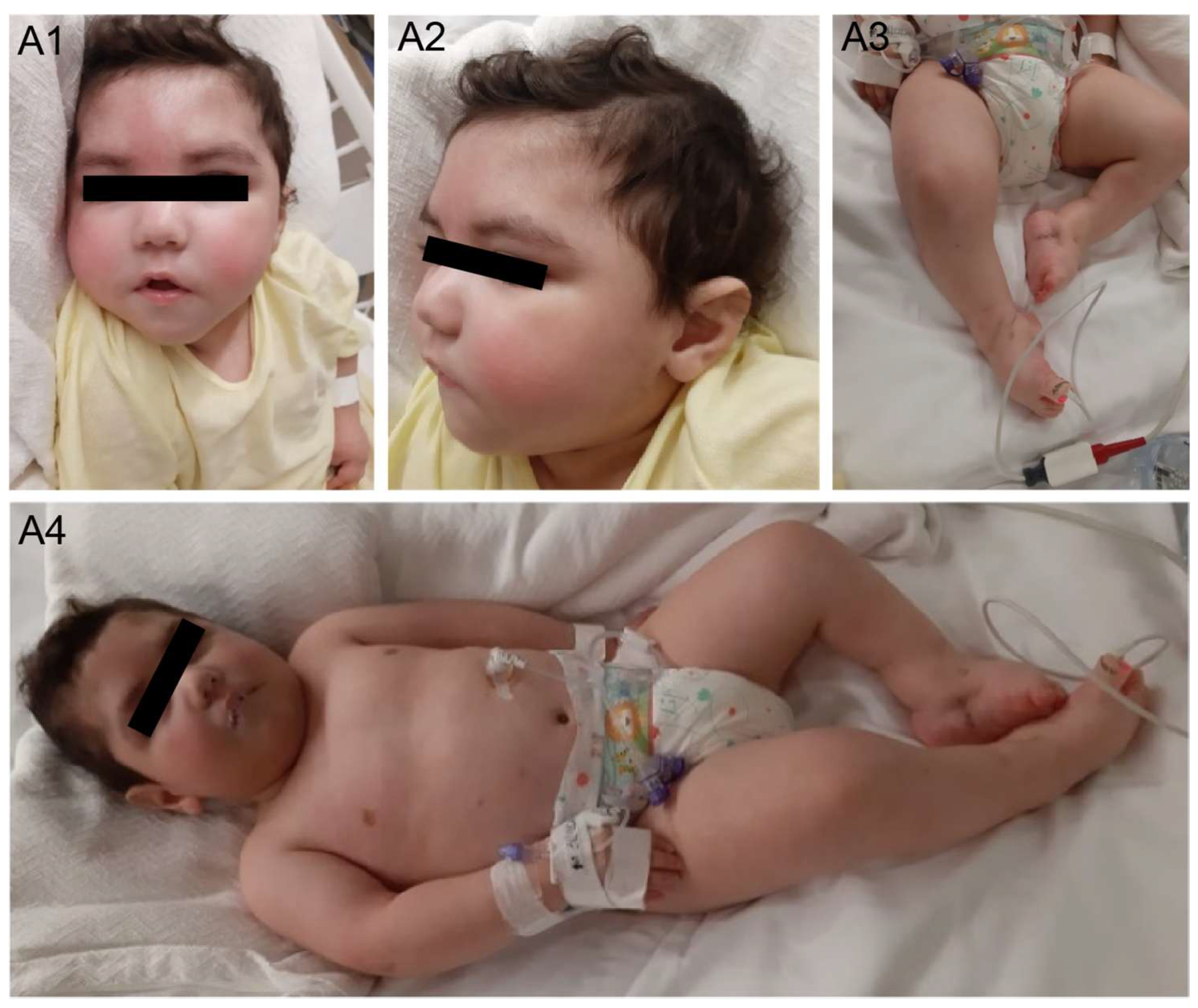
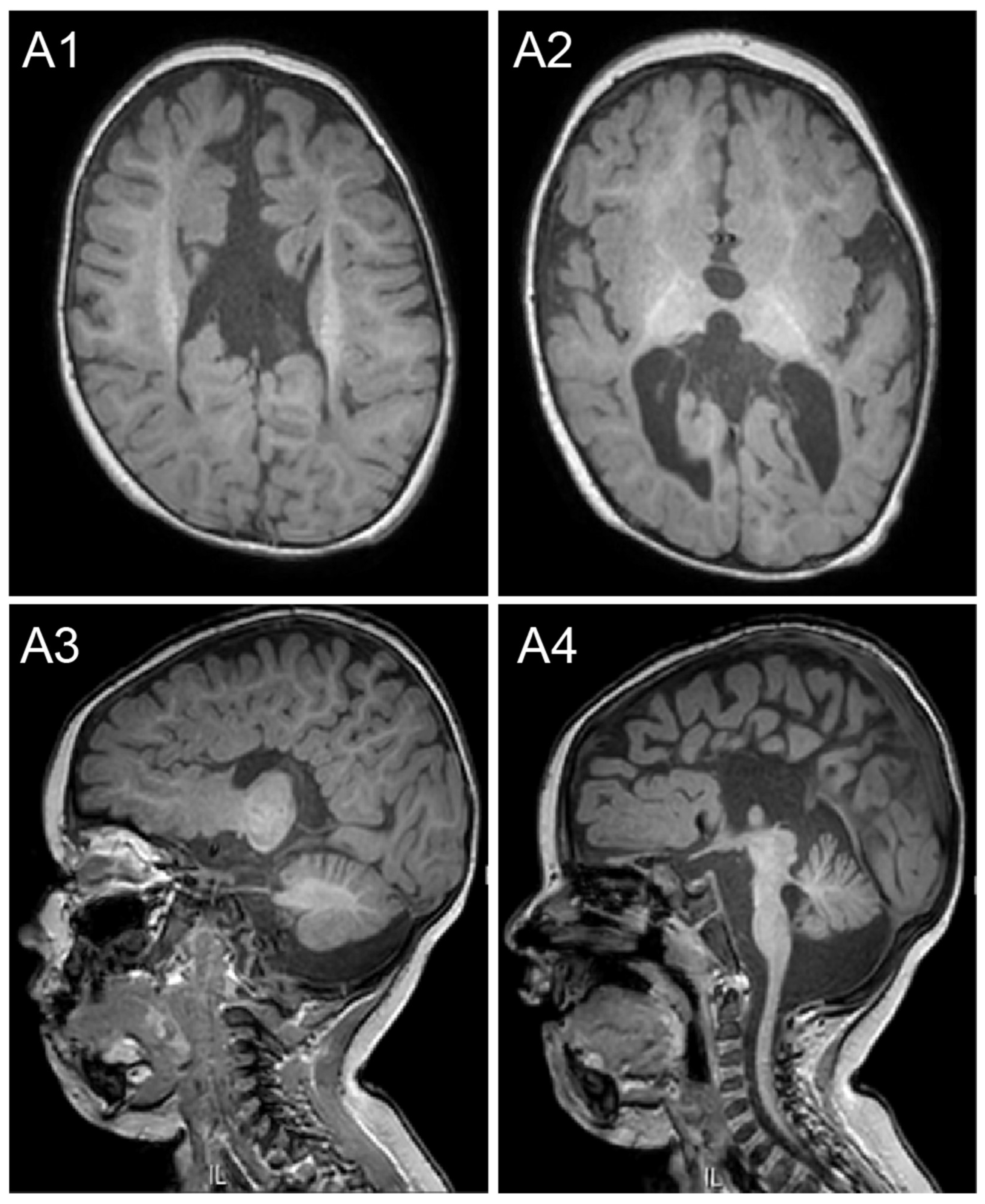
2.1. Case Description
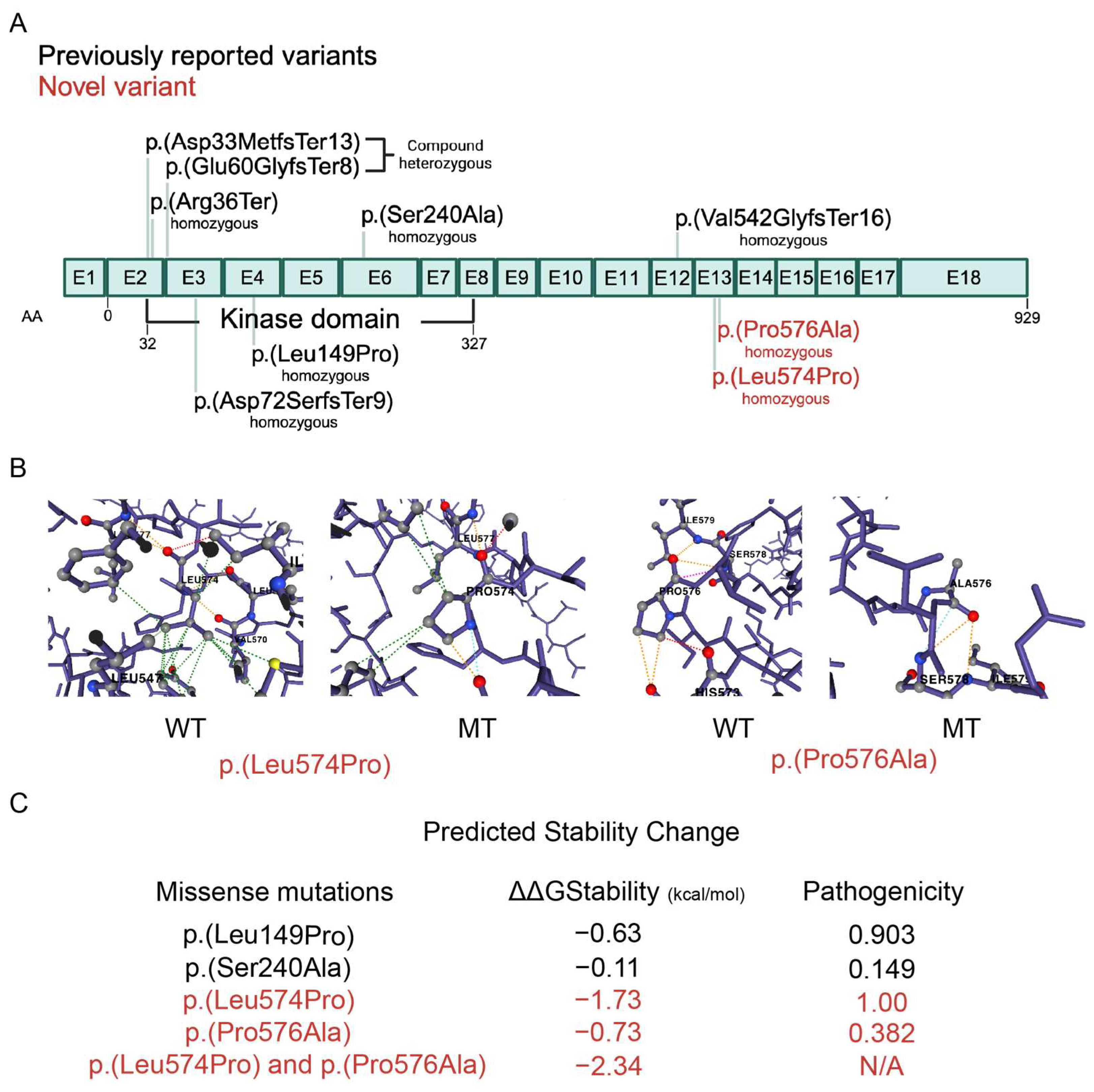
2.2. Genetic Variants
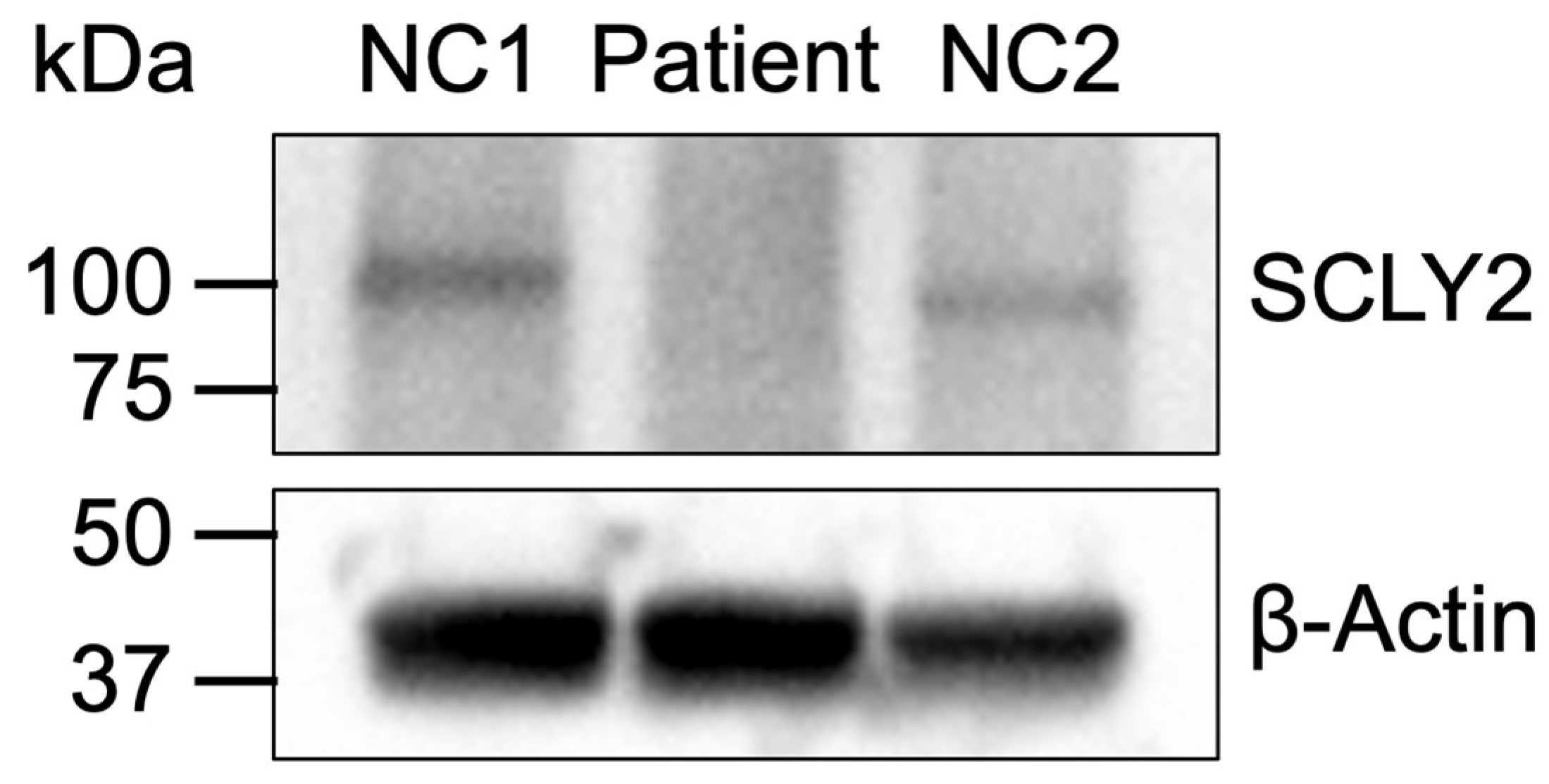
2.3. Protein Expression
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Processing
4.2. Data Processing and Analysis
4.3. Dynamut2 Analysis
4.4. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NDDS | Neurodevelopmental disorders |
| SCYL2 | SCY1-like 2 |
| CNS | Central nervous system |
| WES | Whole-exome sequencing |
| IRB | Institutional review board |
| PBMCs | Peripheral blood mononuclear cells |
| RPMI | Roswell Park Memorial Institute (refers to cell culture medium) |
| TBST | Tris-buffered saline with tween |
| PVDF | Polyvinylidene fluoride |
| CNVs | Copy number variations |
| SNPs | Single-nucleotide polymorphisms |
| DGV | Database of genomic variants |
| MAF | Minor allele frequency |
| SIFT | Sorting intolerant from tolerant (pathogenicity prediction tool) |
| GRCh37 | Genome reference consortium human build 37 |
| GRCh38 | Genome reference consortium human build 38 |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| EGFR | Epidermal growth factor receptor |
| MEFs | Mouse embryonic fibroblasts |
| ESCORT-0 | Endosomal sorting complex required for transport-0 |
| CA3 | Cornu Ammonis 3 (region of the hippocampus) |
| OMIM | Online Mendelian inheritance in man |
| CADD | Combined annotation dependent depletion |
References
