
Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Results
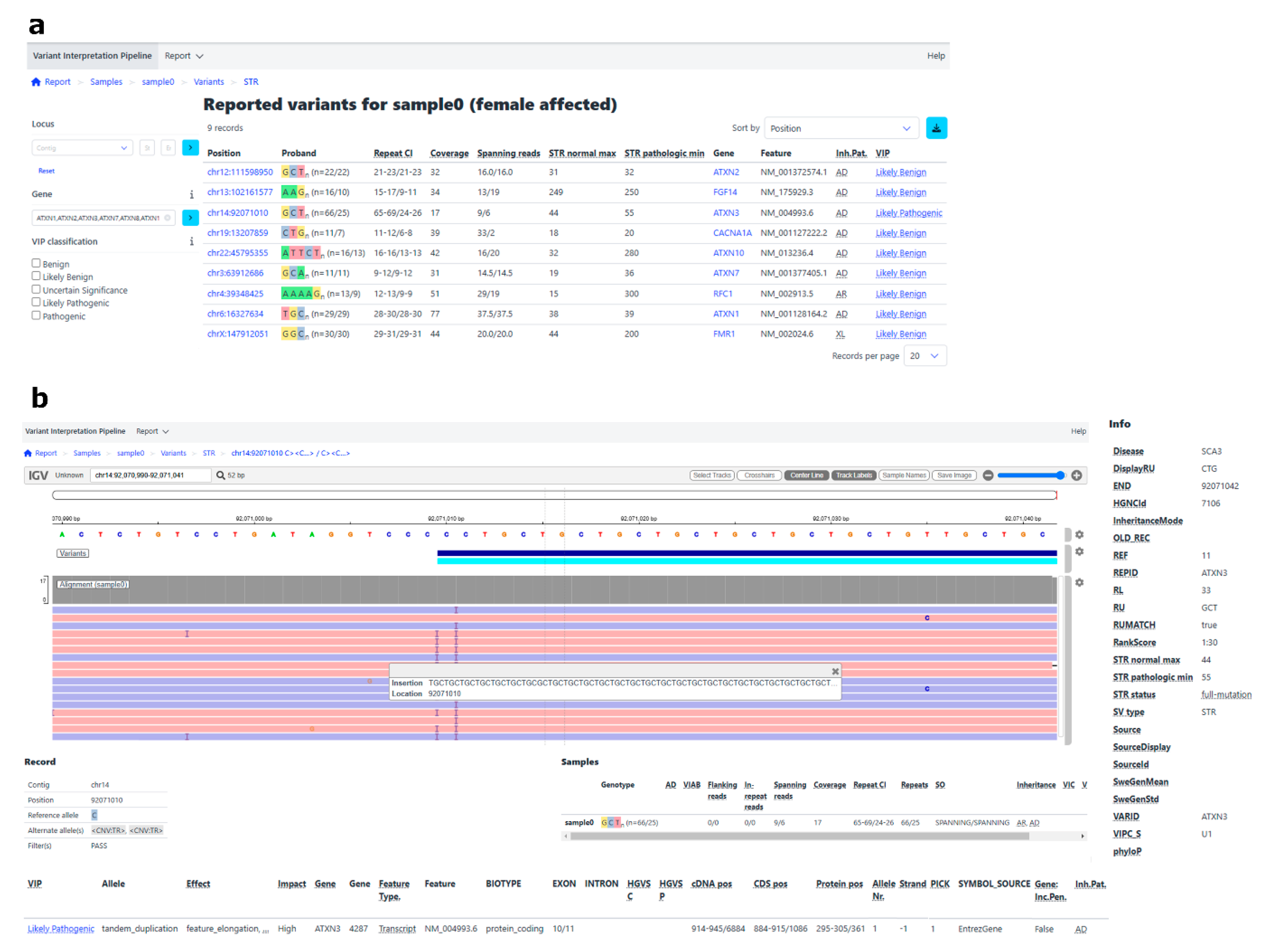
2.1. Validation Phase
2.2. Implementation Phase
2.3. The Accuracy of ONT LRS for SNV- and Indel-Calling and Methylation Detection
2.3.1. SNV and Indel Calling
2.3.2. Methylation Detection
3. Discussion
4. Materials and Methods
4.1. Sample Selection and Study Setup
4.2. Design of the Gene Panel for Adaptive Sampling
4.3. DNA-Extraction and Quality Control
4.4. LRS Sample Preparation
4.5. ONT Sequencing
4.6. Concordance Check
4.7. Data Processing
4.8. STR Expansion Detection: Validation
4.9. STR Expansion Detection: Implementation
4.10. SNV and Indel Detection
4.11. Methylation Calling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | Adaptive Sampling |
| CNV | Copy Number Variant |
| FraX | Fragile X syndrome |
| FXTAS | Fragile X-associated tremor/ataxia syndrome |
| Indel | Insertion and/or deletion |
| LB | Likely Benign |
| LP | Likely Pathogenic |
| LRS | Long-read sequencing |
| ONT | Oxford Nanopore technologies |
| PCR | Polymerase Chain Reaction |
| rpm | Rounds per minute |
| RT | Room temperature |
| RU | Repeat unit |
| SNV | Single Nucleotide Variant |
| SRS | Short-read sequencing |
| STR | Short Tandem Repeat |
| SCA | Spinocerebellar Ataxia |
| SV | Structural Variant |
| VUS | Variant of unknown significance |
References
- De Mattei, F.; Ferrandes, F.; Gallone, S.; Canosa, A.; Calvo, A.; Chiò, A.; Vasta, R. Epidemiology of spinocerebellar ataxias in Europe. Cerebellum 2024, 23, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.; Abbeduto, L.; Hall, S.S.; Hardiman, R.; Hessl, D.; Roberts, J.E.; Scerif, G.; Stanfield, A.C.; Turk, J.; Oliver, C. Fragile X syndrome: An overview of cause, characteristics, assessment and management. Paediatr. Child Health 2020, 30, 400–403. [Google Scholar] [CrossRef]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Willems, T.; Gymrek, M.; Highnam, G.; Mittelman, D.; Erlich, Y. The landscape of human STR variation. Genome Res. 2014, 24, 1894–1904. [Google Scholar] [CrossRef]
- Dolzhenko, E.; Deshpande, V.; Schlesinger, F.; Krusche, P.; Petrovski, R.; Chen, S.; Emig-Agius, D.; Gross, A.; Narzisi, G.; Bowman, B.; et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35, 4754–4756. [Google Scholar] [CrossRef]
