

Expanding the Clinical Spectrum of CRB1-Retinopathies: A Novel Genotype–Phenotype Correlation with Macular Dystrophy and Elevated Intraocular Pressure

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
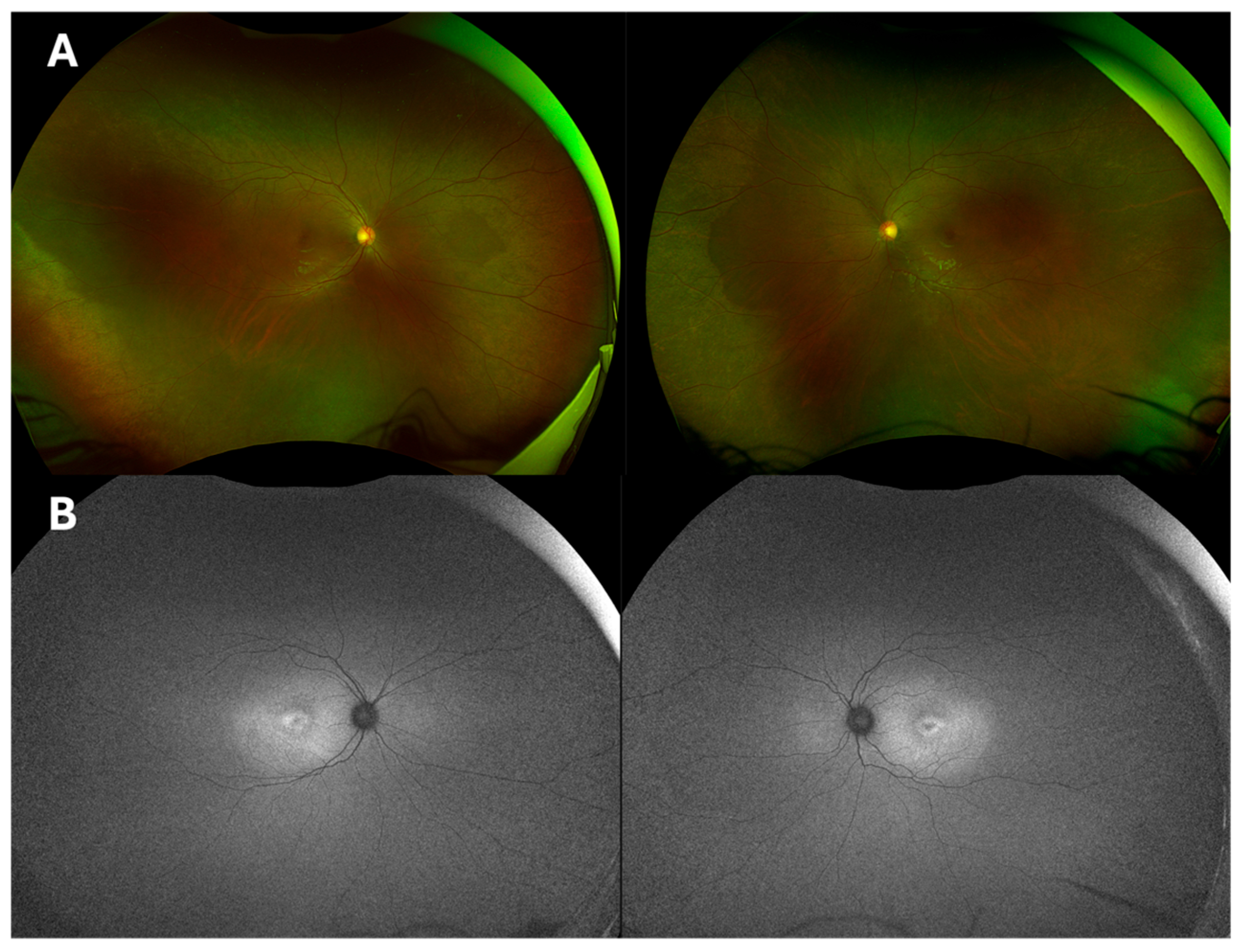
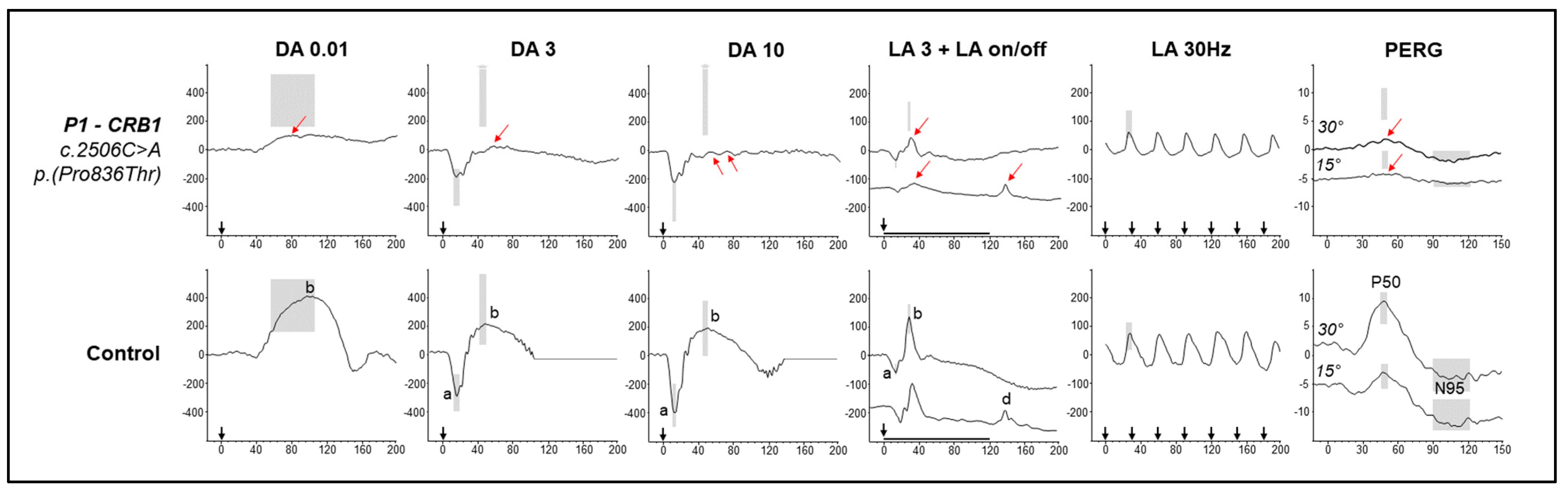
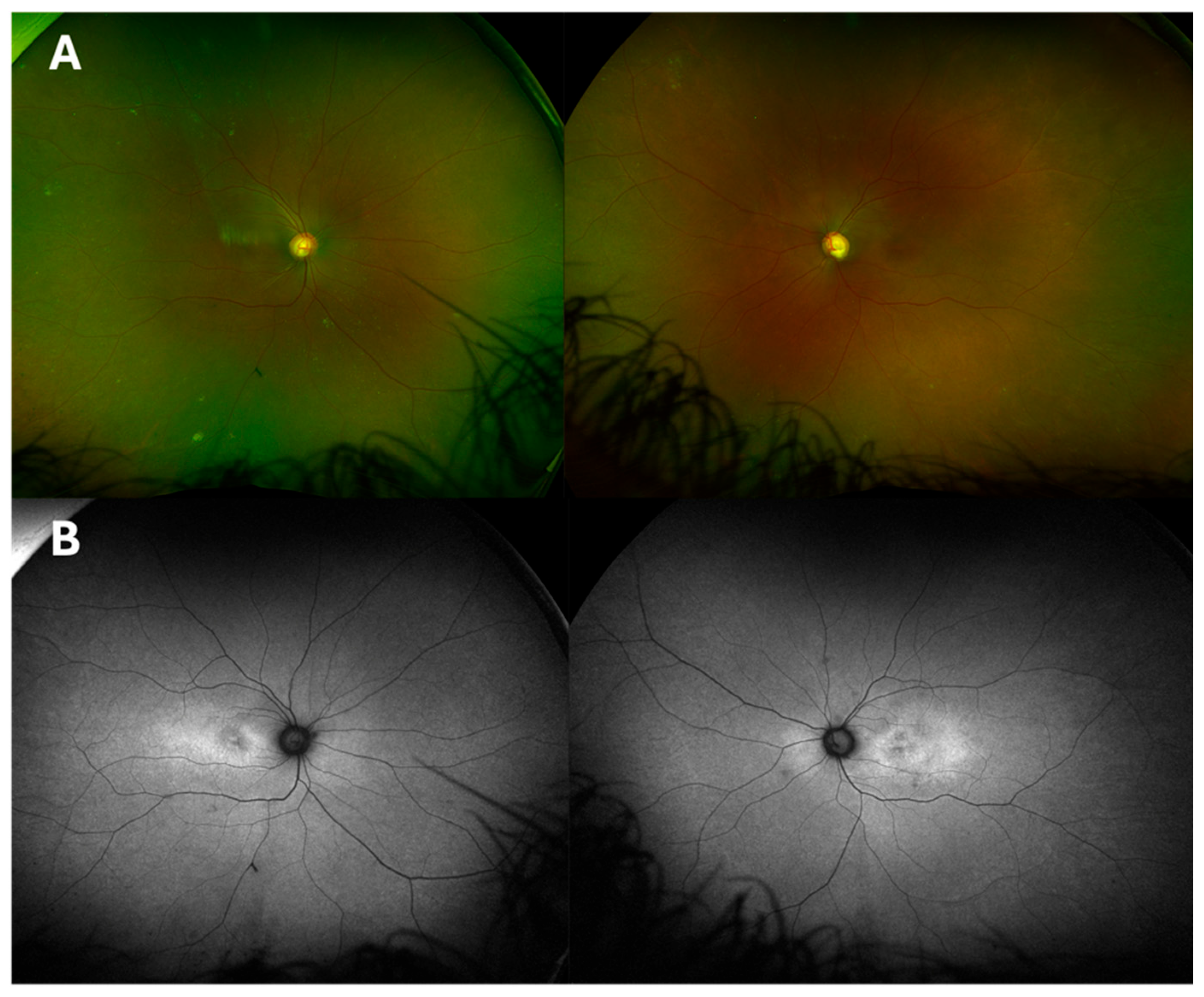
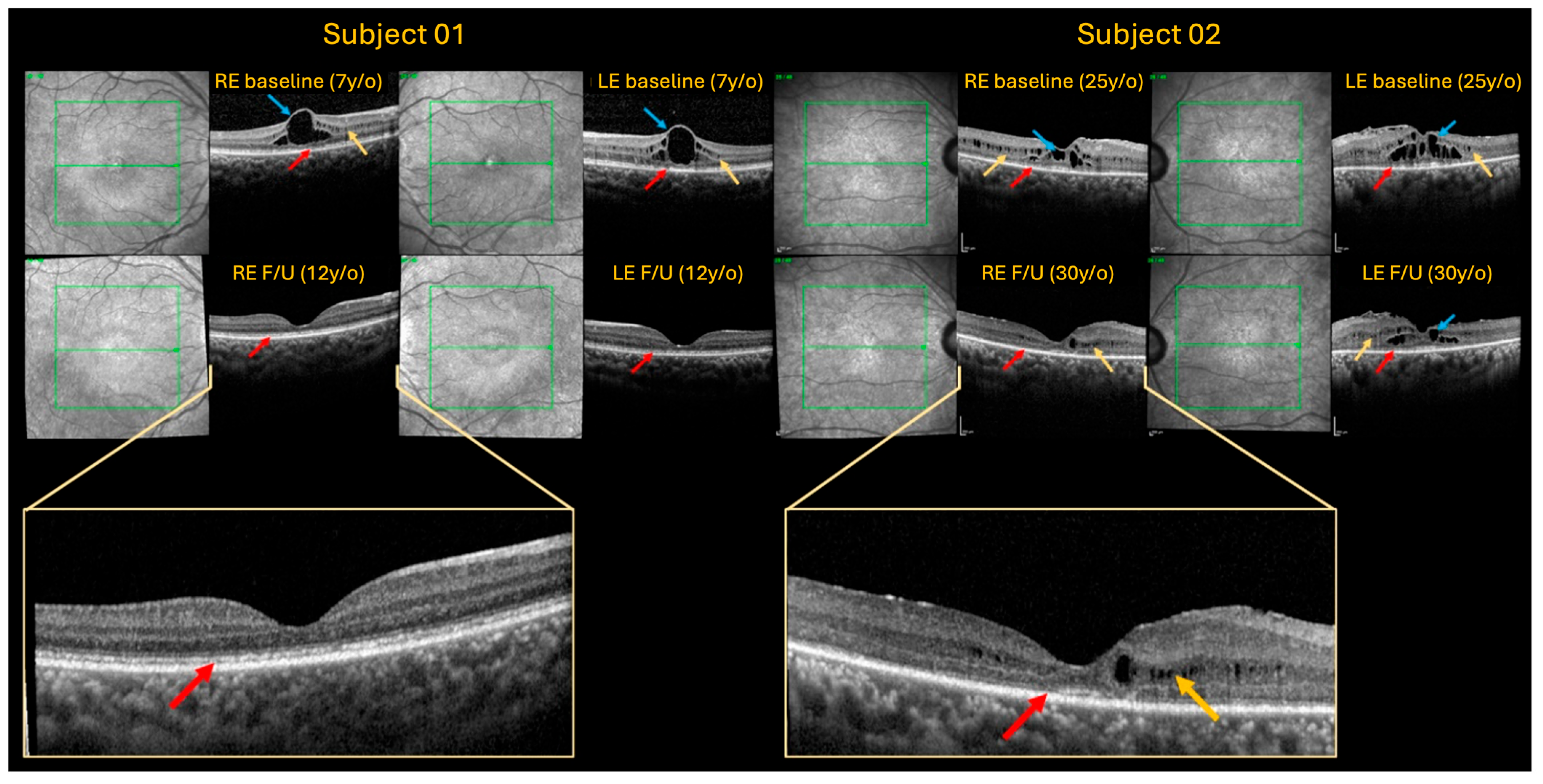
2.1. Case 1
2.2. Case 2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | 1 | 2 |
|---|---|---|
| Family number | Z413096 | 45590 |
| Gender | Female | Female |
| Ethnicity | African decent (Sierra Leone) | African decent (Nigeria) |
| Age | 07 | 25 |
| Age of onset | 07 | 10 |
| Phenotype | MD | MD |
| Zygosity | Homozygous | Homozygous |
| Variant cDNAV ariant protein | c.2506C>A p.Pro836Thr | c.2506C>A p.Pro836Thr |
| Follow-up | 7-year follow-up | 5-year follow-up |
| BCVA LogMAR Baseline Follow-up | RE: 0.66 LogMAR, LE: 0.54 LogMAR RE: 0.50 LogMAR, LE: 0.60 LogMAR | RE: 0.30 LogMAR, LE: 0.30 LogMAR RE: 0.32 LogMAR, LE: 0.32 LogMAR |
| Refractive error | RE: −2.50/−2.25 × 167 LE: −2.50/−2.00 × 30 | RE: −0.75 LE: −0.75 |
| IOP mmHg Baseline Follow-up | RE: 29 mmHg, LE: 44 mmHg RE: 25 mmHg, LE: 25 mmHg | RE: 29 mmHg, LE: 29 mmHg RE: 15 mmHg, LE: 17 mmHg |
| IOP treatment | Brinzolamide BD and Latanoprost once daily | Dorzolamide 2% BD and Ganfort once daily Bilateral laser Iridotomy |
| OCT (CRT) (1 mm3) Baseline Follow-up | RE 448 µm, LE 488 µm RE 117 µm, LE 123 µm | RE 315 µm, LE 374 µm RE 190 µm, LE 258 µm |
| EDTs | Macular dysfunction with mild inner retinal dysfunction affecting rod and cone pathways | Not available |