- Chiurazzi, P.; Pirozzi, F. Advances in understanding—Genetic basis of intellectual disability. F1000Research 2016, 5, 599. [Google Scholar] [CrossRef]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Tarlungeanu, D.C.; Novarino, G. Genomics in neurodevelopmental disorders: An avenue to personalized medicine. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Kurki, M.I.; Saarentaus, E.; Pietilainen, O.; Gormley, P.; Lal, D.; Kerminen, S.; Torniainen-Holm, M.; Hamalainen, E.; Rahikkala, E.; Keski-Filppula, R.; et al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat. Commun. 2019, 10, 410. [Google Scholar] [CrossRef]
- Sudhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef]
- Baudouin, S.J. Heterogeneity and convergence: The synaptic pathophysiology of autism. Eur. J. Neurosci. 2014, 39, 1107–1113. [Google Scholar] [CrossRef]
- Gingras, S.; Earls, L.R.; Howell, S.; Smeyne, R.J.; Zakharenko, S.S.; Pelletier, S. SCYL2 Protects CA3 Pyramidal Neurons from Excitotoxicity during Functional Maturation of the Mouse Hippocampus. J. Neurosci. 2015, 35, 10510–10522. [Google Scholar] [CrossRef]
- Seidahmed, M.Z.; Al-Kindi, A.; Alsaif, H.S.; Miqdad, A.; Alabbad, N.; Alfifi, A.; Abdelbasit, O.B.; Alhussein, K.; Alsamadi, A.; Ibrahim, N.; et al. Recessive mutations in SCYL2 cause a novel syndromic form of arthrogryposis in humans. Hum. Genet. 2020, 139, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Malbos, M.; Vera, G.; Sheth, H.; Schnur, R.E.; Juven, A.; Brehin, A.C.; Sheth, J.; Gandhi, A.; Shapiro, F.L.; Bruel, A.L.; et al. SCYL2-related autosomal recessive neurodevelopmental disorders: Arthrogryposis multiplex congenita-4 and beyond? Clin. Genet. 2024, 106, 757–763. [Google Scholar] [CrossRef]