- Méreaux, J.L.; Davoine, C.S.; Coutelier, M.; Guillot-Noël, L.; Castrioto, A.; Charles, P.; Coarelli, G.; Ewenczyk, C.; Klebe, S.; Heinzmann, A.; et al. Fast and reliable detection of repeat expansions in spinocerebellar ataxia using exomes. J. Med. Genet. 2023, 60, 717–721. [Google Scholar] [CrossRef]
- Ibañez, K.; Polke, J.; Hagelstrom, R.T.; Dolzhenko, E.; Pasko, D.; Thomas, E.R.A.; Daugherty, L.C.; Kasperaviciute, D.; Smith, K.R.; Deans, Z.C.; et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: A retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022, 21, 234–245. [Google Scholar] [CrossRef]
- Perlman, S. Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; Hereditary Ataxia Overview. In GeneReviews® [Internet]; Seattle (WA): University of Washington: Seattle, WA, USA, 2025. [Google Scholar]
- Stevanovski, I.; Chintalaphani, S.R.; Gamaarachchi, H.; Ferguson, J.M.; Pineda, S.S.; Scriba, C.K.; Tchan, M.; Fung, V.; Ng, K.; Cortese, A.; et al. Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci. Adv. 2022, 8, 14–17. [Google Scholar] [CrossRef]
- Miyatake, S.; Koshimizu, E.; Fujita, A.; Doi, H.; Okubo, M.; Wada, T.; Hamanaka, K.; Ueda, N.; Kishida, H.; Minase, G.; et al. Rapid and comprehensive diagnostic method for repeat expansion diseases using nanopore sequencing. Npj Genomic Med. 2022, 7, 62. [Google Scholar] [CrossRef]
- Borgström, E.; Redin, D.; Lundin, S.; Berglund, E.; Andersson, A.F.; Ahmadian, A. Phasing of single DNA molecules by massively parallel barcoding. Nat. Commun. 2015, 6, 7173. [Google Scholar] [CrossRef]
- Gouil, Q.; Keniry, A. Latest techniques to study dna methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Poeta, L.; Drongitis, D.; Verrillo, L.; Miano, M.G. Dna Hypermethylation and unstable repeat diseases: A paradigm of transcriptional silencing to decipher the basis of pathogenic mechanisms. Genes 2020, 11, 684. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.-T.; Espinosa, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar]
- Tassone, F.; Protic, D.; Allen, E.G.; Archibald, A.D.; Baud, A.; Brown, T.W.; Budimirovic, D.B.; Cohen, J.; Dufour, B.; Eiges, R.; et al. Insight and recommendations for fragile X-premutation-associated conditions from the fifth international conference on FMR1 premutation. Cells 2023, 12, 2330. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Tanguy, M.; Giess, A.; Dixit, A.; Wilson, L.C.; Gibbons, R.J.; Twigg, S.R.F.; Elgar, G.; Wilkie, A.O.M. A comparison of structural variant calling from short-read and nanopore-based whole-genome sequencing using optical genome mapping as a benchmark. Genes 2024, 15, 925. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Billingsley, K.J.; Mastoras, M.; Meredith, M.; Monlong, J.; Lorig-Roach, R.; Asri, M.; Alvarez Jerez, P.; Malik, L.; Dewan, R.; et al. Scalable nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat. Methods 2023, 20, 1483–1492. [Google Scholar] [CrossRef]
