
Alterations in RNA Expression Profile Following S. aureus and S. epidermidis Inoculation into Platelet Concentrates

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
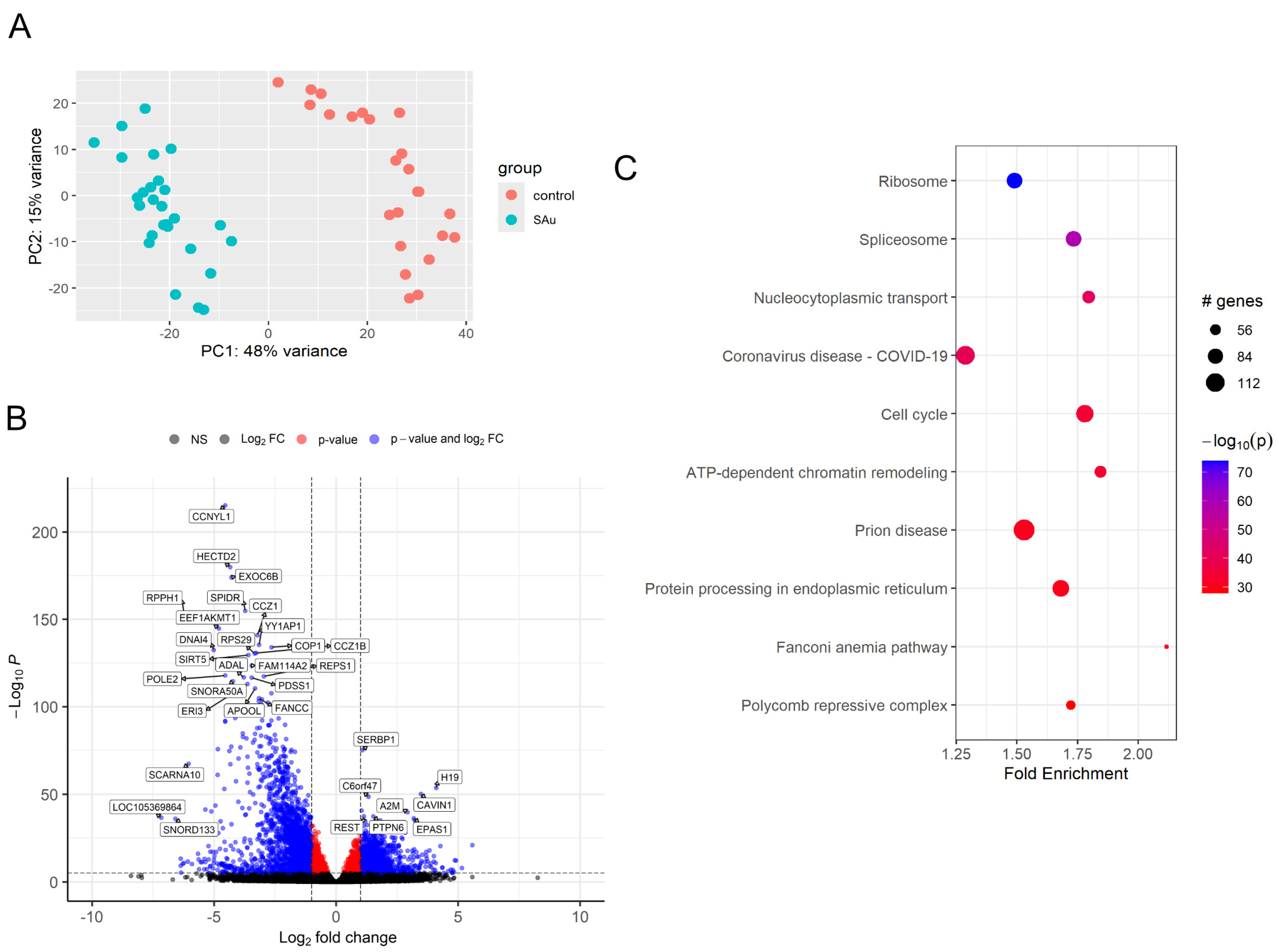
2.1. Quality Control Metrics
2.2. Differentially Expressed Gene (DEG) of Samples After Inoculation of S. aureus
2.3. Comprehensive Gene Enrichment for Pathway Analysis of Samples After Inoculation of S. aureus
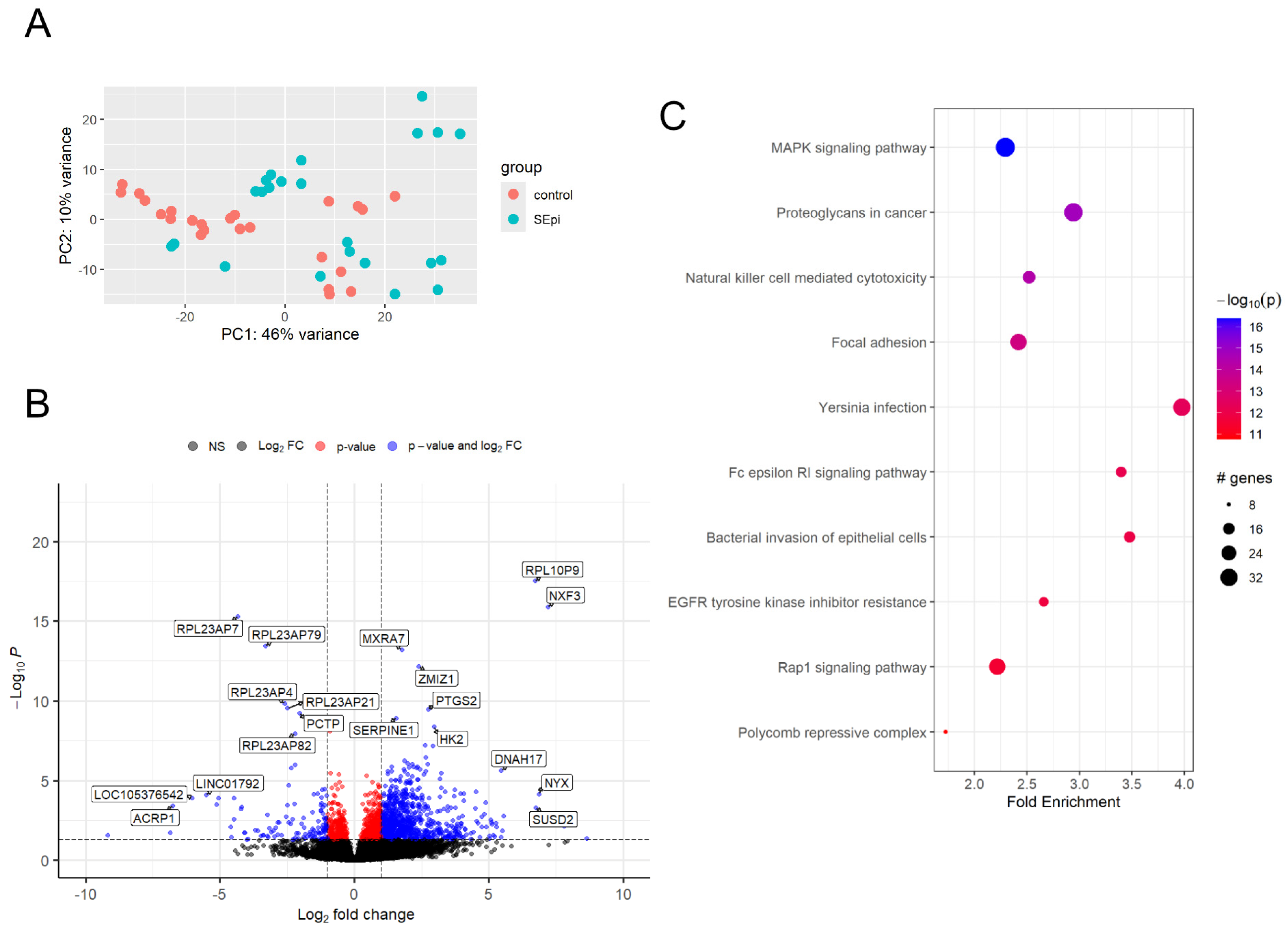
2.4. Differentially Expressed Gene (DEG) of Samples After Inoculation of S. epidermidis
2.5. Comprehensive Gene Set Enrichment for Pathway Analysis of Samples After Inoculation of S. epidermidis
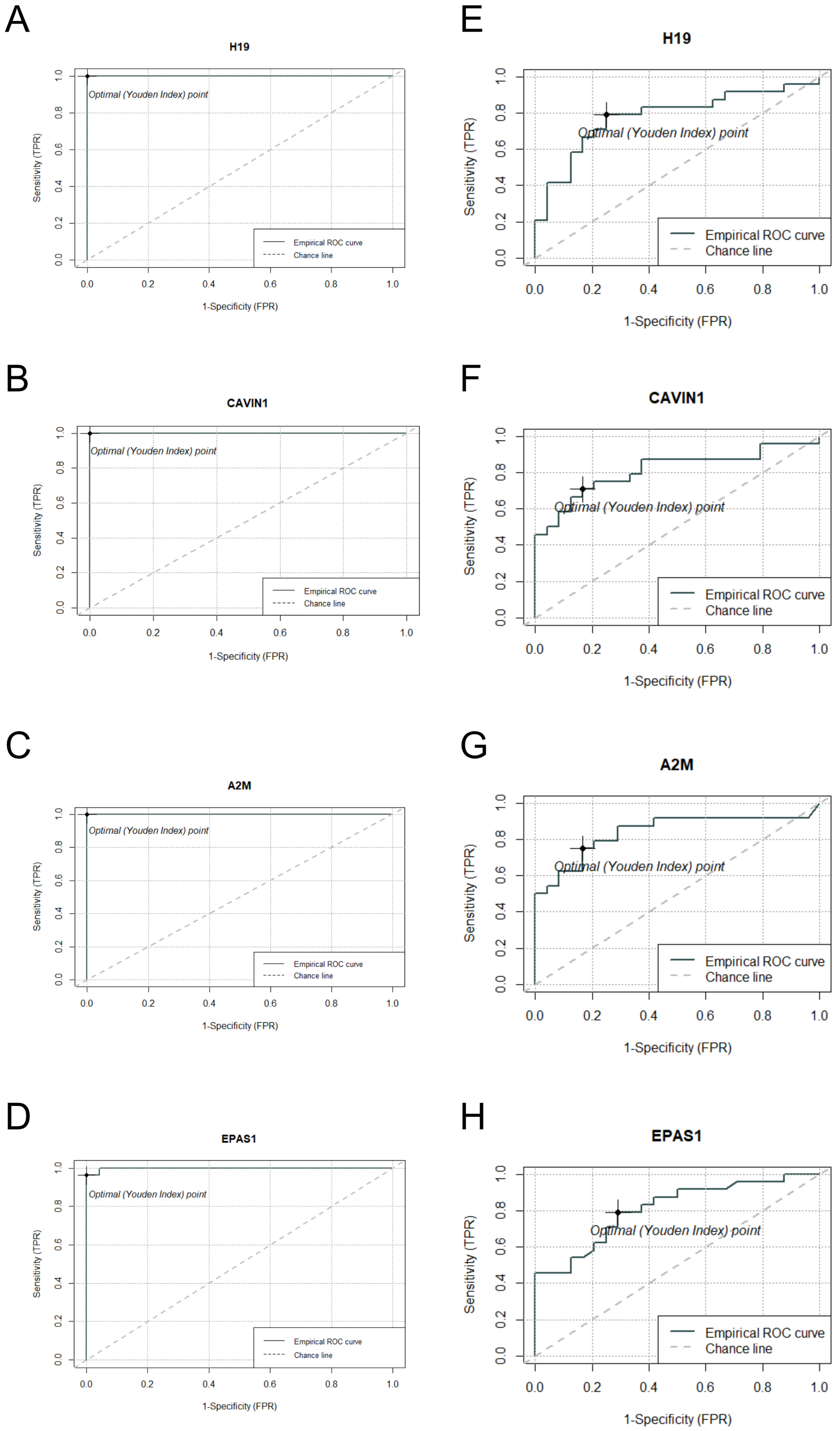
2.6. DEG Analysis and ROC Curve Assessment of Commonly Expressed Genes in S. aureus and S. epidermidis Inoculated Samples
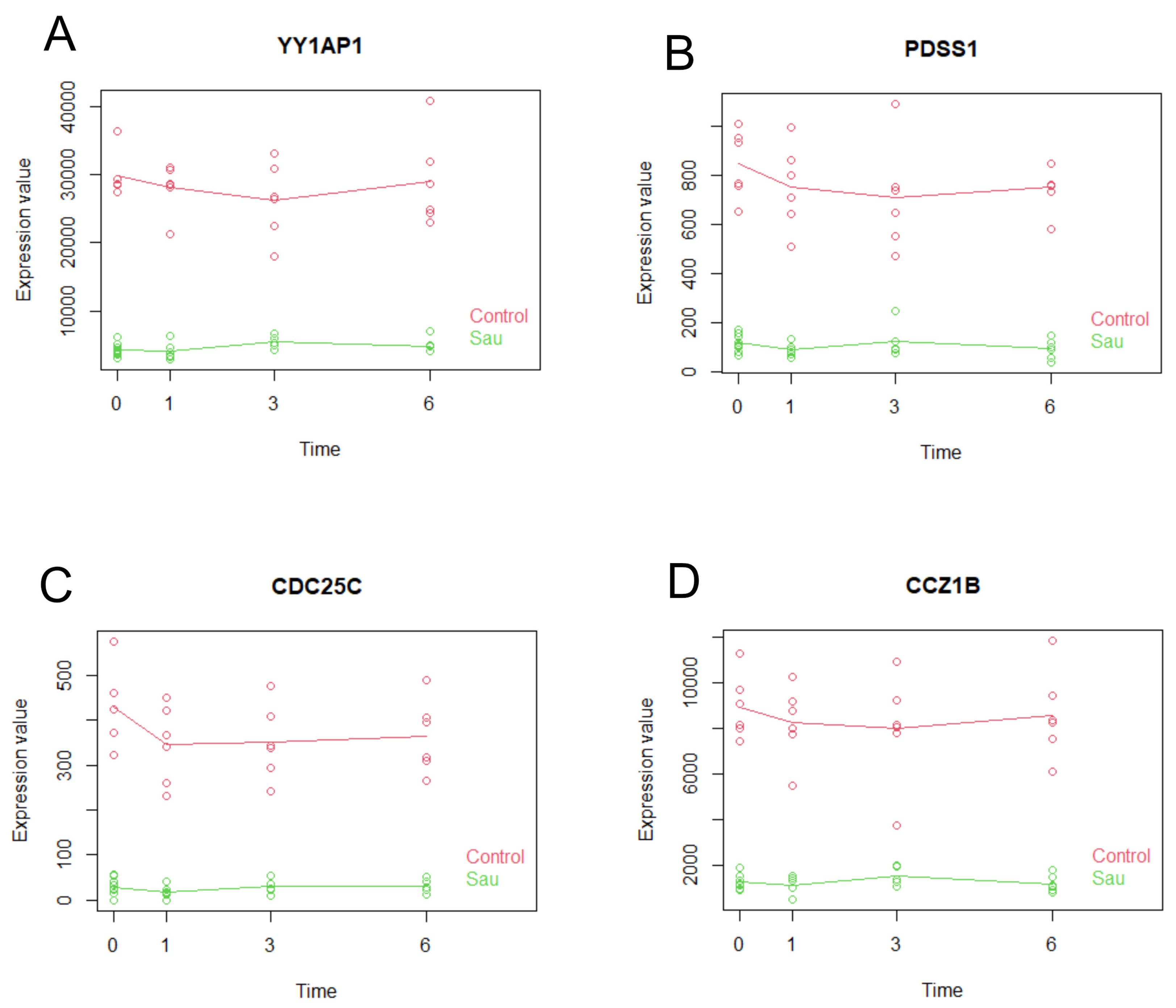
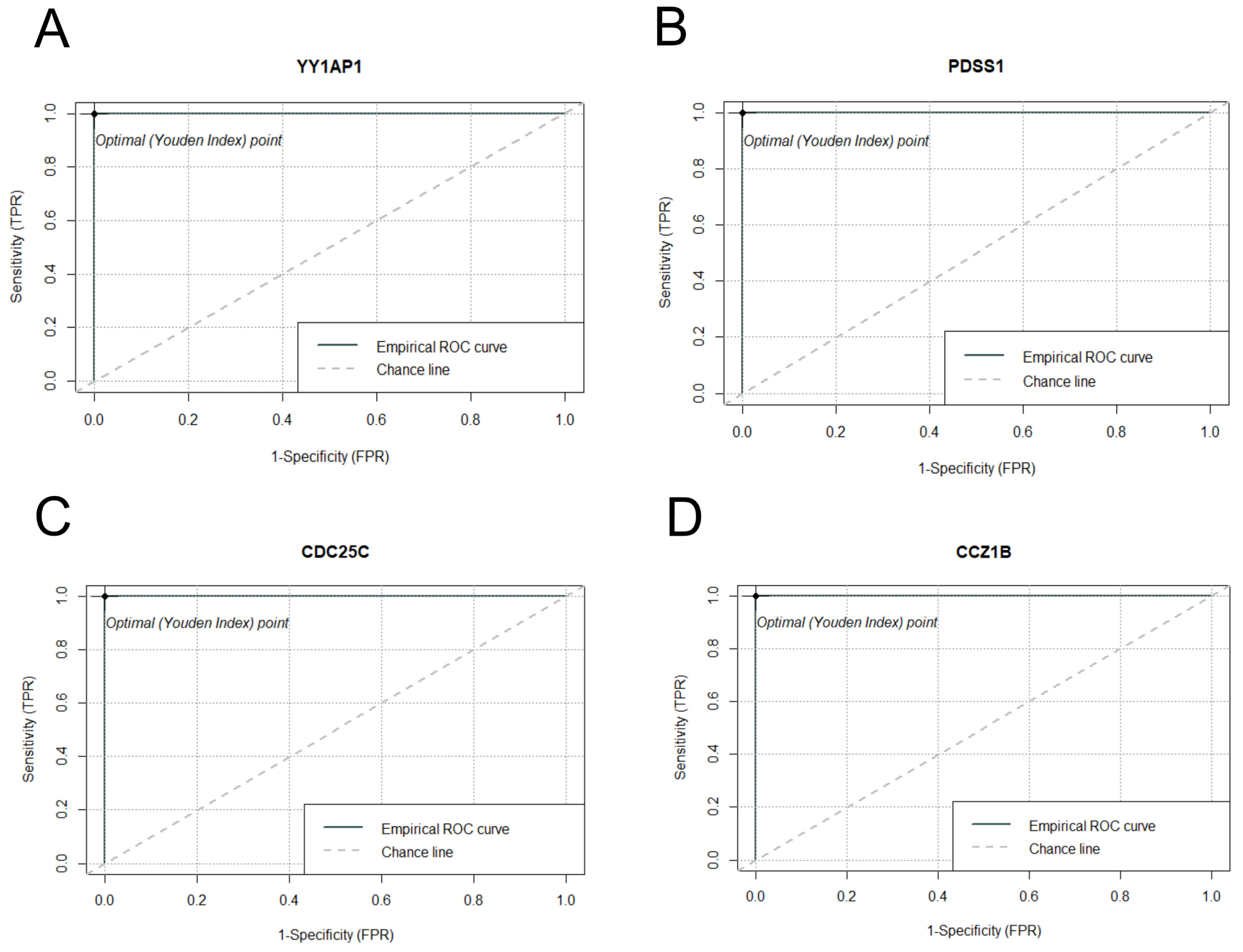
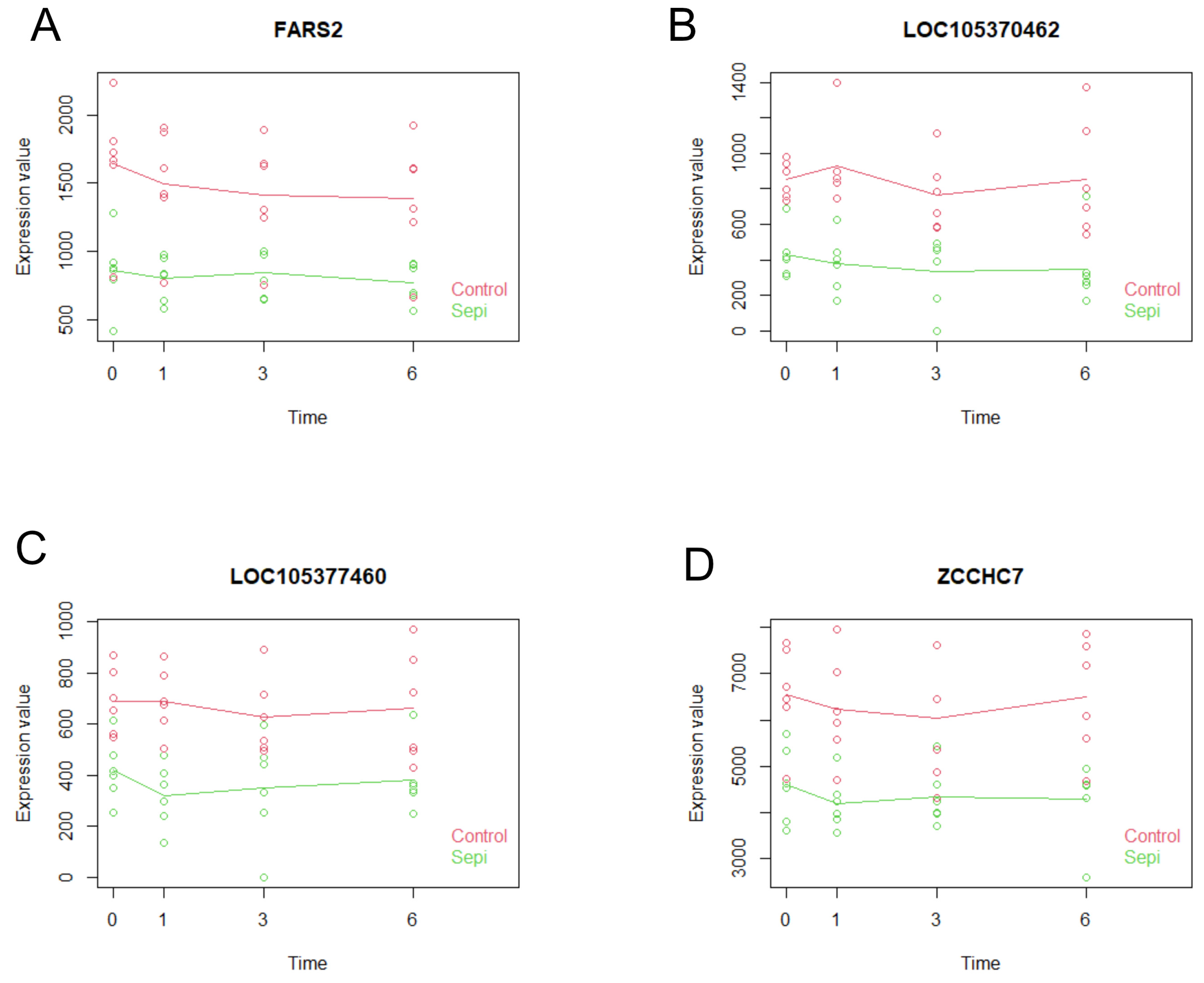
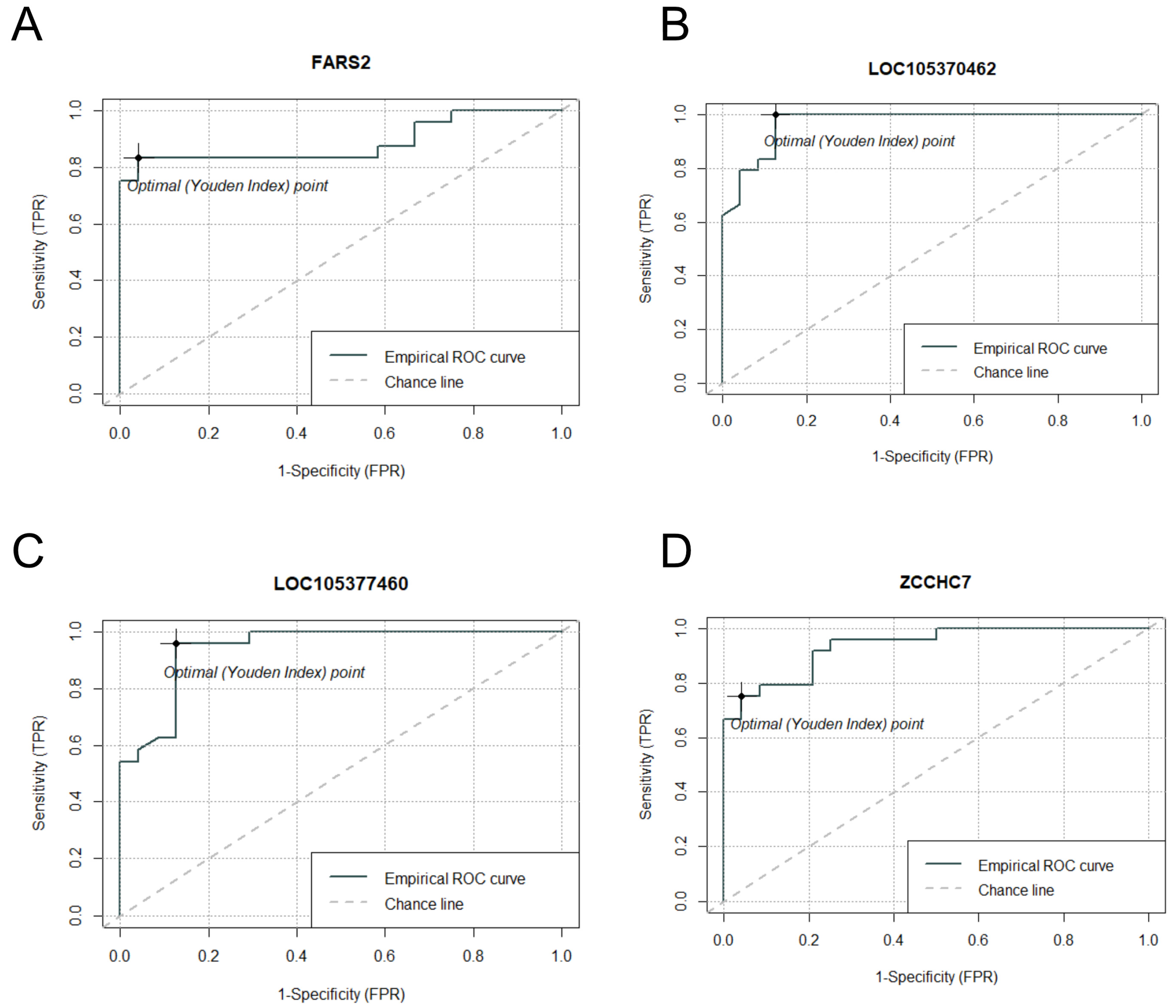