- Schmidt, W.M.; Rutledge, S.L.; Schule, R.; Mayerhofer, B.; Zuchner, S.; Boltshauser, E.; Bittner, R.E. Disruptive SCYL1 Mutations Underlie a Syndrome Characterized by Recurrent Episodes of Liver Failure, Peripheral Neuropathy, Cerebellar Atrophy, and Ataxia. Am. J. Hum. Genet. 2015, 97, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, S.; Gingras, S.; Howell, S.; Vogel, P.; Ihle, J.N. An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J. Neurosci. 2012, 32, 16560–16573. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Miles, R. The CA3 region of the hippocampus: How is it? What is it for? How does it do it? Front. Cell Neurosci. 2015, 9, 19. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Olney, J.W. Role of excitotoxins in developmental neuropathology. APMIS Suppl. 1993, 40, 103–112. [Google Scholar]
- Smeyne, R.J.; Vendrell, M.; Hayward, M.; Baker, S.J.; Miao, G.G.; Schilling, K.; Robertson, L.M.; Curran, T.; Morgan, J.I. Continuous c-fos expression precedes programmed cell death in vivo. Nature 1993, 363, 166–169. [Google Scholar] [CrossRef]
- Duwel, M.; Ungewickell, E.J. Clathrin-dependent association of CVAK104 with endosomes and the trans-Golgi network. Mol. Biol. Cell 2006, 17, 4513–4525. [Google Scholar] [CrossRef]
- Tamai, K.; Toyoshima, M.; Tanaka, N.; Yamamoto, N.; Owada, Y.; Kiyonari, H.; Murata, K.; Ueno, Y.; Ono, M.; Shimosegawa, T.; et al. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am. J. Pathol. 2008, 173, 1806–1817. [Google Scholar] [CrossRef]
