1. Introduction
Epithelial ovarian cancer is a disease with a poor prognosis, often detected at an advanced stage due to the lack of early symptoms and adequate screening tests, with high recurrence and mortality rates. According to 2020 statistics, ovarian cancer kills more than 200,000 people a year worldwide [
1], and epithelial ovarian cancer is the leading cause of death from gynecologic cancer in developed countries [
2]. Epithelial ovarian cancer is classified into low-grade serous carcinoma, high-grade serous carcinoma, mucinous carcinoma, endometrioid carcinoma, clear cell carcinoma, malignant Brenner tumor, mesonephric-like adenocarcinoma, undifferentiated and dedifferentiated carcinoma, carcinosarcoma, and mixed carcinoma according to the WHO classification, of which high-grade serous carcinoma is the most common pathologic type. The standard treatment for newly diagnosed advanced epithelial ovarian cancer is cytoreductive surgery followed by platinum-taxane combination chemotherapy with/without Bevacizumab (anti-VEGF monoclonal antibody), but despite this treatment, the disease has a high recurrence rate and a high mortality rate.
BRCA1 and BRCA2 play an important role in the repair of double-stranded DNA (dsDNA) breaks through homologous recombination, and women with
BRCA1 or
BRCA2 gene mutations are predisposed to cancers such as breast and ovarian cancer. The term ‘BRCAness’ refers to the phenotypic traits with a homologous recombination deficiency (HRD) similar to cells carrying
BRCA1 or
BRCA2 mutations. Poly(ADP-ribose) polymerase (PARP) is an enzyme involved in base excision repair, a key pathway in the repair of single-strand DNA (ssDNA) breaks. PARP inhibitors are agents that inhibit the PARP enzyme and have a pharmacologic mechanism of action that induces cell death through synthetic lethality in cancer cells with HRD such as
BRCA mutations. Synthetic lethality is a phenomenon where a single genetic event is tolerable for cell survival, but the simultaneous occurrence of multiple genetic events leads to cell death [
3]. PARP inhibitors are the first clinically approved drugs designed to exploit synthetic lethality. When the repair of ssDNA breaks is inhibited by PARP inhibitors, ssDNA breaks lead to the occurrence of dsDNA breaks. In the case of cancer cells with HRD, dsDNA break repair involves error-prone DNA repair mechanisms such as Non-Homologous End Joining. Consequently, the accumulation of numerous mutations induces cell death through synthetic lethality.
The genetic concept of synthetic lethality has now been clinically validated through the demonstrated efficacy of PARP inhibitors for the treatment of cancers in patients with HRD. PARP inhibitors, such as olaparib, niraparib, rucaparib, veliparib, and fuzuloparib, have shown encouraging results in the treatment of epithelial ovarian cancer (high-grade serous or endometrioid adenocarcinoma) in phase III clinical trials in recent years [
4,
5,
6,
7,
8,
9,
10]. However, the survival benefit of these PARP inhibitors is primarily seen in patients with HRD, including
BRCA mutations. Although there is some variation across studies and agents, there is no or small survival benefit in patients without HRD compared to patients with HRD.
Approximately 15% of non-mucinous epithelial ovarian cancer patients carry germline mutations in
BRCA1 or
BRCA2, and approximately 5–7% of ovarian cancer patients have somatic
BRCA mutations. This means that approximately 20–25% of patients with epithelial ovarian cancer based on
BRCA mutation may be candidates for PARP inhibitors [
11]. One study revealed that 31 percent of ovarian carcinomas, including serous, endometrioid, clear cell, and carcinosarcoma histologic types, harbored mutations in one or more of the 13 homologous recombination genes [
12]. When considering epithelial ovarian cancer patients with HRD as candidates for PARP inhibitor application, approximately 30% would qualify as eligible recipients. This, in turn, implies that in around 70% of patients, the PARP inhibitor may not be effective or may result in unsatisfactory outcomes. If there is a way to overcome PARP inhibitor resistance in ovarian cancer patients without HRD, it could potentially extend the benefits of PARP inhibitors to a larger number of patients.
Trabectedin (Yondelis), formerly known as ET-743 during its development, is an antitumor agent initially discovered in the Caribbean marine tunicate, Ecteinascidia turbinata, and is now produced synthetically. Trabectedin is currently used for the treatment of patients with soft tissue sarcomas. Trabectedin is known for its unique mechanism of action, binding to the DNA minor groove to disrupt genetic transcription and inhibit cell division. Additionally, it interferes with the nucleotide-excision repair (NER) system, inducing single-strand DNA breaks. Several preclinical studies have demonstrated a synergistic effect when combining PARP inhibitors and trabectedin in sarcomas [
13,
14,
15] or breast cancer [
16]. In a study involving breast cancer cell lines, synergistic effects were observed, regardless of the presence of
BRCA mutations [
16]. These suggest the potential of trabectedin combination therapy as a promising approach to overcome PARP inhibitor resistance in ovarian cancer without
BRCA mutations.
Based on this background, we conducted this study on the synergic effects of the combination therapy of niraparib and trabectedin using epithelial ovarian cancer cell lines without BRCA mutations. The researchers hypothesized that induction of BRCAness is crucial to overcoming PARP inhibitor resistance. Therefore, the main focus of this study was to investigate whether the combination therapy of niraparib and trabectedin could induce BRCAness in ovarian cancer cell lines with wild-type BRCA genes. Additionally, the study aimed to explore the impact on ssDNA repair enzymes such as PARP1 and PARP2 and examine whether it could induce synthetic lethality.
3. Discussion
Epithelial ovarian cancer is diagnosed in 70–80% of patients at advanced stages (Stage 3, 4), and even after undergoing debulking surgery and platinum-taxane combination chemotherapy, it continues to exhibit a poor prognosis with high rates of recurrence and mortality. In reviewing the results of phase III clinical trials reported in recent years, one of the most noteworthy advancements in the treatment of epithelial ovarian cancer is the emergence of PARP inhibitors. Olaparib demonstrated an improvement in progression-free survival in patients with platinum-sensitive, relapsed ovarian cancer and
BRCA1/2 mutations [
17]. Olaparib showed a progression-free survival benefit in maintenance therapy even after primary treatment in newly diagnosed ovarian cancer patients. Through a 7-year follow-up, these improvements in progression-free survival were demonstrated to lead to an overall survival improvement [
4,
18]. Successful phase III clinical trial results have been sequentially reported for other PARP inhibitors such as niraparib, rucaparib, veliparib, and fuzuloparib [
6,
8,
9,
10,
19]. The efficacy of the combination therapy of olaparib and bevacizumab is also noteworthy [
20].
