
Mitochondrial DNA in Exercise-Mediated Innate Immune Responses
Abstract
1. Introduction
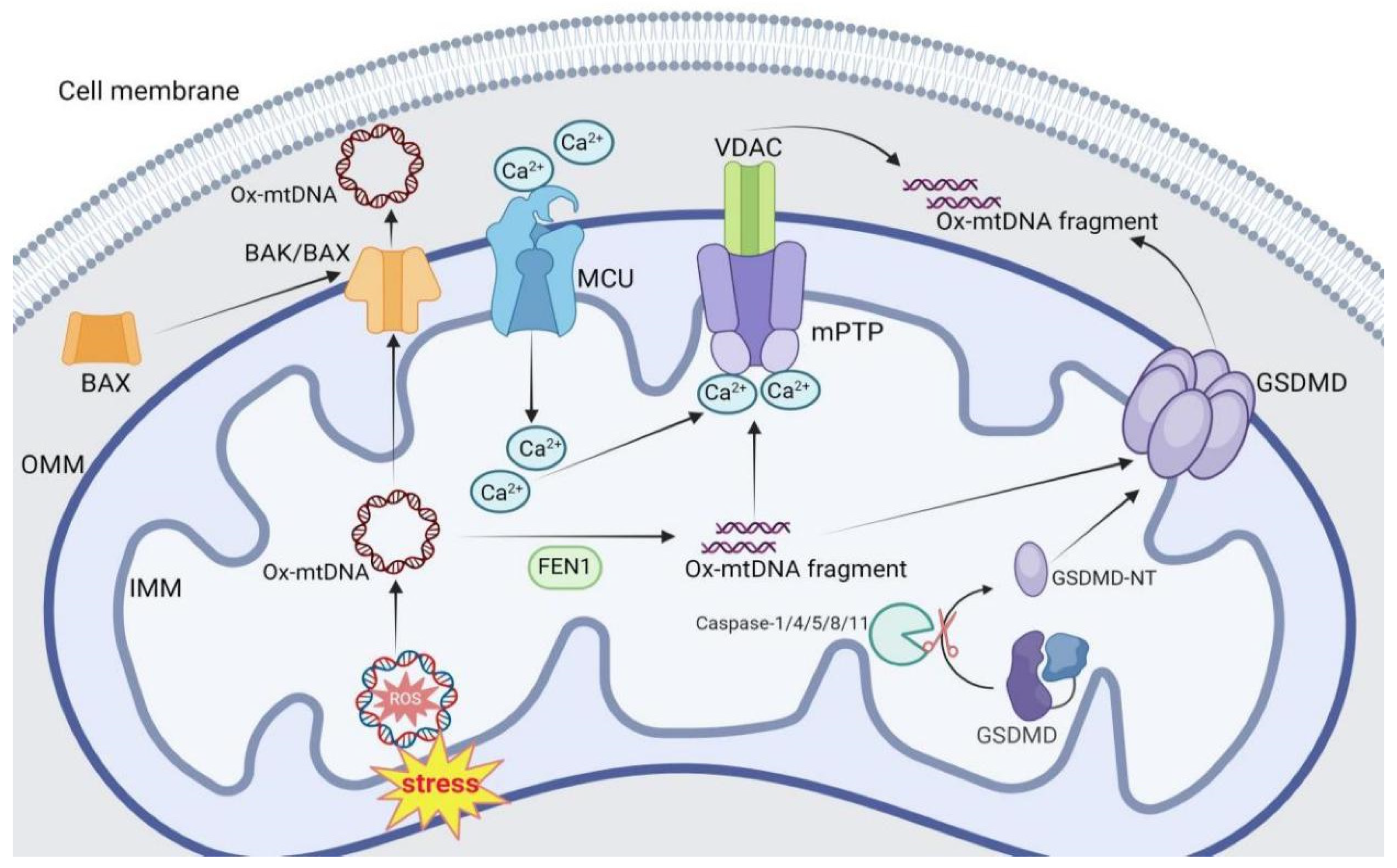
2. Mechanisms of mtDNA Release
2.1. BAK/BAX Pore
2.2. mPTP
2.3. GSDMD Pore
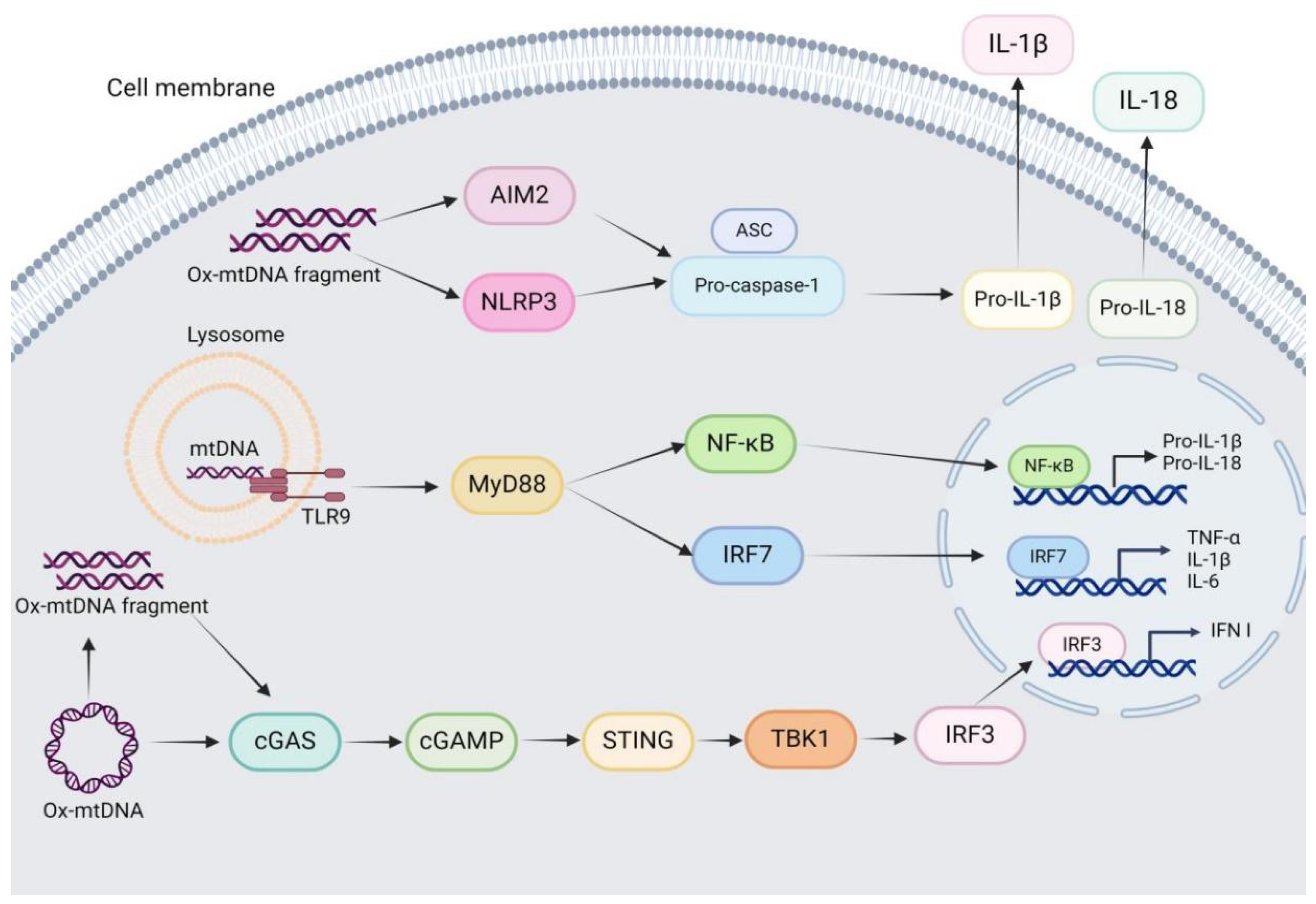
3. MtDNA-Driven Innate Immune Signaling
3.1. mtDNA-TLR9 Signaling
3.2. mtDNA-cGAS–STING Signaling
3.3. mtDNA-NLRP3 Inflammasome Signaling
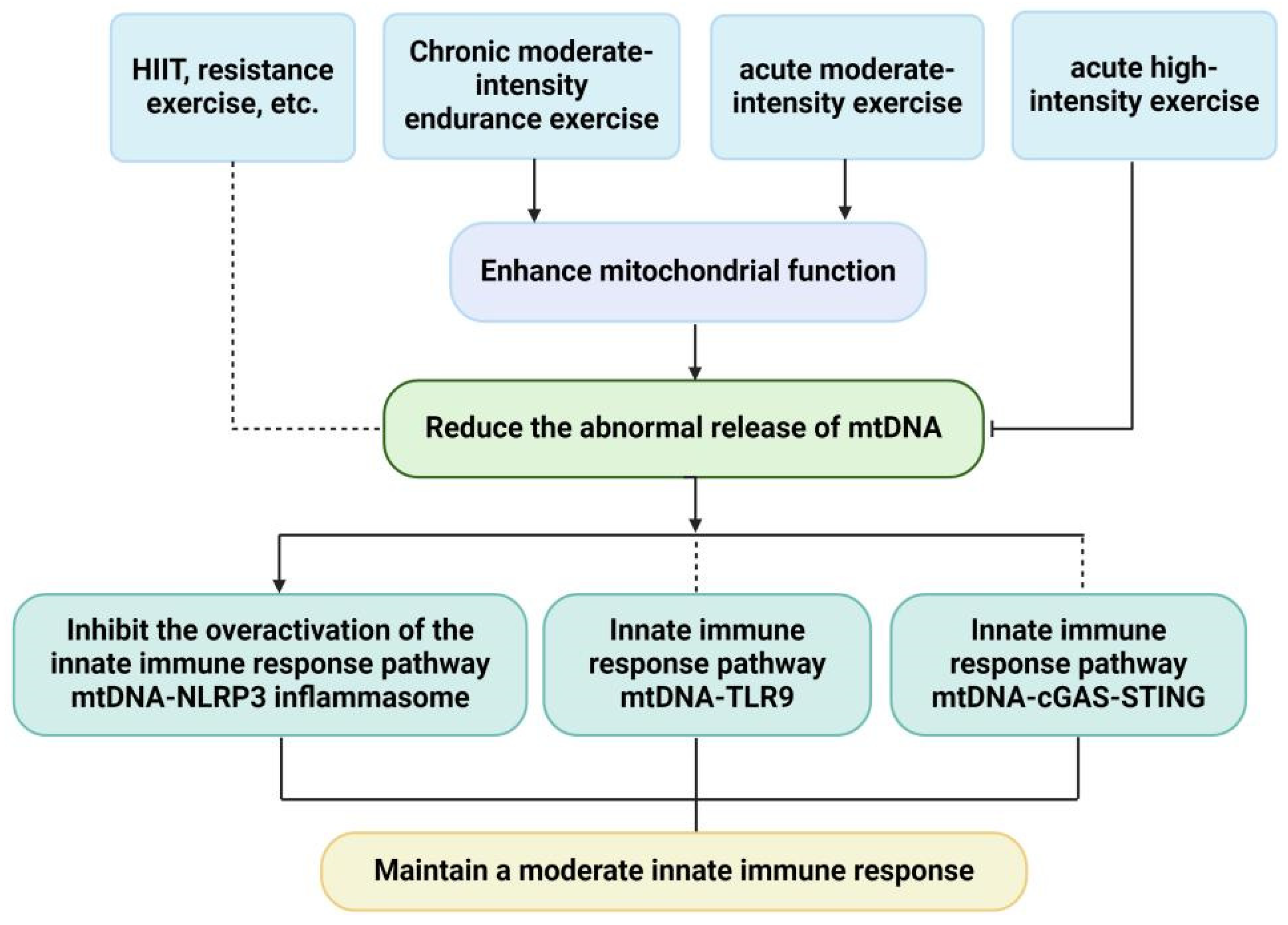
4. Effects of Exercise on mtDNA
5. Effects of Exercise on mtDNA and mtDNA-Driven Innate Immune Response
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PAMPs | pathogen-associated molecular patterns |
| DAMPs | damage-associated molecular patterns |
| PRRs | pattern recognition receptors |
| mtDNA | mitochondrial DNA |
| TFAM | mitochondrial Transcription Factor A |
| ROS | reactive oxygen species |
| ox-mtDNA | oxidized mtDNA |
| TLR9 | toll-like receptor-9 |
| cGAS | cyclic GMP-AMP synthase |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 |
| BAK | Bcl-2-associated K protein |
| BAX | Bcl-2-associated X protein |
| mPTP | mitochondrial permeability transition pore |
| GSDMD | Gasdermin D |
| OMM | mitochondrial outer membrane |
| STING | cGAS-interferon gene stimulating factor |
| ANT | adenine nucleotide translocator |
| CypD | cyclophilin D |
| VDAC | voltage-dependent anion channel |
| MCU | mitochondrial calcium uniporter |
| FEN1 | Flap Endonuclease 1 |
| LPS | lipopolysaccharide |
| XBP1 | X-box binding protein 1 |
| NF-κB | nuclear factor kappa-B |
| MAPK | mitogen-activated protein kinase |
| cGAMP | cyclic guanosine monophosphate-adenosine monophosphate |
| TBK1 | TANK-binding kinase 1 |
| IRF3 | interferon response factor 3 |
| IFN-I | type I interferon |
| ISGs | interferon-stimulated genes |
| PYD | pyrin domain |
| NBD/NACHT | nucleotide-binding oligerization domain |
| LRR | leucine-rich repeat |
| ASC | apoptosis speck-like protein containing a caspase recruitment domain |
| pro-caspase-1 | precursor caspase-1 |
| CARD C | terminal caspase recruitment domain |
| IL-1β | interleukin-1β |
| mtROS | mitochondrial ROS |
| AIM2 | Absent in melanoma 2 |
| MyD88 | myeloid differentiation primary response protein 88 |
| IRF7 | interferon regulatory factor 7 |
| cf-mtDNA | circulating free mtDNA |
| non-IBS | non-irritable bowel syndrome |
| NAFLD | nonalcoholic fatty liver disease |
| CKD | chronic kidney disease |
| T2DM | type 2 diabetes mellitus |
| HIIT | high-intensity interval exercise |
| PVPs | professional volleyball players |
References
- Zhang, T.; Ding, S.; Wang, R. Research Progress of Mitochondrial Mechanism in NLRP3 Inflammasome Activation and Exercise Regulation of NLRP3 Inflammasome. Int. J. Mol. Sci. 2021, 22, 10866. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the Regulation of Innate Immune Responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B. Song Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces in Vivo and in Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Singh, G.; Pachouri, U.C.; Khaidem, D.C.; Kundu, A.; Chopra, C.; Singh, P. Mitochondrial DNA Damage and Diseases. F1000Research 2015, 4, 176. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.-S.; Chung, J.H. Molecular Mechanisms of Mitochondrial DNA Release and Activation of the cGAS-STING Pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and mtDNA Efflux during Apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial Inner Membrane Permeabilisation Enables Mt DNA Release during Apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, H.; Zhang, Y.-L.; Xin, Q.-L.; Guan, Z.-Q.; Chen, X.; Zhang, X.-A.; Li, X.-K.; Xiao, G.-F.; Lozach, P.-Y.; et al. SFTSV Infection Induces BAK/BAX-Dependent Mitochondrial DNA Release to Trigger NLRP3 Inflammasome Activation. Cell Rep. 2020, 30, 4370–4385.e7. [Google Scholar] [CrossRef]