- Sequeiros, J.; Seneca, S.; Martindale, J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur. J. Hum. Genet. 2010, 18, 1188–1195. [Google Scholar] [CrossRef]
- de Leeuw, R.H.; Garnier, D.; Kroon, R.M.J.M.; Horlings, C.G.C.; de Meijer, E.; Buermans, H.; van Engelen, B.G.M.; de Knijff, P.; Raz, V. Diagnostics of short tandem repeat expansion variants using massively parallel sequencing and componential tools. Eur. J. Hum. Genet. 2019, 27, 400–407. [Google Scholar] [CrossRef]
- Bonnet, C.; Pellerin, D.; Roth, V.; Clément, G.; Wandzel, M.; Lambert, L.; Frismand, S.; Douarinou, M.; Grosset, A.; Bekkour, I.; et al. Optimized testing strategy for the diagnosis of GAA-FGF14 ataxia/spinocerebellar ataxia 27B. Sci. Rep. 2023, 13, 9737. [Google Scholar] [CrossRef]
- Mohren, L.; Erdlenbruch, F.; Leitão, E.; Kilpert, F.; Hönes, G.S.; Kaya, S.; Schröder, C.; Thieme, A.; Sturm, M.; Park, J.; et al. Identification and characterization of pathogenic and non-pathogenic FGF14 repeat expansions. Nat. Commun. 2024, 15, 7665. [Google Scholar] [CrossRef]
- Tabolacci, E.; Pietrobono, R.; Maneri, G.; Remondini, L.; Nobile, V.; Monica, M.D.; Pomponi, M.G.; Genuardi, M.; Neri, G.; Chiurazzi, P. Reversion to normal of FMR1 expanded alleles: A rare event in two independent fragile X syndrome families. Genes 2020, 11, 248. [Google Scholar] [CrossRef]
- Brusse, E.; de Koning, I.; Maat-Kievit, A.; Oostra, B.A.; Heutink, P.; van Swieten, J.C. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): A new phenotype. Mov. Disord. 2006, 21, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Danzi, M.C.; Wilke, C.; Renaud, M.; Fazal, S.; Dicaire, M.-J.; Scriba, C.K.; Ashton, C.; Yanick, C.; Beijer, D.; et al. Deep intronic FGF14 GAA repeat expansion in late-onset cerebellar ataxia. N. Engl. J. Med. 2023, 388, 128–141. [Google Scholar]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gillies, G.; Pope, K.; et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am. J. Hum. Genet. 2023, 110, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, K.; Jadhav, B.; Zanovello, M.; Gagliardi, D.; Clarkson, C.; Facchini, S.; Garg, P.; Martin-Trujillo, A.; Gies, S.J.; Deforie, V.G.; et al. G008 Population analysis of repeat expansions indicates increased frequency of pathogenic alleles across different populations. Nat. Med. 2024, 30, A118–A120. [Google Scholar] [CrossRef]