2.7. Multiseries Time-Course Analysis of Gene Expression Patterns and ROC Curve Generation for Curated Genes in S. aureus and S. epidermidis Inoculated Specimens
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. RNA Preparation, Library Construction, and Sequencing
4.3. Data Quality Control and Read Mapping
4.4. Differential Gene Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scridon, A. Platelets and Their Role in Hemostasis and Thrombosis-From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 2772. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, N.M. Does ABO and RhD matching matter for platelet transfusion? Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 512–517. [Google Scholar] [CrossRef]
- Shea, S.M.; Thomas, K.A.; Spinella, P.C. The effect of platelet storage temperature on haemostatic, immune, and endothelial function: Potential for personalised medicine. Blood Transfus. 2019, 17, 321–330. [Google Scholar] [CrossRef]
- Smethurst, P.; Cardigan, R. Current challenges in platelet transfusion. Platelets 2022, 33, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Neal, M.D.; Herman, J.H. Bacterial contamination of platelets for transfusion: Strategies for prevention. Crit. Care 2018, 22, 271. [Google Scholar] [CrossRef] [PubMed]
- Prax, M.; Bekeredjian-Ding, I.; Krut, O. Microbiological Screening of Platelet Concentrates in Europe. Transfus. Med. Hemother. 2019, 46, 76–86. [Google Scholar] [CrossRef]
- Kerantzas, C.A.; Merwede, J.; Snyder, E.L.; Hendrickson, J.E.; Tormey, C.A.; Kazmierczak, B.I.; Peaper, D.R. Assessment of polymicrobial interactions in bacterial isolates from transfused platelet units associated with sepsis. Transfusion 2022, 62, 2458–2463. [Google Scholar] [CrossRef]
- Fridey, J.L.; Stramer, S.L.; Nambiar, A.; Moayeri, M.; Bakkour, S.; Langelier, C.; Crawford, E.; Lu, T.; Lanteri, M.C.; Kamm, J.; et al. Sepsis from an apheresis platelet