- Chen, L.; Dong, J.; Liao, S.; Wang, S.; Wu, Z.; Zuo, M.; Liu, B.; Yan, C.; Chen, Y.; He, H.; et al. Loss of Sam50 in Hepatocytes Induces Cardiolipin-dependent Mitochondrial Membrane Remodeling to Trigger mtDNA Release and Liver Injury. Hepatology 2022, 76, 1389–1408. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.-M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Zhou, L.; Mao, H.-Q.; Yang, F.-H.; Chen, Z.; Zhang, L. Mitochondrial DNA Leakage Exacerbates Odontoblast Inflammation through Gasdermin D-Mediated Pyroptosis. Cell Death Discov. 2021, 7, 381. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Q.; Liu, T.; Yang, S.; Sun, L.; Zhao, Z.-Y.; Li, L.-Y.; She, Y.-C.; Zheng, Y.-Y.; Ye, X.-Y.; Bao, Q.; et al. Perfluoroalkyl Substance Pollutants Activate the Innate Immune System through the AIM2 Inflammasome. Nat. Commun. 2021, 12, 2915. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Peña-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The Interplay between BAX and BAK Tunes Apoptotic Pore Growth to Control Mitochondrial-DNA-Mediated Inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef]
- Newman, L.E.; Shadel, G.S. Mitochondrial DNA Release in Innate Immune Signaling. Annu. Rev. Biochem. 2023, 92, 299–332. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC Oligomers Form Mitochondrial Pores to Release mtDNA Fragments and Promote Lupus-like Disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Xian, H.; Karin, M. Oxidized Mitochondrial DNA: A Protective Signal Gone Awry. Trends Immunol. 2023, 44, 188–200. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA Fragments Exit Mitochondria via mPTP- and VDAC-Dependent Channels to Activate NLRP3 Inflammasome and Interferon Signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Hu, M.-M.; Shu, H.-B. Mitochondrial DNA-Triggered Innate Immune Response: Mechanisms and Diseases. Cell. Mol. Immunol. 2023, 20, 1403–1412. [Google Scholar] [CrossRef]
- Ouyang, W.; Wang, S.; Yan, D.; Wu, J.; Zhang, Y.; Li, W.; Hu, J.; Liu, Z. The cGAS-STING Pathway-Dependent Sensing of Mitochondrial DNA Mediates Ocular Surface Inflammation. Signal Transduct. Target. Ther. 2023, 8, 371. [Google Scholar] [CrossRef]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in Pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lieberman, J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. In Advances in Immunology; Elsevier: Amsterdam, Netherlands, 2017; pp. 81–117. ISBN 978-0-12-812405-5. [Google Scholar]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An Endogenous Caspase-11 Ligand Elicits Interleukin-1 Release from Living Dendritic Cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Jiang, C.; Chang, W.Y.; Zhang, H.; An, J.; Ho, F.; Chen, P.; Zhang, H.; Junqueira, C.; Amgalan, D.; et al. Gasdermin D Permeabilization of Mitochondrial Inner and Outer Membranes Accelerates and Enhances Pyroptosis. Immunity 2023, 56, 2523–2541.e8. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Z.; Wu, X.; Zhang, L.; Cao, Y.; Zhou, P. Distinct Molecular Mechanisms Underlying Potassium Efflux for NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609441. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 Induces Cleavage of Gasdermin D to Elicit Pyroptosis during Yersinia Infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of TAK1 Triggers Caspase-8—Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and Intrinsic Apoptosis Activate Pannexin-1 to Drive NLRP 3 Inflammasome Assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, M.; Wang, X.; Bu, Q.; Wang, Q.; Su, W.; Li, L.; Zhou, H.; Lu, L. XBP1 Deficiency Promotes Hepatocyte Pyroptosis by Impairing Mitophagy to Activate mtDNA-cGAS-STING Signaling in Macrophages during Acute Liver Injury. Redox Biol. 2022, 52, 102305. [Google Scholar] [CrossRef]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e5. [Google Scholar] [CrossRef]