- Burman, J.L.; Bourbonniere, L.; Philie, J.; Stroh, T.; Dejgaard, S.Y.; Presley, J.F.; McPherson, P.S. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J. Biol. Chem. 2008, 283, 22774–22786. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, J.N.; Schroeder, L.K.; Fotouhi, M.; Dokainish, H.; Ioannou, M.S.; Girard, M.; Summerfeldt, N.; Melancon, P.; McPherson, P.S. Scyl1 scaffolds class II Arfs to specific subcomplexes of coatomer through the gamma-COP appendage domain. J. Cell Sci. 2014, 127, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Kamena, F.; Diefenbacher, M.; Kilchert, C.; Schwarz, H.; Spang, A. Ypt1p is essential for retrograde Golgi-ER transport and for Golgi maintenance in S. cerevisiae. J. Cell Sci. 2008, 121, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Bellouze, S.; Schafer, M.K.; Buttigieg, D.; Baillat, G.; Rabouille, C.; Haase, G. Golgi fragmentation in pmn mice is due to a defective ARF1/TBCE cross-talk that coordinates COPI vesicle formation and tubulin polymerization. Hum. Mol. Genet. 2014, 23, 5961–5975. [Google Scholar] [CrossRef]
- Haase, G.; Rabouille, C. Golgi Fragmentation in ALS Motor Neurons. New Mechanisms Targeting Microtubules, Tethers, and Transport Vesicles. Front. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef]
- Martinez-Menarguez, J.A.; Tomas, M.; Martinez-Martinez, N.; Martinez-Alonso, E. Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef]
- Custer, S.K.; Foster, J.N.; Astroski, J.W.; Androphy, E.J. Abnormal Golgi morphology and decreased COPI function in cells with low levels of SMN. Brain Res. 2019, 1706, 135–146. [Google Scholar] [CrossRef]
- Miyakawa, K.; Sawasaki, T.; Matsunaga, S.; Tokarev, A.; Quinn, G.; Kimura, H.; Nomaguchi, M.; Adachi, A.; Yamamoto, N.; Guatelli, J.; et al. Interferon-induced SCYL2 limits release of HIV-1 by triggering PP2A-mediated dephosphorylation of the viral protein Vpu. Sci. Signal. 2012, 5, ra73. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Variant | Bi-Allelic Missense Variants | Bi-Allelic Loss-of-Function Variants | |||||