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, X.; Zhao, L.; Wang, C.; Sun, W.; Jia, B.; Li, D.; Fu, J. Diverse functions and pathogenetic role of Crumbs in retinopathy. Cell Commun. Signal. 2024, 22, 290. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.D. CRB1-Associated Retinal Dystrophies: Genetics, Clinical Characteristics, and Natural History. Am. J. Ophthalmol. 2023, 246, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.J.; Kay, J.N. The Enigma of CRB1 and CRB1 Retinopathies. In Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1185, pp. 251–255. [Google Scholar]
- Mairot, K.; Smirnov, V.; Bocquet, B.; Labesse, G.; Arndt, C.; Defoort-Dhellemmes, S.; Zanlonghi, X.; Hamroun, D.; Denis, D.; Picot, M.-C.; et al. Crb1-related retinal dystrophies in a cohort of 50 patients: A reappraisal in the light of specific Müller cell and photoreceptor Crb1 isoforms. Int. J. Mol. Sci. 2021, 22, 12642. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]
- Boon, N.; Wijnholds, J.; Pellissier, L.P. Research Models and Gene Augmentation Therapy for CRB1 Retinal Dystrophies. Front. Neurosci. 2020, 14, 860. [Google Scholar] [CrossRef]
- Alves, C.H.; Sanz, A.S.; Park, B.; Pellissier, L.P.; Tanimoto, N.; Beck, S.C.; Huber, G.; Murtaza, M.; Richard, F.; Gurubaran, I.S.; et al. Loss of CRB2 in the mouse retina mimics human retinitis pigmentosa due to mutations in the CRB1 gene. Hum. Mol. Genet. 2013, 22, 35–50. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, C.; Yang, D.; Sun, R.; Wang, M.; Sun, H.; Xu, M.; Zhou, L.; Chen, M.; Xie, P.; et al. CRB2 mutation causes autosomal recessive retinitis pigmentosa. Exp. Eye Res. 2019, 180, 164–173. [Google Scholar] [CrossRef]
- Quinn, P.M.; Pellissier, L.P.; Wijnholds, J. The CRB1 Complex: Following the Trail of Crumbs to a Feasible Gene Therapy Strategy. Front. Neurosci. 2017, 11, 175. [Google Scholar] [CrossRef]