- Barnett, K.C.; Ting, J.P.-Y. Mitochondrial GSDMD Pores DAMPen Pyroptosis. Immunity 2020, 52, 424–426. [Google Scholar] [CrossRef]
- Platnich, J.M.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, H.-M.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.e7. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Wang, Z.; Wang, Q.; Xie, H.; Yang, N.; Wang, Y.; Wang, J.; Kang, H.; Bai, W.; Wang, Y.; et al. Oxidized Mitochondrial DNA Induces Gasdermin D Oligomerization in Systemic Lupus Erythematosus. Nat. Commun. 2023, 14, 872. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, J.; Liu, T.; Cheng, W.; Wang, Y.; Ding, S.; Wang, R. A Novel Mechanism for NLRP3 Inflammasome Activation. Metab. Open 2022, 13, 100166. [Google Scholar] [CrossRef]
- Lu, P.; Zheng, H.; Meng, H.; Liu, C.; Duan, L.; Zhang, J.; Zhang, Z.; Gao, J.; Zhang, Y.; Sun, T. Mitochondrial DNA Induces Nucleus Pulposus Cell Pyroptosis via the TLR9-NF-κB-NLRP3 Axis. J. Transl. Med. 2023, 21, 389. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.K.; Bennett, M.R. Mitochondrial DNA Damage and Atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 481–487. [Google Scholar] [CrossRef]
- Julian, M.W.; Shao, G.; VanGundy, Z.C.; Papenfuss, T.L.; Crouser, E.D. Mitochondrial Transcription Factor A, an Endogenous Danger Signal, Promotes TNFα Release via RAGE- and TLR9-Responsive Plasmacytoid Dendritic Cells. PLoS ONE 2013, 8, e72354. [Google Scholar]
- Latz, E.; Schoenemeyer, A.; Visintin, A.; Fitzgerald, K.A.; Monks, B.G.; Knetter, C.F.; Lien, E.; Nilsen, N.J.; Espevik, T.; Golenbock, D.T. TLR9 Signals after Translocating from the ER to CpG DNA in the Lysosome. Nat. Immunol. 2004, 5, 190–198. [Google Scholar]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA Is Released by Shock and Activates Neutrophils via P38 Map Kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Ohto, U.; Shibata, T.; Tanji, H.; Ishida, H.; Krayukhina, E.; Uchiyama, S.; Miyake, K.; Shimizu, T. Structural Basis of CpG and Inhibitory DNA Recognition by Toll-like Receptor 9. Nature 2015, 520, 702–705. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Ritchie, C.; Carozza, J.A.; Li, L. Biochemistry, Cell Biology, and Pathophysiology of the Innate Immune cGAS–cGAMP–STING Pathway. Annu. Rev. Biochem. 2022, 91, 599–628. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Luzwick, J.W.; Dombi, E.; Boisvert, R.A.; Roy, S.; Park, S.; Kunnimalaiyaan, S.; Goffart, S.; Schindler, D.; Schlacher, K. MRE11-Dependent Instability in Mitochondrial DNA Fork Protection Activates a cGAS Immune Signaling Pathway. Sci. Adv. 2021, 7, eabf9441. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, I.; Deng, J.; Lei, Y.; Torres-Odio, S.; Rojas, G.R.; Newman, L.E.; Chung, B.K.; Symes, A.; Zhang, H.; Huang, S.-Y.N.; et al. Activation of the cGAS-STING Innate Immune Response in Cells with Deficient Mitochondrial Topoisomerase TOP1MT. Hum. Mol. Genet. 2023, 32, 2422–2440. [Google Scholar] [CrossRef]
- Hancock-Cerutti, W.; Wu, Z.; Xu, P.; Yadavalli, N.; Leonzino, M.; Tharkeshwar, A.K.; Ferguson, S.M.; Shadel, G.S.; De Camilli, P. ER-Lysosome Lipid Transfer Protein VPS13C/PARK23 Prevents Aberrant mtDNA-Dependent STING Signaling. J. Cell Biol. 2022, 221, e202106046. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and Function of the cGAS–STING Pathway of Cytosolic DNA Sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Wu, Z.; Sainz, A.G.; Shadel, G.S. Mitochondrial DNA: Cellular Genotoxic Stress Sentinel. Trends Biochem. Sci. 2021, 46, 812–821. [Google Scholar] [CrossRef]