The PARP enzyme plays a crucial role in repairing single-strand DNA breaks, and PARP inhibitors impede this repair process by suppressing the enzyme’s activity. In cases of Homologous Recombination Deficiency (HRD), such as those with
BRCA mutations, there is a difficulty in repairing double-strand DNA breaks through homologous recombination. Consequently, DNA repair predominantly relies on the single-strand DNA repair system using PARP enzyme. Administering PARP inhibitors to patients with HRD results in engaging in error-prone double-strand DNA repair mechanisms like Non-Homologous End Joining (NHEJ). This error-prone DNA repair leads to the accumulation of mutations, inducing synthetic lethality in cancer cells [
21,
22].
Considering such an action mechanism, theoretically, PARP inhibitors are expected to be effective when Homologous Recombination Deficiency (HRD) is present. However, clinical research results suggest that effectiveness may be observed irrespective of the HRD status. Niraparib, rucaparib, and fuzuloparib demonstrated DFS benefits regardless of the presence of HRD or
BRCA mutations [
6,
10,
19]. However, upon a more detailed examination of those research findings, the efficacy of PARP inhibitors is less evident in patients without
BRCA mutations or HRD, compared to those with
BRCA mutations or Homologous Recombination Deficiency (HRD). The likely reason for these results may be attributed to the imperfect nature of current HRD tests, raising the possibility of HRD even in cases where the test indicates HRD negativity.
In 20–25% of patients with non-mucinous epithelial ovarian cancer, there are germline or somatic
BRCA mutations [
11], and approximately 30% of patients have one or more mutations in homologous recombination genes [
12]. When considering the patient group expected to respond to PARP inhibitors with the presence of HRD, it is estimated that approximately 70% of epithelial ovarian cancer patients have PARP inhibitor resistance. Expanding the application scope of PARP inhibitors to patients without HRD could be a strategy that contributes to the improved therapeutic outcomes in ovarian cancer. As one of such strategies, we hypothesized that if there were drugs capable of inducing BRCAness in ovarian cancer cells without HRD, combining them with PARP inhibitors could potentially induce synthetic lethality.
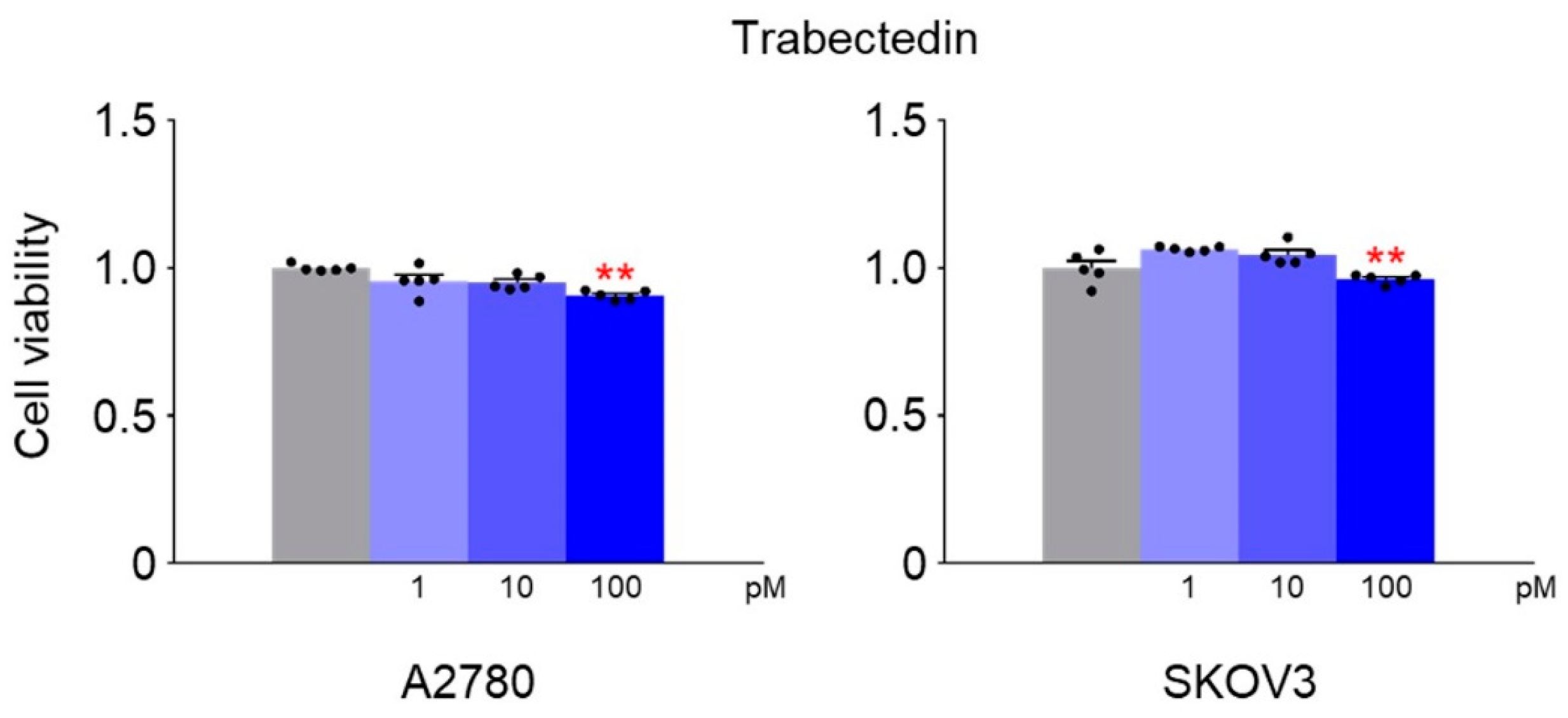
Trabectedin (Yondelis
®, ecteinascidin-743, ET-743) is a natural marine compound with antitumor activity approved for the treatment of unresectable or metastatic soft tissue sarcoma, specifically liposarcoma or leiomyosarcoma. It is used in combination with doxorubicin for patients with uterine leiomyosarcoma [
23]. The structure of trabectedin results in two main effects: the formation of a covalent bond with DNA at the N2-guanine of the minor groove, allowing trabectedin to interact with DNA, and the ability to protrude from the DNA helix, making it accessible to bind with DNA-binding molecules, such as transcriptional factors and DNA repair proteins. Another significant effect of the linkage between trabectedin and the DNA groove is that a segment of the helix becomes recognizable by the nucleotide excision repair (NER) system, resulting in the accumulation of DNA–trabectedin–protein repair complexes. As a result, the formation of DSBs, cell cycle arrest in G2-M phase, and induction of p53-independent apoptosis are primary effects of this phenomenon. Previous in vitro investigations have indicated that trabectedin effectiveness is influenced by the status of both NER and HR DNA repair pathways [
24,
25,
26,
27]. Trabectedin has been observed to be more effective against cells with HR deficiency compared to normal cells [
28]. Although trabectedin’s action mechanism has not been fully elucidated, it is known not to directly cause dsDNA breaks [
29].