- Owen, N.; Toms, M.; Tian, Y.; Toualbi, L.; Richardson, R.; Young, R.; Tracey-White, D.; Dhami, P.; Beck, S.; Moosajee, M. Loss of the crumbs cell polarity complex disrupts epigenetic transcriptional control and cell cycle progression in the developing retina. J. Pathol. 2023, 259, 441–454. [Google Scholar] [CrossRef]
- Boon, N.; Lu, X.; Andriessen, C.A.; Moustakas, I.; Buck, T.M.; Freund, C.; Arendzen, C.H.; Böhringer, S.; Boon, C.J.; Mei, H.; et al. AAV-mediated gene augmentation therapy of CRB1 patient-derived retinal organoids restores the histological and transcriptional retinal phenotype. Stem Cell Rep. 2023, 18, 1123–1137. [Google Scholar] [CrossRef]
- Daher, A.; Banjak, M.; Noureldine, J.; Nehme, J.; El Shamieh, S. Genotype-phenotype associations in CRB1 bi-allelic patients: A novel mutation, a systematic review and meta-analysis. BMC Ophthalmol. 2024, 24, 167. [Google Scholar] [CrossRef]
- Rodriguez-Martinez, A.C.; Higgins, B.E.; Tailor-Hamblin, V.; Malka, S.; Cheloni, R.; Collins, A.M.; Bladen, J.; Henderson, R.; Moosajee, M. Foveal Hypoplasia in CRB1-Related Retinopathies. Int. J. Mol. Sci. 2023, 24, 13932. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.H.; Mackay, D.S.; Li, Z.; Moradi, P.; Sergouniotis, P.; Russell-Eggitt, I.; Thompson, D.A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br. J. Ophthalmol. 2011, 95, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Andrieu-Soler, C.; Kowalczuk, L.; Cortés, M.P.; Berdugo, M.; Dernigoghossian, M.; Halili, F.; Jeanny, J.-C.; Goldenberg, B.; Savoldelli, M.; et al. A new CRB1 rat mutation links Müller glial cells to retinal telangiectasia. J. Neurosci. 2015, 35, 6093–6106. [Google Scholar] [CrossRef]