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC Filament Formation Serves as a Signal Amplification Mechanism for Inflammasomes. Nat. Commun. 2016, 7, 11929. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Xu, J.; Núñez, G. The NLRP3 Inflammasome: Activation and Regulation. Trends Biochem. Sci. 2023, 48, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e11. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.; Chen, J.; Sang, T.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. Overexpression of NAG-1/GDF15 Prevents Hepatic Steatosis through Inhibiting Oxidative Stress-Mediated dsDNA Release and AIM2 Inflammasome Activation. Redox Biol. 2022, 52, 102322. [Google Scholar] [CrossRef]
- Chimienti, G.; Russo, F.; Bianco, A.; Maqoud, F.; De Virgilio, C.; Galeano, G.; Orlando, A.; Riezzo, G.; D’Attoma, B.; Ignazzi, A.; et al. Effect of a 12-Week Walking Program Monitored by Global Physical Capacity Score (GPCS) on Circulating Cell-Free mtDNA and DNase Activity in Patients with Irritable Bowel Syndrome. Int. J. Mol. Sci. 2024, 25, 4293. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Chen, M.; Mu, W.-J.; Luo, H.-Y.; Guo, L. Higd1a Facilitates Exercise-Mediated Alleviation of Fatty Liver in Diet-Induced Obese Mice. Metabolism 2022, 134, 155241. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Morales, J.; Korucu, B.; Pike, M.M.; Lipworth, L.; Stewart, T.; Headley, S.A.E.; Germain, M.; Begue, G.; Roshanravan, B.; Tuttle, K.R.; et al. Effects of Caloric Restriction and Aerobic Exercise on Circulating Cell-Free Mitochondrial DNA in Patients with Moderate to Severe Chronic Kidney Disease. Am. J. Physiol. Renal Physiol. 2022, 322, F68–F75. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Ogino, N.; Tomizuka, K.; Eitoku, M.; Okada, Y.; Tanaka, Y.; Suganuma, N.; Ogino, K. SOD2 mRNA as a Potential Biomarker for Exercise: Interventional and Cross-Sectional Research in Healthy Subjects. J. Clin. Biochem. Nutr. 2021, 69, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Oo, S.M.; Htut, M.T.; Htun, Y.W.; Mon, A.A.; Kyaw, M.P. Lower Plasma Selenoprotein P Levels in Regularly Exercising Young Adults. J. ASEAN Fed. Endocr. Soc. 2023, 38, 6–12. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. J. Gerontol. A. Biol. Sci. Med. Sci. 2006, 61, 534–540. [Google Scholar] [CrossRef]
- Latimer, L.E.; Constantin-Teodosiu, D.; Popat, B.; Constantin, D.; Houchen-Wolloff, L.; Bolton, C.E.; Steiner, M.C.; Greenhaff, P.L. Whole-Body and Muscle Responses to Aerobic Exercise Training and Withdrawal in Ageing and COPD. Eur. Respir. J. 2022, 59, 2101507. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.S.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of Weight Loss and Physical Activity on Skeletal Muscle Mitochondrial Function in Obesity. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Petzinger, G.M.; Jakowec, M.W.; Cadenas, E. Treadmill Exercise Rescues Mitochondrial Function and Motor Behavior in the CAG (140) Knock-in Mouse Model of Huntington’s Disease. Chem. Biol. Interact. 2020, 315, 108907. [Google Scholar] [CrossRef]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise Training Increases Mitochondrial Biogenesis in the Brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar]
- Jaroslawska, J.; Gospodarska, E.; Korytko, A. Increasing Energy Expenditure through Exercise and Low Ambient Temperature Offers Oxidative Protection to the Hypothalamus after High-Fat Feeding to Mice. J. Neuroendocrinol. 2022, 34, e13095. [Google Scholar] [CrossRef]
- Shin, J.; Hong, S.-G.; Choi, S.Y.; Rath, M.E.; Saredy, J.; Jovin, D.G.; Sayoc, J.; Park, H.-S.; Eguchi, S.; Rizzo, V.; et al. Flow-Induced Endothelial Mitochondrial Remodeling Mitigates Mitochondrial Reactive Oxygen Species Production and Promotes Mitochondrial DNA Integrity in a P53-Dependent Manner. Redox Biol. 2022, 50, 102252. [Google Scholar] [CrossRef]