Combined therapy with PARP inhibitors and trabectedin has been reported to demonstrate a synergistic effect in several preclinical studies conducted on sarcoma and breast cancer models [
13,
14,
15,
16]. In a study using breast cancer cell lines [
16], the combination of trabectedin and olaparib induces an artificial synthetic lethality effect, regardless of
BRCA1 status. The study showed that the combination treatment was associated with a strong accumulation of double-stranded DNA breaks, G2/M arrest, and apoptotic cell death. However, synergistic effects were not observed when trabectedin was combined with veliparib or iniparib [
16].
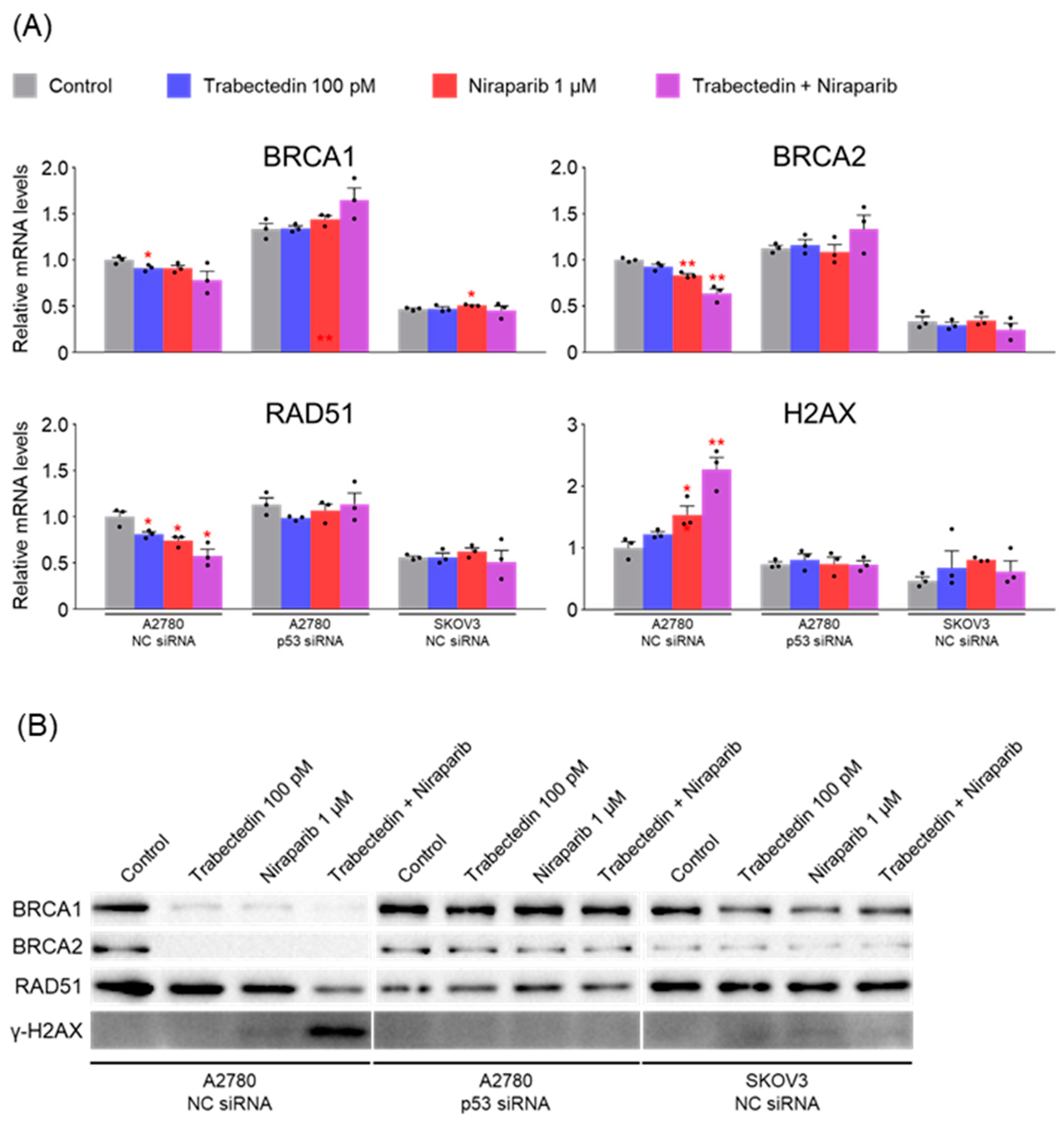
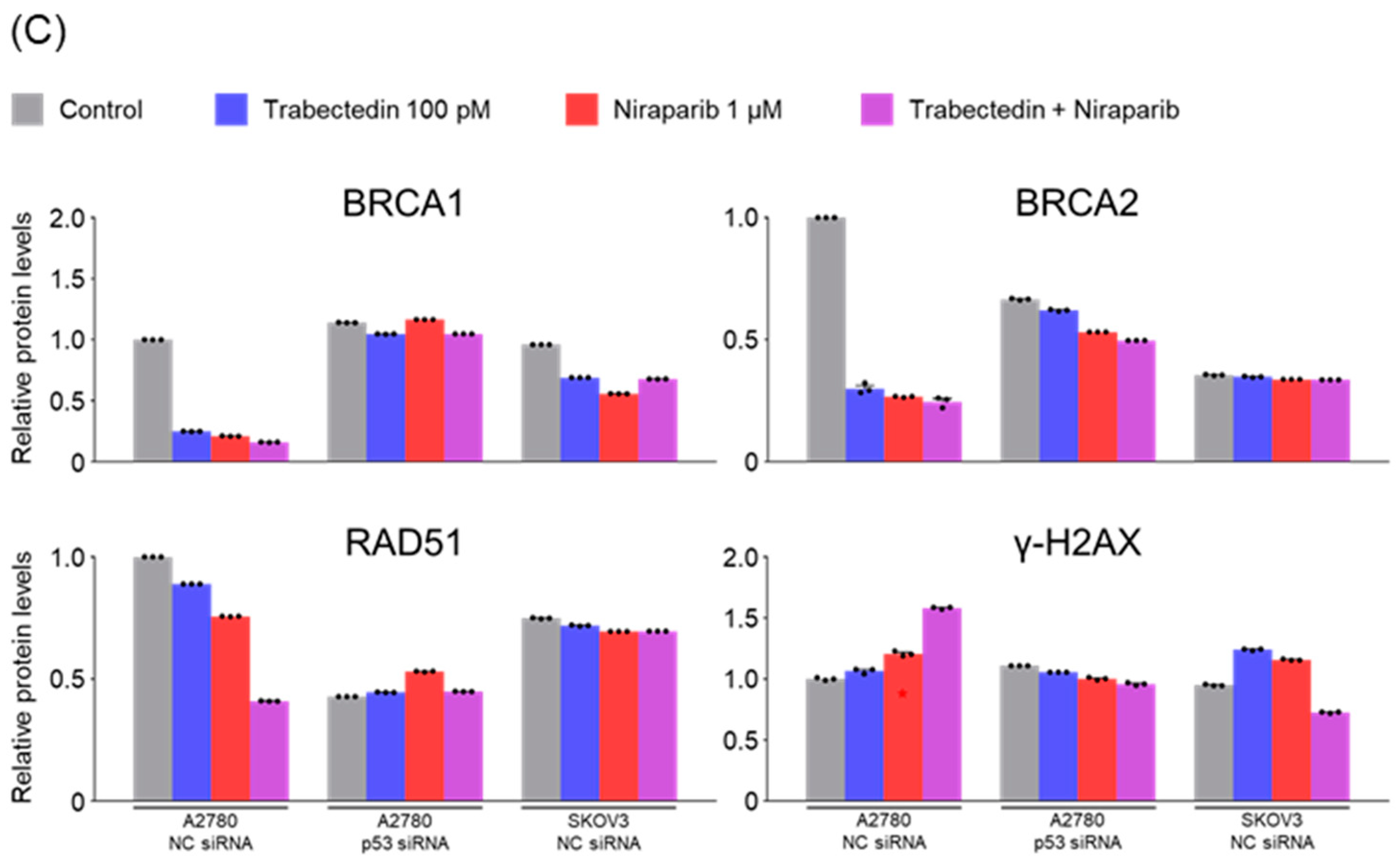
The primary mechanism known for trabectedin to date is the inhibition of the nucleotide excision repair (NER) system. Therefore, when used in conjunction with PARP inhibitors, it results in dual inhibition of systems involved