- Khan, K.N.; Robson, A.; Mahroo, O.A.R.; Arno, G.; Inglehearn, C.F.; Armengol, M.; Waseem, N.; Holder, G.E.; Carss, K.J.; Raymond, L.F.; et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur. J. Hum. Genet. 2018, 26, 687–694. [Google Scholar] [CrossRef]
- Shamsnajafabadi, H.; Kaukonen, M.; Bellingrath, J.S.; MacLaren, R.E.; Cehajic-Kapetanovic, J. In Silico CRISPR-Cas-Mediated Base Editing Strategies for Early-Onset, Severe Cone–Rod Retinal Degeneration in Three Crumbs homolog 1 Patients, including the Novel Variant c.2833G>A. Genes 2024, 15, 625. [Google Scholar] [CrossRef]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef]
- Thompson, D.A.; Bach, M.; McAnany, J.J.; Habjan, M.Š.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical pattern electroretinography (2024 update). Doc. Ophthalmol. 2024, 148, 75–85. [Google Scholar] [CrossRef]
- International Society for Clinical Electrophysiology of Vision; Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc. Ophthalmol. 2016, 133, 1–9. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Schalij-Delfos, N.E.; Talsma, H.E.; Florijn, R.J.; Brink, J.B.T.; Cremers, F.P.; Thiadens, A.A.; et al. Defining inclusion criteria and endpoints for clinical trials: A prospective cross-sectional study in CRB1-associated retinal dystrophies. Acta Ophthalmol. 2021, 99, e402–e414. [Google Scholar] [CrossRef]
- Talib, M.; Van Cauwenbergh, C.; De Zaeytijd, J.; Van Wynsberghe, D.; De Baere, E.; Boon, C.J.F.; Leroy, B.P. CRB1-associated retinal dystrophies in a Belgian cohort: Genetic characteristics and long-term clinical follow-up. Br. J. Ophthalmol. 2022, 106, 696–704. [Google Scholar] [CrossRef]