- Ghorbani, F.; De Boer, E.N.; Benjamins-Stok, M.; Verschuuren-Bemelmans, C.C.; Knapper, J.; De Boer-Bergsma, J.; De Vries, J.J.; Sikkema-Raddatz, B.; Verbeek, D.S.; Westers, H.; et al. Copy number variant analysis of spinocerebellar ataxia genes in a cohort of dutch patients with cerebellar ataxia. Neurol. Genet. 2023, 9, e200050. [Google Scholar] [CrossRef]
- Maassen, W.T.K.; Johansson, L.F.; Charbon, B.; Hendriksen, D.; van den Hoek, S.; Slofstra, M.K.; Mulder, R.; Meems-Veldhuis, M.T.; Sietsma, R.; Lemmink, H.H.; et al. MOLGENIS VIP: An open-source and modular pipeline for high-throughput and integrated DNA variant analysis. medRxiv 2024. preprint. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse Genomes. Genome Res. 2009, 19, 1316–1323. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.; Rajan-Babu, I.S.; Friedman, J.M.; Birol, I. Straglr: Discovering and genotyping tandem repeat expansions using whole genome long-read sequences. Genome Biol. 2021, 22, 224. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genome viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- De Coster, W.; Stovner, E.B.; Strazisar, M. Methplotlib: Analysis of modified nucleotides from nanopore sequencing. Bioinformatics 2020, 36, 3236–3238. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sample | ATXN2 | ATXN8 | FGF14 | ATXN3 | CACNA1A | ATXN10 | ATXN7 | RFC1 | ATXN1 | FMR1 | Average (Autosomal Genes) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 (m) | 28 | 38 | 29 | 31 | 26 | 41 | 37 | 18 | 22 | 11 | 30 |
| S1_1 | 16 | 29 | 18 | 14 | 13 | 24 | 17 | 11 | 13 | 6 | 17 |
| S1_2 | 12 | 9 | 11 | 17 | 13 | 17 | 20 | 7 | 9 | 5 | 13 |
| S1 (r) | 21 | 12 | 15 | 22 | 17 | 16 | 13 | 8 | 18 | 15 | 16 |
| S2 (m) | 38 | 30 | 30 | 40 | 14 | 37 | 32 | 33 | 27 | 16 | 32 |
| S2_1 | 14 | 14 | 15 | 21 | 5 | 17 | 13 | 15 | 11 | 9 | 14 |
| S2_2 | 24 | 16 | 15 | 19 | 9 | 20 | 19 | 18 | 16 | 7 | 18 |
| S2 (r) | 18 | 22 | 17 | 13 | 15 | 24 | 26 | 16 | 32 | 11 | 21 |
| S3 (m) | 15 | 22 | 20 | 16 | 18 | 28 | 17 | 22 | 30 | 22 | 20 |
| S3_1 | 10 | 15 | 14 | 20 | 15 | 13 | 16 | 18 | 22 | 16 | 16 |
| S3_2 | 15 | 22 | 20 | 16 | 18 | 28 | 17 | 22 | 30 | 22 | 20 |