in ssDNA break repair (inhibition of NER by trabectedin and inhibition of PARP by olaparib). Unrepaired ssDNA breaks can progress to dsDNA breaks, leading to the accumulation of dsDNA breaks. In such a scenario, it would be predicted that BRCA-proficient cancer cells need to enhance the activation of homologous recombination factors such as BRCA1, BRCA2, and RAD51 to repair dsDNA breaks.
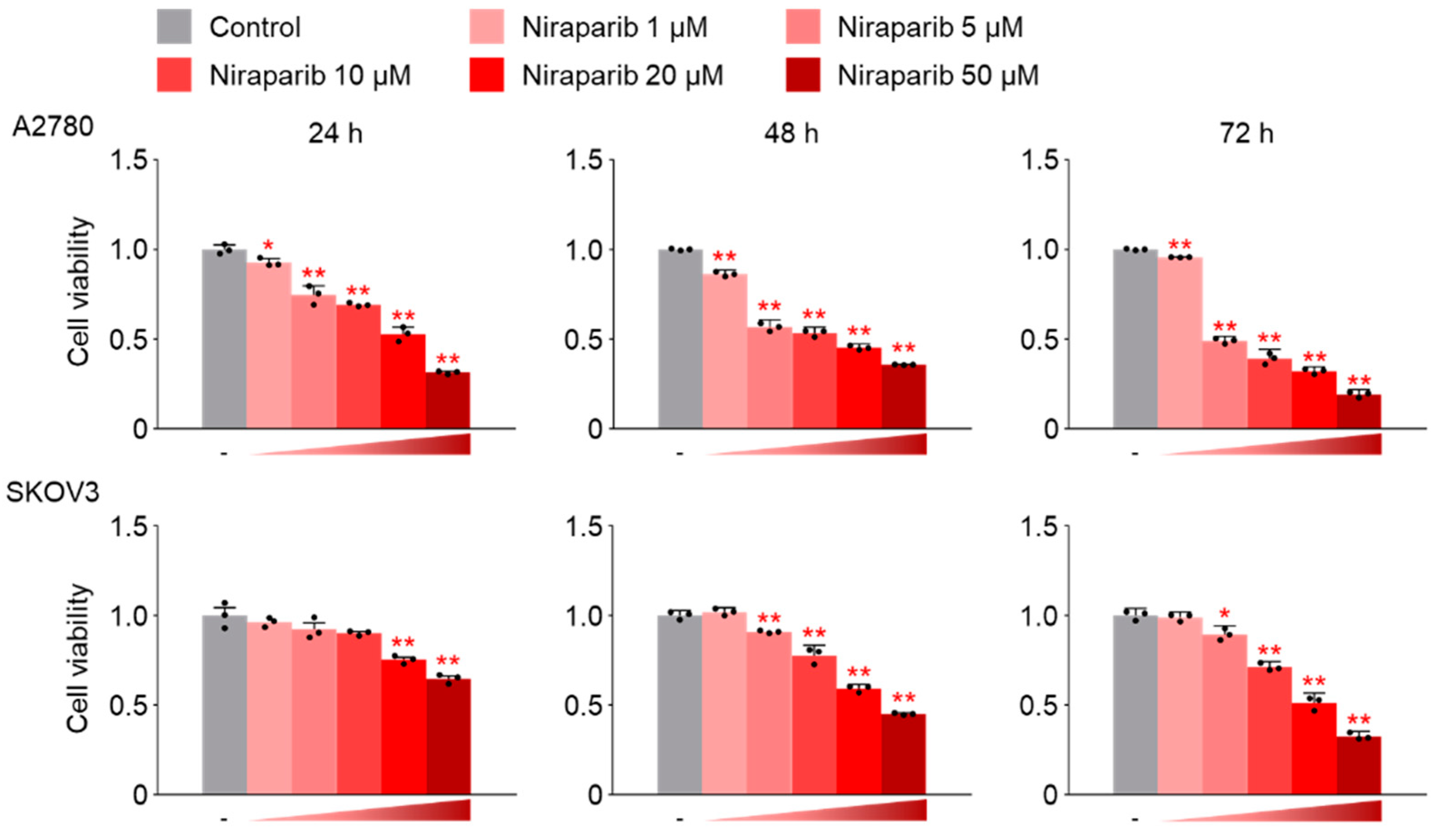
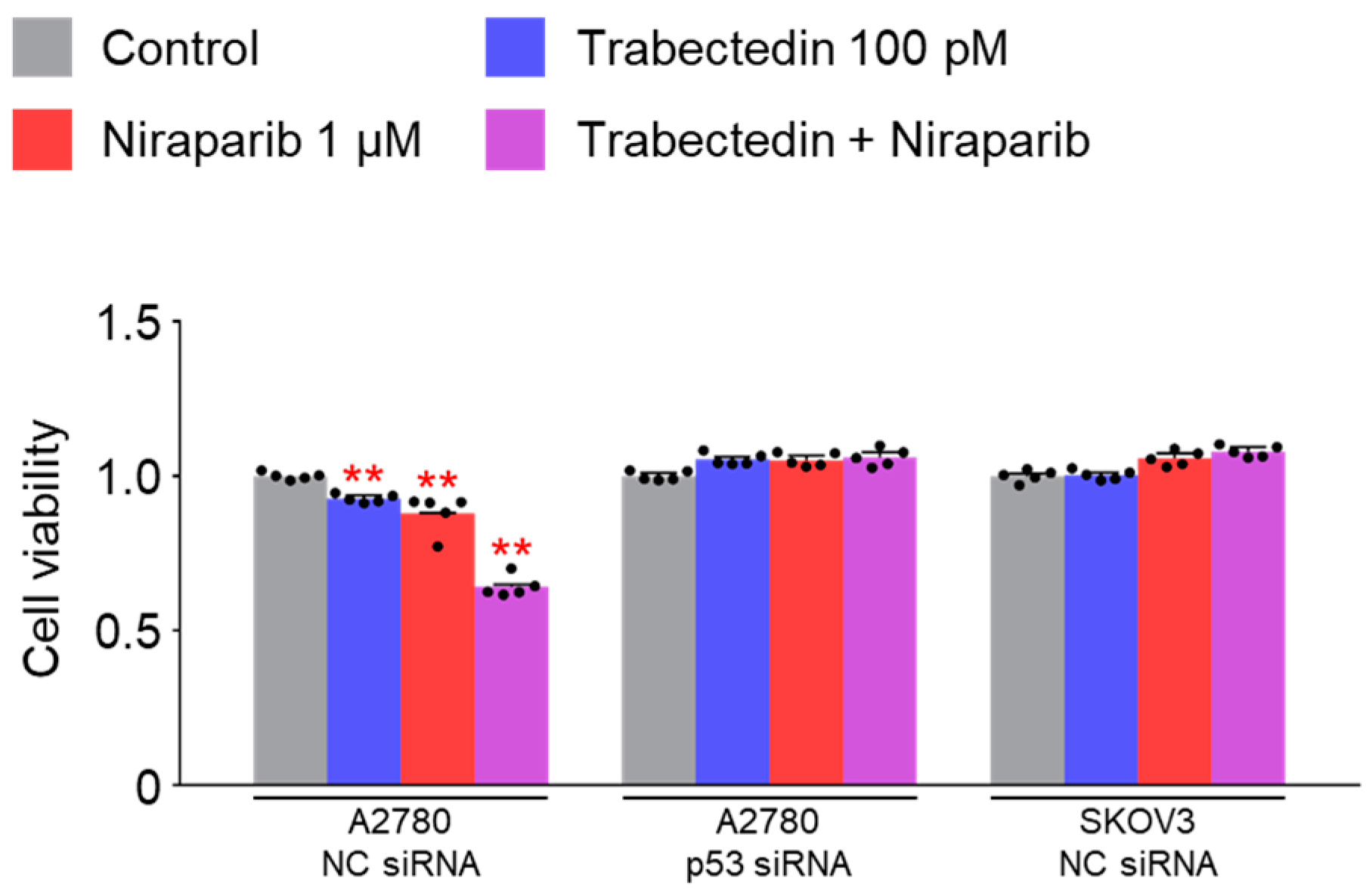
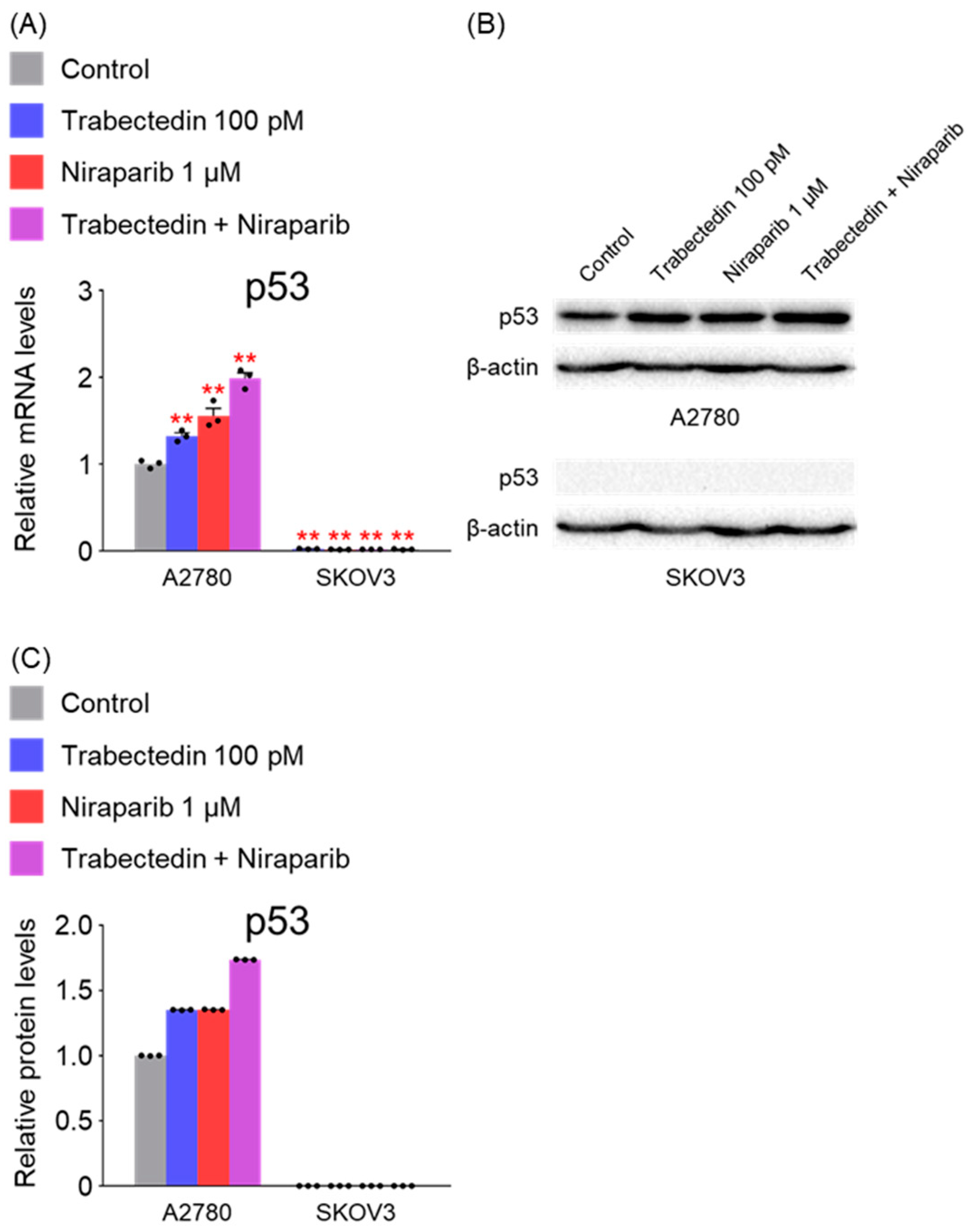
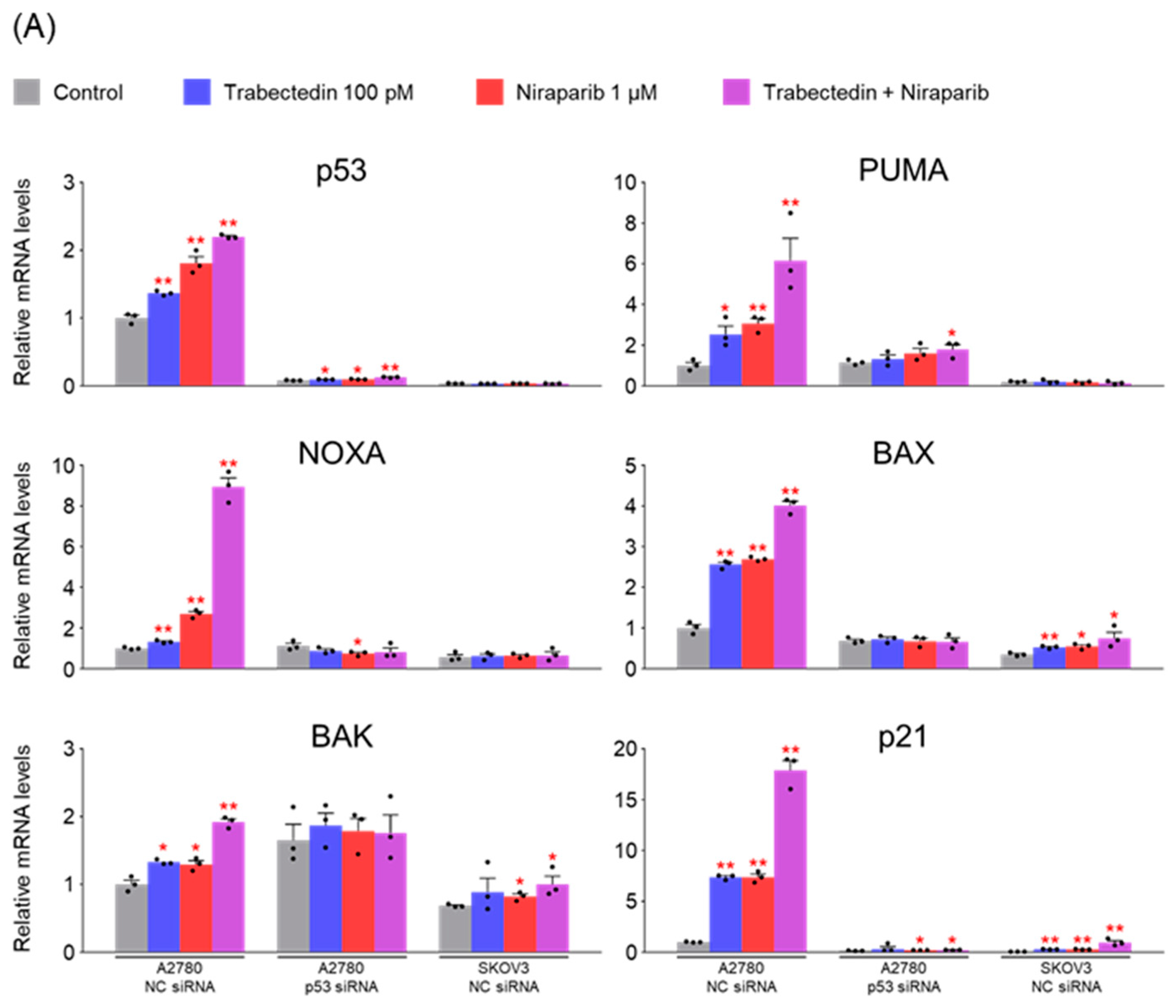
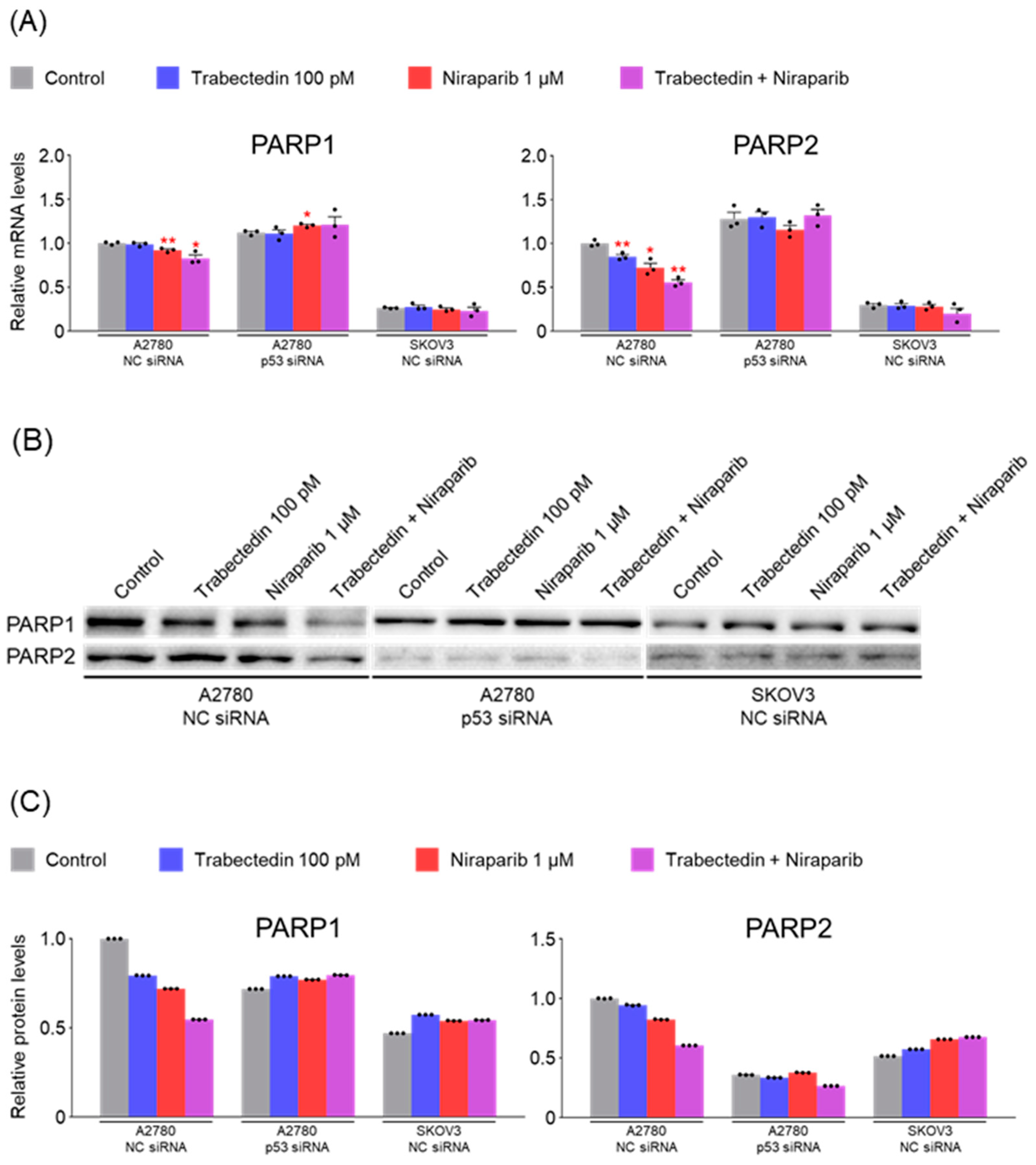
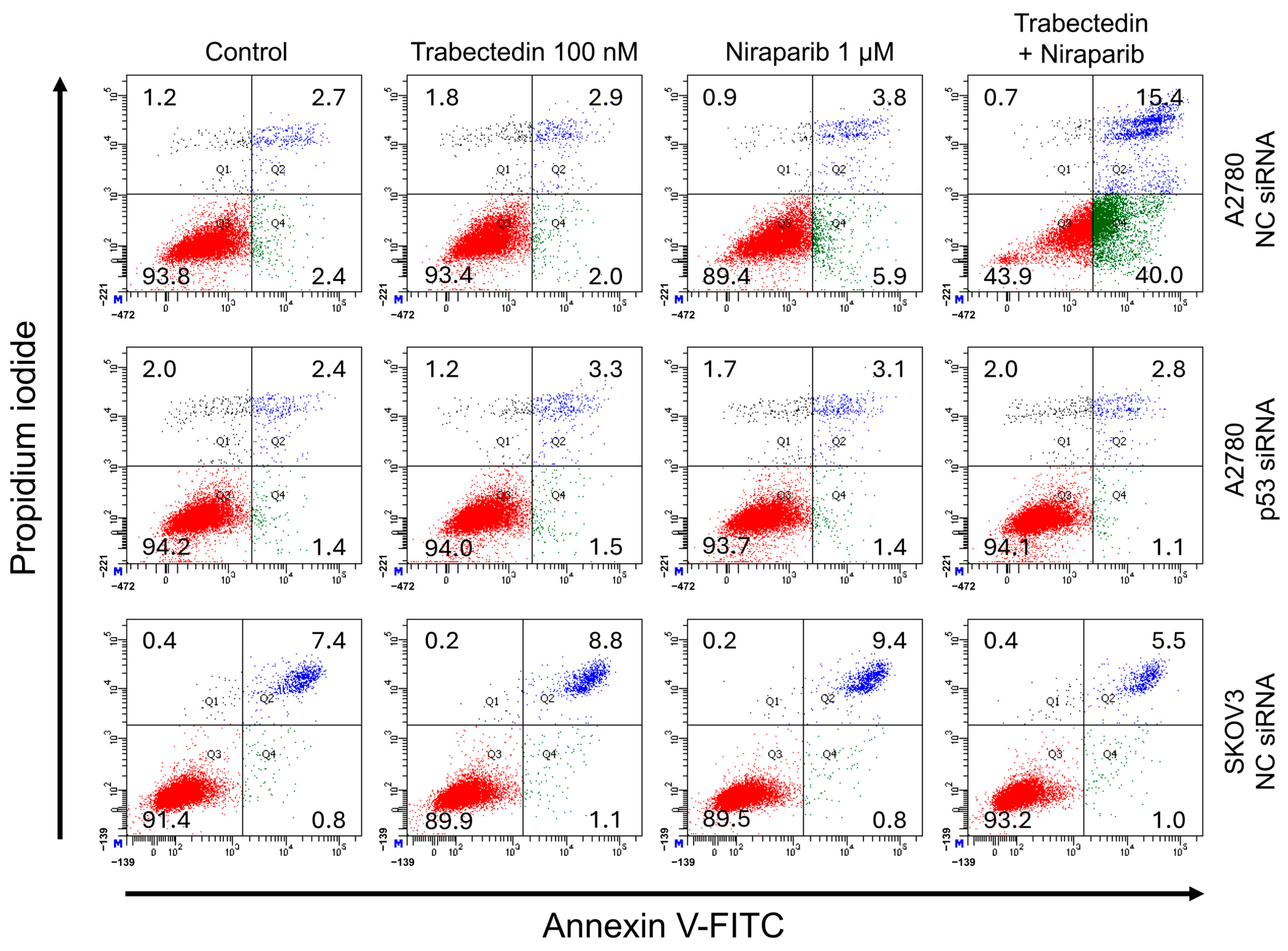
However, the results of this study did not support this prediction. In the findings of this study, not only were PARP1 and PARP2, involved in ssDNA break repair, more inhibited in the combination therapy of trabectedin and niraparib than in either agent alone, but factors associated with dsDNA break homologous