- Maclaine, K.D.; Stebbings, K.A.; Llano, D.A.; Rhodes, J.S. Voluntary Wheel Running Has No Impact on Brain and Liver Mitochondrial DNA Copy Number or Mutation Measures in the PolG Mouse Model of Aging. PLoS ONE 2020, 15, e0226860. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Bouzourène, K.; Aubert, J.-F.; Bielmann, C.; Gruetter, R.; Rosenblatt-Velin, N.; Poitry-Yamate, C.; Mazzolai, L. Impact of Aerobic Exercise Type on Blood Flow, Muscle Energy Metabolism, and Mitochondrial Biogenesis in Experimental Lower Extremity Artery Disease. Sci. Rep. 2020, 10, 14048. [Google Scholar] [CrossRef]
- Gureev, A.P.; Sadovnikova, I.S.; Shaforostova, E.A.; Starkov, A.A.; Popov, V.N. Mildronate Protects Heart mtDNA from Oxidative Stress Toxicity Induced by Exhaustive Physical Exercise. Arch. Biochem. Biophys. 2021, 705, 108892. [Google Scholar] [CrossRef]
- Lee, H.; Kim, K.; Kim, B.; Shin, J.; Rajan, S.; Wu, J.; Chen, X.; Brown, M.D.; Lee, S.; Park, J.-Y. A Cellular Mechanism of Muscle Memory Facilitates Mitochondrial Remodelling Following Resistance Training. J. Physiol. 2018, 596, 4413–4426. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, V.A.; Baena-García, L.; Sánchez-González, C.; Acosta-Manzano, P.; Varela-López, A.; Quiles, J.L. Influence of a Concurrent Exercise Training Program during Pregnancy on the Placenta Mitochondrial DNA Integrity and Content of Minerals with Enzymatic Relevance. The GESTAFIT Project. Placenta 2023, 139, 19–24. [Google Scholar] [CrossRef]
- Yang, S.Y.; Mirabal, C.S.; Newcomb, C.E.; Stewart, K.J.; Arking, D.E. Mitochondrial DNA Copy Number, Metabolic Syndrome, and Insulin Sensitivity: Insights from the Sugar, Hypertension, and Physical Exercise Studies. PLoS ONE 2022, 17, e0270951. [Google Scholar] [CrossRef]
- Al-Rawaf, H.A.; Gabr, S.A.; Iqbal, A.; Alghadir, A.H. High-Intensity Interval Training Improves Glycemic Control, Cellular Apoptosis, and Oxidative Stress of Type 2 Diabetic Patients. Med. Kaunas Lith. 2023, 59, 1320. [Google Scholar] [CrossRef]
- Fritzen, A.M.; Thøgersen, F.B.; Thybo, K.; Vissing, C.R.; Krag, T.O.; Ruiz-Ruiz, C.; Risom, L.; Wibrand, F.; Høeg, L.D.; Kiens, B.; et al. Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle. Cells 2019, 8, 237. [Google Scholar] [CrossRef]
- Rivera-Alvarez, I.; Pérez-Treviño, P.; Chapoy-Villanueva, H.; Vela-Guajardo, J.E.; Nieblas, B.; Garza-González, S.; García-Rivas, G.; García, N. A Single Session of Physical Activity Restores the Mitochondrial Organization Disrupted by Obesity in Skeletal Muscle Fibers. Life Sci. 2020, 256, 117965. [Google Scholar] [CrossRef]
- Shockett, P.E.; Khanal, J.; Sitaula, A.; Oglesby, C.; Meachum, W.A.; Castracane, V.D.; Kraemer, R.R. Plasma Cell-Free Mitochondrial DNA Declines in Response to Prolonged Moderate Aerobic Exercise. Physiol. Rep. 2016, 4, e12672. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Grzybowska-Adamowicz, J.; Stawski, R.; Brzezińska, O.; Zmysłowska, A.; Nowak, D. Response of Circulating Free Cellular DNA to Repeated Exercise in Men with Type 1 Diabetes Mellitus. J. Clin. Med. 2024, 13, 5859. [Google Scholar] [CrossRef]
- Stawski, R.; Walczak, K.; Kosielski, P.; Meissner, P.; Budlewski, T.; Padula, G.; Nowak, D. Repeated Bouts of Exhaustive Exercise Increase Circulating Cell Free Nuclear and Mitochondrial DNA without Development of Tolerance in Healthy Men. PLoS ONE 2017, 12, e0178216. [Google Scholar]
- Hummel, E.M.; Hessas, E.; Müller, S.; Beiter, T.; Fisch, M.; Eibl, A.; Wolf, O.T.; Giebel, B.; Platen, P.; Kumsta, R.; et al. Cell-Free DNA Release under Psychosocial and Physical Stress Conditions. Transl. Psychiatry 2018, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Stawski, R.; Perdas, E.; Brzezinska, O.; Kosielski, P.; Galczynski, S.; Budlewski, T.; Padula, G.; Nowak, D. Circulating Cell Free DNA Response to Exhaustive Exercise in Average Trained Men with Type I Diabetes Mellitus. Sci. Rep. 2021, 11, 4639. [Google Scholar] [CrossRef]