|---|---|---|---|---|---|---|---|
| Cases | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Cases 6–9 | Cases 10–11 |
| Gender | Male | Male | Male | Male | Male | 3 males and 1 female | 2 females |
| Arthrogryposis | Yes | No | No | Yes | Yes | Yes (4/4) | Yes (2/2) |
| Corpus callosum agenesis | Yes | No | Not available | Yes | Yes | Yes (4/4) | Yes (2/2) |
| Cerebral findings | Pronto-cerebellar volume reduction and white matter abnormalities | No | Not available | Polymicrogyria in the insula, agenesis of the posterior corpus callosum, and pyramidal tract hypoplasia | Cerebral and cerebellar cortical atrophy | Frontoparietal cortical atrophy (3/3) | No (2/2) |
| Psychomotor delay | Yes, severe global developmental delay affecting motor, language, and social/communicative spheres | Yes, speech delay and poor understanding but normal motor development | Yes, speech delay, particularly for expressive language with a better receptive language | Not applicable | Yes | Yes (4/4) | Yes (2/2) |
| Ventriculomegaly | Yes | No | Not available | No | No | Yes (2/3) | Yes (2/2) |
| Reduced fetal movements | Yes | No | No | Yes | No | Yes (4/4) | Yes (2/2) |
| Polyhydramnios | No | No | No | No | No | Yes (4/4) | Yes (2/2) |
| Microcephaly | Yes | No | Yes <1st percentile | No | No | Yes (2/3) | Yes (2/2) |
| Cryptorchidism | No | Yes, unilateral | No | Testicles not present in the scrotum (at 27 weeks of gestational age) | Yes | Yes (3/3) | Not applicable |