recombination repair (BRCA1, BRCA2, and RAD51) were also suppressed. This was accompanied by an increase in γ-H2AX, indicating DNA damage, along with a decrease in cell viability, ultimately leading to apoptosis. These results were observed in A2780 cells (with wild-type p53), but not in SKOV3 cells (p53 knockout). Additionally, the synergistic effect observed in A2780 was found to be attenuated upon the loss of p53 through p53 siRNA. This study demonstrated that the combined therapy of trabectedin and PARP inhibitors can induce BRCAness and elicit a synthetic lethality effect in BRCA-proficient epithelial ovarian cancer cells. It also highlighted the significance of the p53-mediated apoptotic pathway in such a synergistic effect. This result suggests that there may be an as-yet-unknown mechanism by which trabectedin, in combination with niraparib, induces homologous recombination deficiency.
There are likely to be numerous hurdles in demonstrating the effectiveness of the combined therapy in HR-proficient epithelial ovarian cancer patients. One of them is because
TP53 mutations are commonly found in most epithelial ovarian cancers. The mutations are predominantly in the order of missense, frameshift, and nonsense, with the majority exhibiting abnormalities in p53 function [
30]. Therefore, these research findings emphasize the importance of p53 function recovery therapy in the treatment of epithelial ovarian cancer. Research on the combination therapy with p53 function recovery agents such as PRIMA-1, PRIMA-1Met [
31], and ReACp53 [
32] is considered necessary.
The SKOV3 cell line, with its p53 gene knockout, cannot be considered representative of epithelial ovarian cancer, given that the majority of TP53 mutations in this type of cancer are missense mutations. Therefore, additional research using a broader range of epithelial ovarian cancer cell lines is necessary, and the