- Nguyen, X.-T.; Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Schalij-Delfos, N.E.; Klaver, C.C.; Talsma, H.E.; Fiocco, M.; Florijn, R.J.; et al. CRB1-Associated Retinal Dystrophies: A Prospective Natural History Study in Anticipation of Future Clinical Trials. Am. J. Ophthalmol. 2022, 234, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Pennica, C.; Hanna, G.; Islam, S.A.; Sternberg, M.J.; David, A. Missense3D-PPI: A Web Resource to Predict the Impact of Missense Variants at Protein Interfaces Using 3D Structural Data. J. Mol. Biol. 2023, 435, 168060. [Google Scholar] [CrossRef]
- Sun, J.-X.; Yan, H.-X.; Hu, D.; Zhou, J.; Wang, Y.-S.; Wu, J.; Song, X.-J.; Hou, X. Biallelic Heterozygous Mutations in Crumbs Homolog-1 Gene Associated with Macular Retinoschisis and Angle-Closure Glaucoma: A Case Report and Literature Review. Front. Ophthalmol. 2022, 2, 902898. [Google Scholar] [CrossRef]
- Abe, R.Y.; Makarczyk, L.d.S.Q.; de Ávila, M.P.; Sallum, J.M.F. Early occurrence of primary angle-closure glaucoma in a patient with retinitis pigmentosa and CRB1 gene variations. Arq. Bras. Oftalmol. 2023, 85, 74–78. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Genderen, M.M.; Wijnholds, J.; Florijn, R.J.; Brink, J.B.T.; Schalij-Delfos, N.E.; Dagnelie, G.; Cremers, F.P.M.; Wolterbeek, R.; et al. Genotypic and phenotypic characteristics of CRB1-associated retinal dystrophies: A long-term follow-up study. Ophthalmology 2017, 124, 884–895. [Google Scholar]
- Vincent, A.; Ng, J.; Gerth-Kahlert, C.; Tavares, E.; Maynes, J.T.; Wright, T.; Tiwari, A.; Tumber, A.; Li, S.; Hanson, J.V.M.; et al. Biallelic Mutations in CRB1 Underlie Autosomal Recessive Familial Foveal Retinoschisis. Investig Ophthalmol. Vis. Sci. 2016, 57, 2637–2646. [Google Scholar]