contaminated with Acinetobacter calcoaceticus/baumannii complex bacteria and Staphylococcus saprophyticus after pathogen reduction. Transfusion 2020, 60, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; De Korte, D. Isbt Transfusion-Transmitted Infectious Diseases Working Party SoB. Residual risks of bacterial contamination for pathogen-reduced platelet components. Vox Sang. 2022, 117, 879–886. [Google Scholar] [CrossRef]
- Abela, M.A.; Fenning, S.; Maguire, K.A.; Morris, K.G. Bacterial contamination of platelet components not detected by BacT/ALERT®. Transfus. Med. 2018, 28, 65–70. [Google Scholar] [CrossRef]
- White, S.K.; Schmidt, R.L.; Walker, B.S.; Metcalf, R.A. Bacterial contamination rate of platelet components by primary culture: A systematic review and meta-analysis. Transfusion 2020, 60, 986–996. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Allen, J.; Brailsford, S.; Roy, A.; Ball, J.; Moule, R.; Vasconcelos, M.; Morrison, R.; Pitt, T. Bacterial screening of platelet components by National Health Service Blood and Transplant, an effective risk reduction measure. Transfusion 2017, 57, 1122–1131. [Google Scholar] [CrossRef]
- Jacobs, M.R.; Zhou, B.; Tayal, A.; Maitta, R.W. Bacterial Contamination of Platelet Products. Microorganisms 2024, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; Savelkoul, P.H.; Pietersz, R.N.; Reesink, H.W. Applications of real-time PCR in the screening of platelet concentrates for bacterial contamination. Expert. Rev. Mol. Diagn. 2006, 6, 865–872. [Google Scholar] [CrossRef]
- El Mehdaoui, F.; Benajiba, M.; Boulahdid, S.; El Hattimy, F.; Soulaymani, A.; Alami, R. Skin flora and bacterial contamination of diversion pouch and recovered platelet components in Moroccan blood donors. Transfus. Med. 2020, 30, 384–390. [Google Scholar] [CrossRef]
- Szczepiorkowski, Z.M.; Pagano, M.B. Platelet components and bacterial contamination: Hospital perspective 2022. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 430–436. [Google Scholar] [CrossRef]
- Gammon, R.R.; Reik, R.A.; Stern, M.; Vassallo, R.R.; Waxman, D.A.; Young, P.P.; Benjamin, R.J. Acquired platelet storage container leaks and contamination with environmental bacteria: A preventable cause of bacterial sepsis. Transfusion 2022, 62, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Ramirez-Arcos, S.; Stiller, L.; McDonald, C. Isbt Transfusion-Transmitted Infectious Diseases Working Party SoB. Current status of rapid bacterial detection methods for platelet components: A 20-year review by the ISBT Transfusion-Transmitted Infectious Diseases Working Party Subgroup on Bacteria. Vox Sang. 2022, 117, 983–988. [Google Scholar] [CrossRef]
- AABB. Clinical Recognition and Investigation of Suspected Bacterial Contamination of Platelets. Available online: http://www.aabb.org/programs/publications/bulletins/Documents/ab14-04.pdf (accessed on 24 May 2024).