| S4 | 30 | 29 | 32 | 27 | 16 | 33 | 34 | 13 | 36 | 22 | 27 |
| S5 | 26 | 32 | 27 | 27 | 25 | 29 | 26 | 36 | 33 | 6 | 29 |
| S5 (r) | 26 | 32 | 27 | 27 | 25 | 29 | 26 | 36 | 33 | 6 | 29 |
| S6 | 25 | 23 | 27 | 29 | 39 | 27 | 33 | 31 | 31 | 11 | 30 |
| S7 | 23 | 14 | 8 | 15 | 16 | 19 | 16 | 20 | 19 | 10 | 16 |
| S8 | 22 | 23 | 19 | 17 | 18 | 18 | 21 | 17 | 17 | 11 | 19 |
| S9 | 22 | 35 | 19 | 31 | 35 | 24 | 27 | 20 | 18 | 20 | 25 |
| S10 | 29 | 35 | 35 | 30 | 23 | 38 | 23 | 32 | 30 | 13 | 30 |
| S11 (m) | 83 | 79 | 101 | 89 | 74 | 91 | 99 | 88 | 75 | 59 | 86 |
| S11_1 | 40 | 40 | 54 | 46 | 30 | 43 | 44 | 42 | 46 | 34 | 42 |
| S11_2 | 43 | 39 | 47 | 43 | 44 | 48 | 55 | 46 | 29 | 25 | 44 |
| S12 | 25 | 30 | 36 | 34 | 23 | 29 | 34 | 30 | 23 | 25 | 29 |
| Sample | FGF14 | ATXN1 | ATXN2 | ATXN3 | CACNA1A | ATXN7 | FMR1 | Note |
|---|---|---|---|---|---|---|---|---|
| S1 | 304/348 ^ | 12/12 | ||||||
| S2 | 31/54 | |||||||
| S3 | 30/42 ^# | 21/22 ^ | 15/29 ^ | 7/11 | 10/10 | |||
| S4 | 30/63 | |||||||
| S5 | 28/30 +^ | 22/22 | 23/23 | 12/12 | 11/11 | |||
| S6 | 223 † | FMR1 range 147–397 | ||||||
| S7 | 28/69 * | |||||||
| S8 | ||||||||
| S9 | 31/456 | |||||||
| S10 | ||||||||
| S11 | 29/29 | 22/22 | 23/28^ | 11/11 | 10/12 ^ | |||
| S12 | 31/393 |
| Reported Variant 1 | Reported Variant 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Gene | STR Length (Allele1/Allele2) | Classification | Repeat Motif or Presence of Interruptions? | Gene | STR Length (Allele1/Allele2) | Classification | Repeat Motif |
| S1 (m) | FGF14 | 303/348 | LP/LB * | GAA/GAAGGA | RFC1 | 134/134 | VUS/VUS | AAAGG/AAGGG |
| S1_1 | FGF14 | 300/350 | LP/LB * | GAA/GAAGGA | RFC1 | 136/136 | VUS/VUS | AAAGG/AAGGG |
| S1_2 | FGF14 | 304/342 | LP/LB * | GAA/GAAGGA | RFC1 | 129/129 | VUS/VUS | AAAGG/AAGGG |
| S1 (r) | FGF14 | 305/348 | LP/LB * | GAA/GAAGGA | RFC1 | 134/134 | VUS/VUS | AAAGG/AAGGG |
| S2 (m) | ATXN1 | 31/54 | LB/LP | RFC1 | 105/374 | LB †/LP | AAAG, AAAAG/AAGGG | |
| S2_1 | ATXN1 | 31/54 | LB/LP | RFC1 | 104/362 | LB †/LP | AAAG, AAAAG/AAGGG | |
| S2_2 | ATXN1 | 31/54 | LB/LP | RFC1 | 107/374 | LB †/LP | AAAG, AAAAG/AAGGG | |
| S2 (r) | ATXN1 | 31/54 | LB/LP | RFC1 | 106/378 | LB †/LP | AAAG, AAAAG/AAGGG | |
| S3 (m) | ATXN1 | 30/41 | LB/LP | RFC1 | 11/528 | LB/LP | Allele 2 AAGGG | |
| S3_1 | ATXN1 | 30/42 | LB/LP | RFC1 | 11/524 | LB/LP | Allele 2 AAGGG | |
| S3_2 | ATXN1 | 30/41 | LB/LP | RFC1 | 11/528 | LB/LP | Allele 2 AAGGG | |