- Wolfson, Y.; Applegate, C.D.; Strauss, R.W.; Han, I.C.; Scholl, H.P. CRB1-Related Maculopathy with Cystoid Macular Edema. JAMA Ophthalmol. 2015, 133, 1357–1360. [Google Scholar] [CrossRef]
- Gupta, S.K.; Chakraborty, R.; Verkicharla, P.K. Electroretinogram responses in myopia: A review. Doc. Ophthalmol. 2022, 145, 77–95. [Google Scholar] [CrossRef]
- Jiang, X.; Mahroo, O.A. Negative electroretinograms: Genetic and acquired causes, diagnostic approaches and physiological insights. Eye 2021, 35, 2419–2437. [Google Scholar] [CrossRef]
- Mantel, I.; Ramchand, K.V.; Holder, G.E.; Ohbayashi, M.; Morohoshi, K.; Patel, N.; Toda, M.; Fitzke, F.W.; Bird, A.C.; Ono, S.J. Macular and retinal dysfunction of unknown origin in adults with normal fundi: Evidence for an autoimmune pathophysiology. Exp. Mol. Pathol. 2008, 84, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Holder, G.E.; Robson, A.G.; Pavesio, C.; Graham, E.M. Electrophysiological characterisation and monitoring in the management of birdshot chorioretinopathy. Br. J. Ophthalmol. 2005, 89, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, A.H.; de Wit, G.C.; Dam, N.H.T.; Wijnhoven, R.; van Genderen, M.M.; de Boer, J.H. Electroretinogram abnormalities in non-infectious uveitis often persist. Acta Ophthalmol. 2020, 98, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Hettinga, Y.M.; van Genderen, M.M.; Wieringa, W.; Norel, J.O.-V.; de Boer, J.H. Retinal Dystrophy in 6 Young Patients Who Presented with Intermediate Uveitis. Ophthalmology 2016, 123, 2043–2046. [Google Scholar] [CrossRef]
- Verhagen, F.; Kuiper, J.; Nierkens, S.; Imhof, S.M.; Radstake, T.; de Boer, J. Systemic inflammatory immune signatures in a patient with CRB1 linked retinal dystrophy. Expert Rev. Clin. Immunol. 2016, 12, 1359–1362. [Google Scholar] [CrossRef]