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.; Chhugani, K.; Chang, Y.; Karlsberg, A.; Loeffler, C.; Zhang, J.; Muszyńska, A.; Munteanu, V.; Yang, H.; Rotman, J.; et al. RNA-seq data science: From raw data to effective interpretation. Front. Genet. 2023, 14, 997383. [Google Scholar] [CrossRef]
- Best, M.G.; In’t Veld, S.; Sol, N.; Wurdinger, T. RNA sequencing and swarm intelligence-enhanced classification algorithm development for blood-based disease diagnostics using spliced blood platelet RNA. Nat. Protoc. 2019, 14, 1206–1234. [Google Scholar] [CrossRef]
- Heililahong, H.; Jin, P.; Lei, H.; Gu, H.; Qian, B.; Wang, X.; Dai, J.; Cai, X. Whole transcriptome analysis of platelet concentrates during storage. Blood Transfus. 2023, 21, 146–156. [Google Scholar] [CrossRef]
- Supernat, A.; Popeda, M.; Pastuszak, K.; Best, M.G.; Grešner, P.; Veld, S.I.T.; Siek, B.; Bednarz-Knoll, N.; Rondina, M.T.; Stokowy, T.; et al. Transcriptomic landscape of blood platelets in healthy donors. Sci. Rep. 2021, 11, 15679. [Google Scholar] [CrossRef] [PubMed]
- Nuhrenberg, T.G.; Stockle, J.; Marini, F.; Zurek, M.; Grüning, B.A.; Benes, V.; Hein, L.; Neumann, F.J.; Stratz, C.; Cederqvist, M.; et al. Impact of high platelet turnover on the platelet transcriptome: Results from platelet RNA-sequencing in patients with sepsis. PLoS ONE 2022, 17, e0260222. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.M.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef]
- Kim, J.T.; Kim, Y.; Kim, J.Y.; Lee, S.; Kim, M.; Jekarl, D.W. Regulatory Mechanism between Ferritin and Mitochondrial Reactive Oxygen Species in Spinal Ligament-Derived Cells from Ossification of Posterior Longitudinal Ligament Patient. Int. J. Mol. Sci. 2023, 24, 2872. [Google Scholar] [CrossRef]
- Wang, B.; Suen, C.W.; Ma, H.; Wang, Y.; Kong, L.; Qin, D.; Lee, Y.W.W.; Li, G. The Roles of H19 in Regulating Inflammation and Aging. Front. Immunol. 2020, 11, 579687. [Google Scholar] [CrossRef]
- Vandooren, J.; Itoh, Y. Alpha-2-Macroglobulin in Inflammation, Immunity and Infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef]
- Kelly, B.J.; Lautenbach, E.; Nachamkin, I.; Coffin, S.E.; Gerber, J.S.; Fuchs, B.D.; Garrigan, C.; Han, X.; Bilker, W.B.; Wise, J.; et al. Centers for Disease Control and Prevention (CDC) Prevention Epicenters Program. Combined biomarkers discriminate a low likelihood of bacterial infection among surgical intensive care unit patients with suspected sepsis. Diagn. Microbiol. Infect. Dis. 2016, 85, 109–115. [Google Scholar] [CrossRef]
- Schweizer, R.M.; Velotta, J.P.; Ivy, C.M.; Jones, M.R.; Muir, S.M.; Bradburd, G.S.; Storz, J.F.; Scott, G.R.; Cheviron, Z.A. Physiological and genomic evidence that selection on the transcription factor Epas1 has altered cardiovascular function in high-altitude deer mice. PLoS Genet. 2019, 15, e1008420. [Google Scholar] [CrossRef]
- Djumagulov, M.; Demeshkina, N.; Jenner, L.; Rozov, A.; Yusupov, M.; Yusupova, G. Accuracy mechanism of eukaryotic ribosome translocation. Nature 2021, 600, 543–546. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, Y.; Yu, X.Y.; Pan, X.; Zhang, Y.; Tu, J.; Song, Y.H.; Li, Y. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Turi, Z.; Lacey, M.; Mistrik, M.; Moudry, P. Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 2019, 11, 2512–2540. [Google Scholar] [CrossRef]
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstadt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Elemam, N.M.; Ramakrishnan, R.K.; Hundt, J.E.; Halwani, R.; Maghazachi, A.A.; Hamid, Q. Innate Lymphoid Cells and Natural Killer Cells in Bacterial Infections: Function, Dysregulation, and Therapeutic Targets. Front. Cell Infect. Microbiol. 2021, 11, 733564. [Google Scholar] [CrossRef]
- Schmidt, S.; Ullrich, E.; Bochennek, K.; Zimmermann, S.Y.; Lehrnbecher, T. Role of natural killer cells in antibacterial immunity. Expert. Rev. Hematol. 2016, 9, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, G.R.; Niesel, D.W.; Klimpel, K.D. Natural cytotoxic effector cell activity against Shigella flexneri-infected HeLa cells. J. Immunol. 1986, 136, 1081–1086. [Google Scholar]