- Ohlsson, L.; Hall, A.; Lindahl, H.; Danielsson, R.; Gustafsson, A.; Lavant, E.; Ljunggren, L. Increased Level of Circulating Cell-Free Mitochondrial DNA Due to a Single Bout of Strenuous Physical Exercise. Eur. J. Appl. Physiol. 2020, 120, 897–905. [Google Scholar] [CrossRef]
- Helmig, S.; Frühbeis, C.; Krämer-Albers, E.-M.; Simon, P.; Tug, S. Release of Bulk Cell Free DNA during Physical Exercise Occurs Independent of Extracellular Vesicles. Eur. J. Appl. Physiol. 2015, 115, 2271–2280. [Google Scholar] [CrossRef]
- Beiter, T.; Fragasso, A.; Hudemann, J.; Nieß, A.M.; Simon, P. Short-Term Treadmill Running as a Model for Studying Cell-Free DNA Kinetics In Vivo. Clin. Chem. 2011, 57, 633–636. [Google Scholar] [CrossRef]
- Blatteau, J.-E.; Gaillard, S.; De Maistre, S.; Richard, S.; Louges, P.; Gempp, E.; Druelles, A.; Lehot, H.; Morin, J.; Castagna, O.; et al. Reduction in the Level of Plasma Mitochondrial DNA in Human Diving, Followed by an Increase in the Event of an Accident. Front. Physiol. 2018, 9, 1695. [Google Scholar] [CrossRef]
- Nasi, M.; Cristani, A.; Pinti, M.; Lamberti, I.; Gibellini, L.; De Biasi, S.; Guazzaloca, A.; Trenti, T.; Cossarizza, A. Decreased Circulating mtDNA Levels in Professional Male Volleyball Players. Int. J. Sports Physiol. Perform. 2016, 11, 116–121. [Google Scholar] [CrossRef]
- Ubaida-Mohien, C.; Spendiff, S.; Lyashkov, A.; Moaddel, R.; MacMillan, N.J.; Filion, M.-E.; Morais, J.A.; Taivassalo, T.; Ferrucci, L.; Hepple, R.T. Unbiased Proteomics, Histochemistry, and Mitochondrial DNA Copy Number Reveal Better Mitochondrial Health in Muscle of High-Functioning Octogenarians. eLife 2022, 11, e74335. [Google Scholar] [CrossRef] [PubMed]
- Hagman, M.; Fristrup, B.; Michelin, R.; Krustrup, P.; Asghar, M. Football and Team Handball Training Postpone Cellular Aging in Women. Sci. Rep. 2021, 11, 11733. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Object | Exercise Type/Intensity/Time | Effects on mtDNA | Reference |
|---|---|---|---|
| Endurance Training | |||
| Non-IBS Controls (n = 17); Patients with IBS (n = 26) | Walking, 60/75% of HRmax, 5–10 km/h, 60 min/time, 3 times/week, 12 weeks | Exercise reduces cf-mtDNA in controls but not in patients with IBS | [61] |
| NAFLD mice | Treadmill training, 12 + 2 m/min/2 weeks, 1 h/day, 6 days/week, 8 weeks | Exercise decreases the cytosolic ox-mtDNA in hepatocytes of NAFLD mice | [62] |
| Patients with CKD (n = 99) | Treadmill, elliptical cross-trainer, NuStep cross-trainer, stationary recumbent bicycle, 60–80% VO2max, 30–45 min for 3 times/week for 4 months | Endurance exercise for 4 month increases plasma cf-mtDNA levels in patients with moderate to severe CKD | [63] |
| Healthy workers (n = 169); Healthy workers with exercise (n = 187) | Jogging for 30 min, 5 days a week for 2 weeks | Exercise increases mtDNA in peripheral blood mononuclear cells | [64] |
| Regularly exercising young adults (n = 44); non-exercising controls (n = 44) | The regular-exercising person is defined: a minimum of 300 min per month for more than 6 months | The regular-exercising group has higher leucocyte mtDNA copy numbers than the non-exercising group | [65] |
| Healthy elderly volunteers (3 women and 5 men) | Treadmill, stationary bicycles or outdoor walking, 4–6 sessions/week, 12 week. 30 min at 50–60% VO2max for the first 4 weeks; 40 min at 50–60% VO2max for next 4 weeks; ≥40 min at ~70% VO2max for last 4 weeks | Exercise increases skeletal muscle mtDNA copy number | [66] |
| Healthy young (HY, n = 10); Healthy older (HO, n = 10); COPD (n = 20) | Cycling, 65% VO2max, 3 sessions of 30 min per week, 8 weeks | Exercise increases muscle mtDNA copy number in HY group, but not HO or COPD group | [67] |