| Micrognathia | Yes | Yes | No | Yes | No | Yes (4/4) | Yes (2/2) |
| Optic atrophy | Yes, bilateral with mild esotropia | No | No | Not available | No | Yes (3/3) | Yes (1/1) |
| Spasticity | Yes | No | No | Not applicable | No | Yes (4/4) | Yes (2/2) |
| Seizures | Febrile status epilepticus and epilepsy | No | No | Not applicable | No | Yes (3/3) | Yes (1/1) |
| Other morphological features | Prominent forehead, depressed nasal bridge, bulbous nose, low-set abnormally folded ears, small mouth, thin lips, and short neck | Synophrys, mild slant eyes, lop undifferentiated ears, and thick lower lip | Brachycephaly, low anterior hairline, heavy eyebrows, long eyelashes, large upper central incisors, clinodactyly, mild 2–3–4 finger syndactyly, and narrow 5th toenails | Square forehead, depressed nasal bridge, broad nasal tip, low-set slightly dysplastic ears, hypoplastic helix, thin lips, and short neck | No | Prominent forehead (4/4), depressed nasal bridge (3/3), bulbous nose (4/4), low-set abnormally folded ears (4/4), small mouth (4/4), thin lips (4/4), and short neck (4/4) | Prominent forehead (2/2), depressed nasal bridge (2/2), bulbous nose (2/2), low-set abnormally folded ears (2/2), small mouth (2/2), thin lips (2/2), and short neck (1/2) |
| Pathological fractures | No | No | No | No | No | Yes (3/4) | Yes (2/2) |
| Other signs | Dysphagia, gastroenteritis, chest infections, febrile seizures, neutropenic episodes, and died at 22 months | Some findings of autism spectrum disorder and unilateral inguinal hernia repair | Autism spectrum disorder | Mild calf muscle hypoplasia | Inguinal hernia and bilateral hip dysplasia | Feeding difficulties (4/4) and death between 8 and 30 months (4/4) | Feeding difficulties (2/2) and death at 3 years (1/2) |