- Li, A.S.; Pasricha, M.V.; Mishra, K.; Nguyen, Q.D.; Beres, S.J.; Wood, E.H. CRB1-associated retinal dystrophy presenting as self-resolving opsoclonus and posterior uveitis. Am. J. Ophthalmol. Case Rep. 2022, 26, 101444. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Martinez, A.C.; Marmoy, O.R.; Prise, K.L.; Henderson, R.H.; Thompson, D.A.; Moosajee, M. Expanding the Clinical Spectrum of CRB1-Retinopathies: A Novel Genotype–Phenotype Correlation with Macular Dystrophy and Elevated Intraocular Pressure. Int. J. Mol. Sci. 2025, 26, 2836. https://doi.org/10.3390/ijms26072836
Rodriguez-Martinez AC, Marmoy OR, Prise KL, Henderson RH, Thompson DA, Moosajee M. Expanding the Clinical Spectrum of CRB1-Retinopathies: A Novel Genotype–Phenotype Correlation with Macular Dystrophy and Elevated Intraocular Pressure. International Journal of Molecular Sciences. 2025; 26(7):2836. https://doi.org/10.3390/ijms26072836
Chicago/Turabian StyleRodriguez-Martinez, Ana Catalina, Oliver R. Marmoy, Katrina L. Prise, Robert H. Henderson, Dorothy A. Thompson, and Mariya Moosajee. 2025. "Expanding the Clinical Spectrum of CRB1-Retinopathies: A Novel Genotype–Phenotype Correlation with Macular Dystrophy and Elevated Intraocular Pressure" International Journal of Molecular Sciences 26, no. 7: 2836. https://doi.org/10.3390/ijms26072836
APA StyleRodriguez-Martinez, A. C., Marmoy, O. R., Prise, K. L., Henderson, R. H., Thompson, D. A., & Moosajee, M. (2025). Expanding the Clinical Spectrum of CRB1-Retinopathies: A Novel Genotype–Phenotype Correlation with Macular Dystrophy and Elevated Intraocular Pressure. International Journal of Molecular Sciences, 26(7), 2836. https://doi.org/10.3390/ijms26072836