| 14 middle-aged participants (7 male/7 female) | Stationary bicycles, treadmill, or walking, 4–6 sessions/week, 30 min at 50–60% VO2max for the first 4 weeks; 40 min at 50–60% VO2max for next 4 weeks; ≥40 min at ~70% VO2max for last 4 weeks | Exercise increases mtDNA content of skeletal muscle mildly without significance | [68] |
| Male ICR mice | Treadmill training, 25 m/min, 1 h/day, 6 days/week, and a 5% incline | Exercise increases mtDNA in most brain regions and soleus | [70] |
| Mice | Treadmill training, 8 m/min, 20 min for the first week; 12 m/min, 30 min for the second week; 4 m/min, 45 min for the third week; 16 m/min, 60 min for the fourth week; 18 m/min, 90 min for the fifth week | Exercise increases mtDNA content in hypothalamus | [71] |
| CAG140 knockin mice | Treadmill training, 8 ± 0.5 m/min for first day; 10 ± 1.5 m/min for 8 weeks; 20 ± 1.5 m/min, 40 min/time, 3 times/week, 12 weeks | Exercise elevates the mtDNA/nDNA ratio in brain | [69] |
| Mice | Voluntary wheel running, 1.8 km/h, 10 km/day, 7 weeks | Exercise enhances mtDNA in endothelial cells | [72] |
| Mice | Exhausting forced swimming, 1 h/day, 7 days | continuous exhaustive forced swimming for 7 days results in mtDNA damage in heart tissue of mice | [75] |
| PolG mutant mice | Voluntary wheel running, 10 months | Exercise has no impact on brain and liver mtDNA copy number | [73] |
| Apolipoprotein E knock-out mice with lower extremity artery disease | Treadmill training, 9 m/min for 3 min, with an increase of 2 m/min every 3 min until 19 m/min, 5 days/week; voluntary wheel running, 7 days/week; forced swimming, 60 min/day, 5 days/week; 4 weeks | Exercise has no obvious effect on hindlimb muscle mtDNA content | [74] |
| Resistance training | |||
| SD rats (untrained, training, pre-training, re-training) | Weight loaded-ladder climbing, The amount of weight load was initially set at 50% of the body weight and gradually increased up to 300%. Each training session consisted of 3 sets of 5 climbing repetition, each rat was trained twice a day every third day for 8 weeks. | mtDNA copy numbers are significantly higher in re-trained muscles compared to the others | [76] |
| Concurrent endurance and resistance training | |||
| Women (n = 47) | Concurrent endurance and resistance training, three 60-min sessions/week, from the 17th gestational week until birth | Exercise increases mtDNA copy number in placentas | [77] |
| Sugar, hypertension, and physical exercise cohorts (n = 105) | Endurance and resistance training for 45 min, 3 times/week, 6 month | Exercise has no significant effect on mtDNA copy number in blood | [78] |
| HIIT | |||
| Healthy volunteer (n = 20); T2DM male patients (n = 30) | Treadmill training, 4 × 4 min intervals at 80–85% of HRmax, with 3-min active recovery at 70% of HRmax between intervals, 40 min/time, 3 times/week, 12 weeks | Exercise enhances mtDNA content of skeletal muscle of T2DM patients | [79] |
| HIIT and moderate-high continuous exercise | |||
| Healthy, sedentary male subjects (n = 10) | One-legged knee-extensor exercise, containing two HIIT and two moderate-high continuous exercise per week, 4 times/week, 6 weeks. | Exercise has no effect on mtDNA in trained leg | [80] |
| Acute exercise | |||
| Male Zucker lean and Zucker obese rats | A single session swimming test, swam freely for first 30 min, and the next 30 min swimming was stimulated with the manual movement of water. | Acute exercise decreases mtDNA levels in gastrocnemius of lean and obese rats | [81] |
| Healthy moderately trained young men (n = 7) | Treadmill training, 60% VO2max, 90 min | Cf-mtDNA is declined when exercised for 54 min and immediately after exercise. | [82] |