- Kim, K.S.; Kang, T.; Jekarl, D.W. Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Biomedicines 2024, 12, 628. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Up-regulated genes at S. aureus inoculation | |
| 1 h | EPAS1, CDKN1C, ZNF761, TYMP, CAVIN1, NADK |
| 3 h | TYMP, H19, CAVIN1, URB2, IGF2, A2M |
| 6 h | CAVIN1, IGF2, A2M, ARHGAP29, EPAS1, H19 |
| Down-regulated genes at S. aureus inoculation | |
| 1 h | CCNYL1, EEF1AKMT1, DNAI4, HECTD2, RPPH1, SPIDR |
| 3 h | CCNYL1, RPS29, EXOC6B, EEF1AKMT1, SNORD133, SPIDR |
| 6 h | EEF1AKMT1, CCNYL1, HECTD2, EXOC6B, ADAL, AGAP4 |
| Up-regulated genes at S. epidermidis inoculation | |
| 1 h | SERPINE1, BPI, IL1R1, PTGS2, RPL10P9, DNAH17 |
| 3 h | SLC34A3, RPL10P9 |
| 6 h | LMO3, RPL10P9, NXF3, PTGS2, MXRA7 |
| Down-regulated genes at S. epidermidis inoculation | |
| 1 h | HNRNPUL2-BSCL2, LOC105370464, BIVM-ERCC5, RPL23AP7, RPL23AP79, LINC00964 |
| 3 h | TMX2-CTNND1, TRIM6-TRIM34, LOC101927613 |
| 6 h | PRSS1, RPL23AP7, RPL23AP79 |
| Gene Name | log2FC.Sau | padj.Sau | AUC | log2FC.Sepi | padj.Sepi | AUC |
|---|---|---|---|---|---|---|
| H19 | 4.104081 | 2.38 × 10−54 | 1 | 1.389704 | 0.004315 | 0.7795 |
| CAVIN1 | 3.47354 | 6.56 × 10−51 | 1 | 1.477886 | 0.000469 | 0.8125 |
| A2M | 2.925464 | 1.82 × 10−40 | 1 | 1.178996 | 0.008684 | 0.8385 |
| EPAS1 | 3.189499 | 4.96 × 10−37 | 0.9985 | 1.233558 | 0.000808 | 0.8090 |
| LOC105376872 | −4.28848 | 1.36 × 10−33 | 1 | −1.04865 | 0.010769 | 0.8359 |
| SNORA53 | −4.63863 | 2.32 × 10−31 | 0.9598 | 1.510768 | 0.010982 | 0.6354 |
| PPM1F | 1.802254 | 1.14 × 10−26 | 0.9869 | 1.100995 | 0.008567 | 0.7118 |
| CDKN1C | 2.279073 | 3.42 × 10−26 | 0.9830 | 1.157439 | 0.001252 | 0.8056 |
| HK3 | 2.435473 | 5.62 × 10−26 | 0.9923 | 1.328566 | 0.002777 | 0.7682 |
| RXRA | 2.338701 | 6.99 × 10−26 | 0.9799 | 1.5613 | 0.0002 | 0.7448 |
| p-Value | R-Squared | AUC | ciAUC | Cutoff | |
|---|---|---|---|---|---|
| YY1AP1 | 1.45 × 10−29 | 0.9274 | 1 | 1–1 | 17,985 |
| PDSS1 | 8.69 × 10−26 | 0.8965 | 1 | 1–1 | 470 |
| CDC25C | 1.12 × 10−25 | 0.8955 | 1 | 1–1 | 231 |
| CCZ1B | 1.29 × 10−25 | 0.8949 | 1 | 1–1 | 4065 |
| p-Value | R-Squared | AUC | ciAUC | Cutoff | |
|---|---|---|---|---|---|
| FARS2 | 4.67 × 10−9 | 0.5294 | 0.8854 | 0.7875–0.9833 | 1219 |
| LOC105370462 | 9.83 × 10−11 | 0.6011 | 0.9696 | 0.9191–1 | 544 |
| LOC105377460 | 1.01 × 10−8 | 0.5137 | 0.9418 | 0.8718–1 | 494 |
| ZCCHC7 | 5.09 × 10−9 | 0.5277 | 0.9358 | 0.8622–1 | 5575 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.K.; Kang, T.; Kweon, Y.; Yoo, I.Y.; Oh, E.-J.; Park, Y.-J.; Kim, Y.; Kim, H.S.; Jekarl, D.W. Alterations in RNA Expression Profile Following S. aureus and S. epidermidis Inoculation into Platelet Concentrates. Int. J. Mol. Sci. 2025, 26, 3009. https://doi.org/10.3390/ijms26073009
Kim JK, Kang T, Kweon Y, Yoo IY, Oh E-J, Park Y-J, Kim Y, Kim HS, Jekarl DW. Alterations in RNA Expression Profile Following S. aureus and S. epidermidis Inoculation into Platelet Concentrates. International Journal of Molecular Sciences. 2025; 26(7):3009. https://doi.org/10.3390/ijms26073009
Chicago/Turabian StyleKim, Jae Kwon, Taewon Kang, Youngeun Kweon, In Young Yoo, Eun-Jee Oh, Yeon-Joon Park, Yonggoo Kim, Hoon Seok Kim, and Dong Wook Jekarl. 2025. "Alterations in RNA Expression Profile Following S. aureus and S. epidermidis Inoculation into Platelet Concentrates" International Journal of Molecular Sciences 26, no. 7: 3009. https://doi.org/10.3390/ijms26073009
APA StyleKim, J. K., Kang, T., Kweon, Y., Yoo, I. Y., Oh, E.-J., Park, Y.-J., Kim, Y., Kim, H. S., & Jekarl, D. W. (2025). Alterations in RNA Expression Profile Following S. aureus and S. epidermidis Inoculation into Platelet Concentrates. International Journal of Molecular Sciences, 26(7), 3009. https://doi.org/10.3390/ijms26073009