| Variant (GRCh38) | chr12: 100329279T>C and chr12: 100329284C>G | chr12: g.100298141T>C | chr12: g.100312519T>G | chr12: g.100283067del and chr12: g.100283146dup | chr12: g.100291539_100291559delinsTC | chr12: g.100283076C>T | chr12: g.100326736dup |
| Variant (NM_017988.6) | c.1721T>C and c.1726C>G | c.446T>C | c.718T>G | c.97del and c.176dup | c.214_234delinsTC | c.106C>T | c.1624dup |
| Amino acid variant | p.(Leu574Pro) and p.(Pro576Ala) | p.(Leu149Pro) | p.(Ser240Ala) | p.(Asp33MetfsTer13) and p.(Glu60GlyfsTer8) | p.(Asp72SerfsTer9) | p.(Arg36Ter) | p.(Val542GlyfsTer16) |
| Inheritance | Both parents heterozygous | Both parents heterozygous | Both parents heterozygous | Paternal and maternal, respectively | Both parents heterozygous | Both parents heterozygous | Both parents heterozygous |
| Classification | Uncertain significance | Uncertain significance | Uncertain significance | Both pathogenic | Pathogenic | Pathogenic | Pathogenic |
| ACMG criteria and REVEL score (if applicable) | c.1721T>C PM2 PP3 REVEL score: 0.850 c.1726C>G PM2 REVEL score: 0.202 | PM2 PP3 REVEL score: 0.918 | PM2 REVEL score: 0.205 | Both PVS1 PM2 PM3 | PVS1 PM2 PM3 | PVS1 PM2 PM3 | PVS1 PM2 PM3 |
| Other variants | See Table 2 | FLNA: NM_001110556.2: c.5975G>C p.(Gly1992Ala) Hemizygous VUS | RBPJ: NM_005349.4: c.173del p. (Lys58SerfsTer48) Heterozygous De novo VUS | No | No | Not available | Not available |
| Genes | Transcript ID | HGVSp | HGVSc | Genotype | Pop. Allele Freq % | PolyPhen Prediction | SIFT Prediction | dbSNP | Chromosome Region | Varsome |