| Healthy men (n = 11); T1DM patients (n = 14) | Treadmill run to exhaustion at 70%VO2max at three consecutive days, separated by a 72 h resting period. | Each bout of exhaustive exercise increases cf-mtDNA. | [83] |
| Healthy controls (n = 11); T1DM men (n = 14) | Treadmill training, 1.5% incline, 70%VO2max to exhaustion | The increase in cf-mtDNA concentration is significantly different between groups only in the second bout. | [86] |
| Young, healthy men (n = 20) | Exhaustive treadmill exercise, 15% incline, starting with a 5 min walking period at 1 m/s, increased by 0.2 m/s every 30 s afterwards until subjective exhaustion. | Circulating cf-mtDNA increases with peak levels at 15 min after exercise, and then rapidly drops to baseline levels. | [85] |
| Average-trained men (n = 11) | Three treadmill exercise tests to exhaustion at 70%VO2max separated by 72 h of resting. | Cf-mtDNA rises significantly after the second and third bout of exercise, and decreases during recovery. | [84] |
| Healthy volunteers (n = 8) | Controlled ergo-spirometry cycle test, the resistance began at 30 W and 50 W for female and male, and increased by 10 W/min and 15 W/min respectively until exhaustion. | Cf-mtDNA significantly increases during exercise, compared to baseline values and after 30 and 90 min of rest | [87] |
| Healthy, physically active, non-smoking men (n = 5) | An incremental treadmill exercise test with a starting speed of 6 km/h and increased by 2 km/h every 3 min with 1.5% incline until exhaustion. | There is no difference in mitochondrial cf-DNA before and after exercise | [88] |
| Well-trained male athletes | Incremental treadmill exercise, 1% incline, the speed was increased by 2 km/h every 3 min until exhaustion. | Cf-mtDNA concentrations are not affected by exercise | [89] |
| Specialized sports | |||
| Healthy nonathlete volunteers (n = 20); PVPs (n = 12) | Volleyball, 2 consecutive seasons (from fall to spring) from 2010 to 2012, 15 h per week. | mtDNA levels are lower in plasma of PVPs than in nonathletes, cf-mtDNA is decreased only in the first session, with no variations in the second session. | [91] |
| Non-divers (n = 22); accident-free divers (n = 8) | diving | Accident-free divers have less cf-mtDNA than non-divers. | [90] |
| Non-athlete controls (n = 14); World-class track and field master athletes (n = 15) | Track and field | World-class track and field master athletes have higher mtDNA copy numbers in muscle than non-athletes. | [92] |
| Young untrained controls (n = 30); young elite football players (n = 29) | football | Young elite football players have higher mtDNA copy numbers in lymphocytes and mononuclear cells compared to young untrained controls. | [93] |
| Elderly untrained controls (n = 35); elderly team handball players (n = 35) | Team handball | Elderly team handball players have lower mtDNA copy numbers in lymphocytes compared to elderly untrained controls. | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Fan, J.; Duan, X.; Zhu, X.; Bai, J.; Zhang, T. Mitochondrial DNA in Exercise-Mediated Innate Immune Responses. Int. J. Mol. Sci. 2025, 26, 3069. https://doi.org/10.3390/ijms26073069
Wen X, Fan J, Duan X, Zhu X, Bai J, Zhang T. Mitochondrial DNA in Exercise-Mediated Innate Immune Responses. International Journal of Molecular Sciences. 2025; 26(7):3069. https://doi.org/10.3390/ijms26073069
Chicago/Turabian StyleWen, Xin, Jingcheng Fan, Xuemei Duan, Xinyi Zhu, Jianzheng Bai, and Tan Zhang. 2025. "Mitochondrial DNA in Exercise-Mediated Innate Immune Responses" International Journal of Molecular Sciences 26, no. 7: 3069. https://doi.org/10.3390/ijms26073069
APA StyleWen, X., Fan, J., Duan, X., Zhu, X., Bai, J., & Zhang, T. (2025). Mitochondrial DNA in Exercise-Mediated Innate Immune Responses. International Journal of Molecular Sciences, 26(7), 3069. https://doi.org/10.3390/ijms26073069