efficacy needs to be verified through animal experiments using xenografts. Furthermore, there is a need for research on the effectiveness of trabectedin in combination with other PARP inhibitors, apart from niraparib.
For actual clinical use as a treatment, a survival benefit must be demonstrated through clinical trials. However, there is currently a scarcity of clinical trials investigating the combined use of trabectedin and PARP inhibitors. There was a phase 1b study using trabectedin and olaparib in patients with advanced and non-resectable bone and soft-tissue sarcomas. If future clinical trials are conducted, the results of this study may serve as a reference for drug dosage determination [
33].
A limitation of our study is that we did not directly assess the involvement of ATM and ATR pathways in the observed DNA damage response. Given that niraparib is a PARP inhibitor that induces replication stress and trabectedin interferes with transcription-coupled repair, it is plausible that both ATR and ATM signaling may contribute to the accumulation of DNA damage. Future studies should investigate the activation status of these pathways using phosphorylation markers (e.g., p-ATR, p-ATM, p-CHK1, p-CHK2) and pharmacological inhibitors to further elucidate the molecular mechanisms underlying the synergistic effects observed in this study.
In conclusion, the combination therapy of niraparib and trabectedin inhibits PARP1 and PARP2 involved in ssDNA break repair, and suppresses BRCA1, BRCA2, and RAD51 involved in dsDNA break homologous recombination. This leads to an increase in DNA damage, inhibiting cell viability and enhancing p53-mediated apoptosis. Trabectedin, when used in combination with PARP inhibitors, could be a strategy to induce BRCAness and trigger synthetic lethality, particularly in HRD-proficient ovarian cancer.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}