|---|---|---|---|---|---|---|---|---|---|---|
| SCN5A | NM_198056.2 | p.(Val1202Met) | c.3604G>A | Hom | 0.006 | Probably damaging | Deleterious | rs375509048 | chr3:38,575,359 | VUS |
| KLHL40 | NM_152393.3 | p.(Val452Ala) | c.1355T>C | Hom | 0.065 | Benign | Deleterious | rs140389534 | chr3:42,686,651 | VUS |
| STAB1 | NM_015136.2 | p.(Pro1978Ser) | c.5932C>T | Hom | 0.003 | Probably damaging | Deleterious | rs752987636 | chr3:52,521,384 | Likely pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamel, K.; Al-Subaiey, A.A.; Alsabbagh, M.; Fadda, A.; Saeed, A.; Mourao Pacheco, B.; Lo, B.; Benini, R. Novel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature. Int. J. Mol. Sci. 2025, 26, 3079. https://doi.org/10.3390/ijms26073079
Zamel K, Al-Subaiey AA, Alsabbagh M, Fadda A, Saeed A, Mourao Pacheco B, Lo B, Benini R. Novel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature. International Journal of Molecular Sciences. 2025; 26(7):3079. https://doi.org/10.3390/ijms26073079
Chicago/Turabian StyleZamel, Khaled, Abdulrahman Ahmed Al-Subaiey, Mohamed Alsabbagh, Abeer Fadda, Amira Saeed, Bruno Mourao Pacheco, Bernice Lo, and Ruba Benini. 2025. "Novel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature" International Journal of Molecular Sciences 26, no. 7: 3079. https://doi.org/10.3390/ijms26073079
APA StyleZamel, K., Al-Subaiey, A. A., Alsabbagh, M., Fadda, A., Saeed, A., Mourao Pacheco, B., Lo, B., & Benini, R. (2025). Novel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature. International Journal of Molecular Sciences, 26(7), 3079. https://doi.org/10.3390/ijms26073079