| S4 | FMR1 | 30/63 | LB/VUS | RFC1 | 142/142 | VUS | AAAAG with AAAG interruptions | |
| S5 | ||||||||
| S5 (r) | ||||||||
| S6 | FMR1 | 223 | LP | GGT interruptions | RFC1 | 15/141 | LB/LB † | AAAAG with AAAG interruptions |
| S7 | ATXN3 | 25/66 | LB/LP | |||||
| S8 | RFC1 | 11/122 | LB/LB † | Allele 2 AAAAG | ||||
| S9 | FMR1 | 31/456 | LB/LP | GC, GCA and GAC interruptions | RFC1 | 12/114 | LB/VUS | AAAAG |
| S10 | ||||||||
| S11 (m) | ||||||||
| S11_1 | ||||||||
| S11_2 | ||||||||
| S12 | FMR1 | 31/393 | LB/LP | RFC1 | 12/112 | LB/VUS | Allele 2 AAAAG | |
| Sample | Number of Indels | Number of SNVs | ||||
|---|---|---|---|---|---|---|
| Concordant Both Platforms | LRS Only | WES Only | Concordant Both Platforms | LRS Only | WES Only | |
| S1 | 4 | 1 | 0 | 21 | 12 * | 1 |
| S3 | 4 | 0 | 0 | 41 | 1 | 1 |
| S4 | 5 | 0 | 0 | 28 | 0 | 0 |
| S5 | 5 | 0 | 0 | 25 | 0 | 0 |
| S8 | 3 | 0 | 0 | 33 | 0 | 2 |
| S11 | 4 | 0 | 0 | 32 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Boer, E.N.; Scheper, A.J.; Hendriksen, D.; Charbon, B.; van der Vries, G.; ten Berge, A.M.; Grootscholten, P.M.; Lemmink, H.H.; Jongbloed, J.D.H.; Bosscher, L.; et al. Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders. Int. J. Mol. Sci. 2025, 26, 2850. https://doi.org/10.3390/ijms26072850
de Boer EN, Scheper AJ, Hendriksen D, Charbon B, van der Vries G, ten Berge AM, Grootscholten PM, Lemmink HH, Jongbloed JDH, Bosscher L, et al. Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders. International Journal of Molecular Sciences. 2025; 26(7):2850. https://doi.org/10.3390/ijms26072850
Chicago/Turabian Stylede Boer, Eddy N., Arjen J. Scheper, Dennis Hendriksen, Bart Charbon, Gerben van der Vries, Annelies M. ten Berge, Petra M. Grootscholten, Henny H. Lemmink, Jan D. H. Jongbloed, Laura Bosscher, and et al. 2025. "Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders" International Journal of Molecular Sciences 26, no. 7: 2850. https://doi.org/10.3390/ijms26072850
APA Stylede Boer, E. N., Scheper, A. J., Hendriksen, D., Charbon, B., van der Vries, G., ten Berge, A. M., Grootscholten, P. M., Lemmink, H. H., Jongbloed, J. D. H., Bosscher, L., Knoers, N. V. A. M., Swertz, M. A., Sikkema-Raddatz, B., Dijkstra, D. J., Johansson, L. F., & van Diemen, C. C. (2025). Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders. International Journal of Molecular Sciences, 26(7), 2850. https://doi.org/10.3390/ijms26072850