
Extracellular Vesicles Analysis as Possible Signatures of Antiphospholipid Syndrome Clinical Features


Abstract
1. Introduction
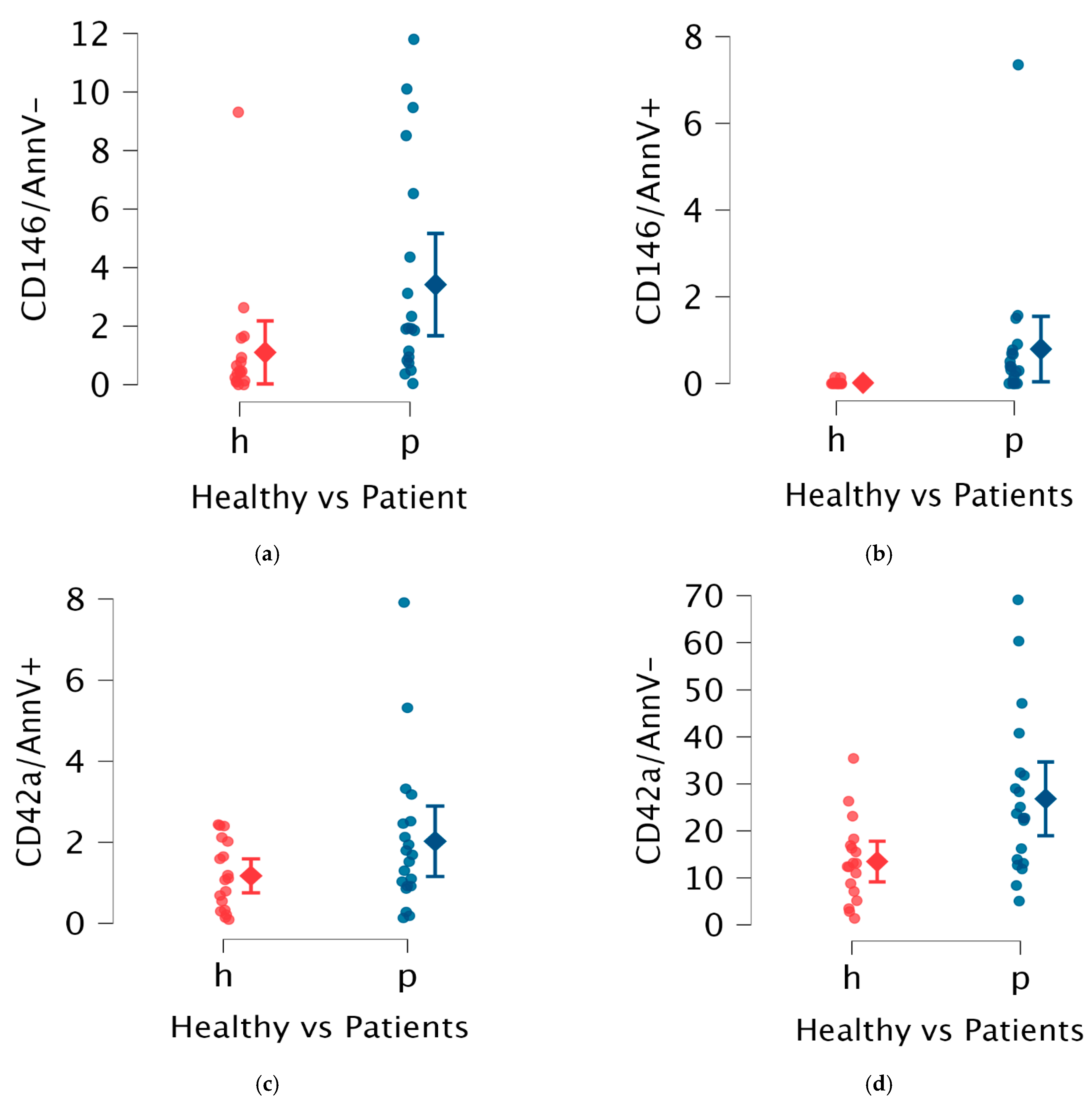
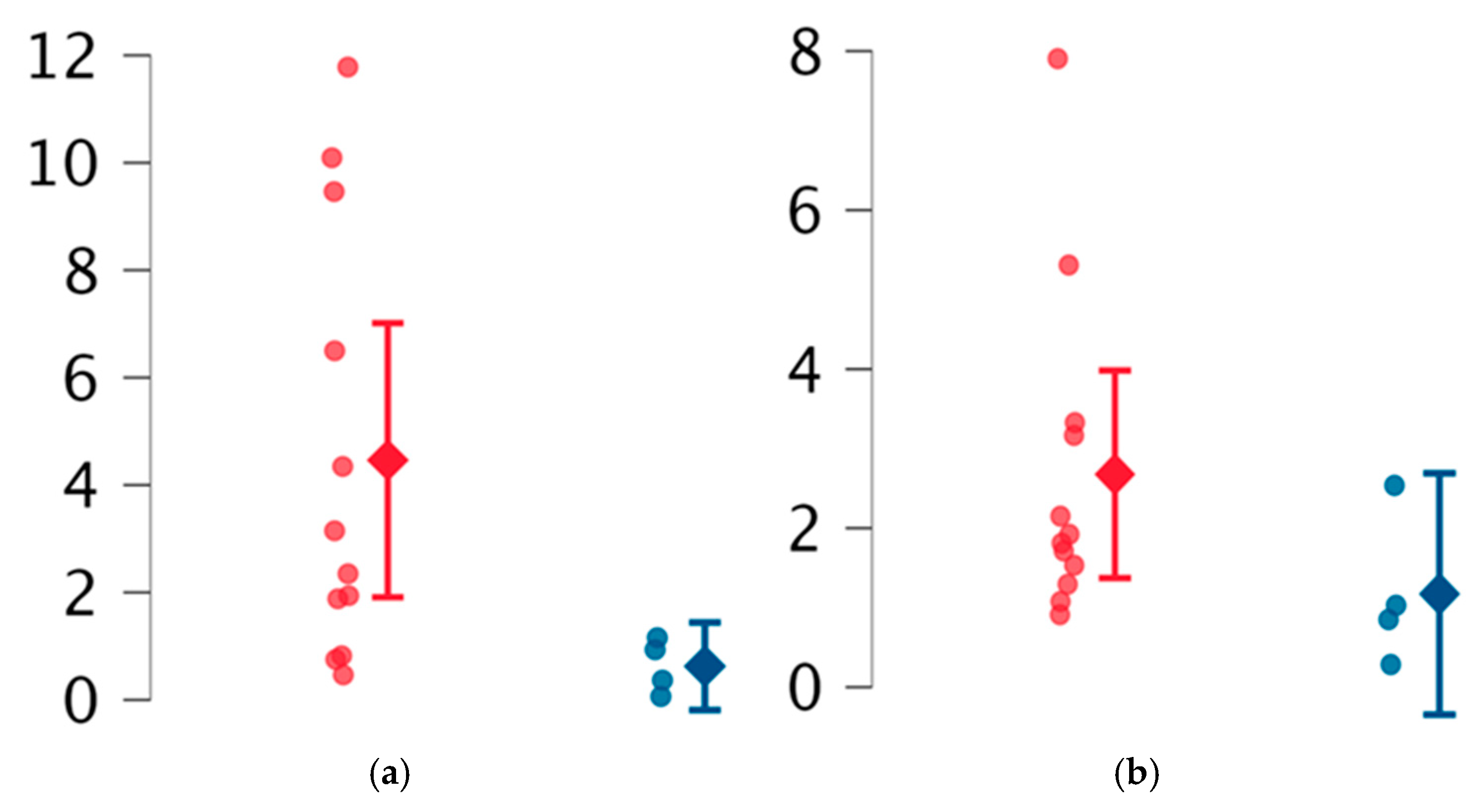
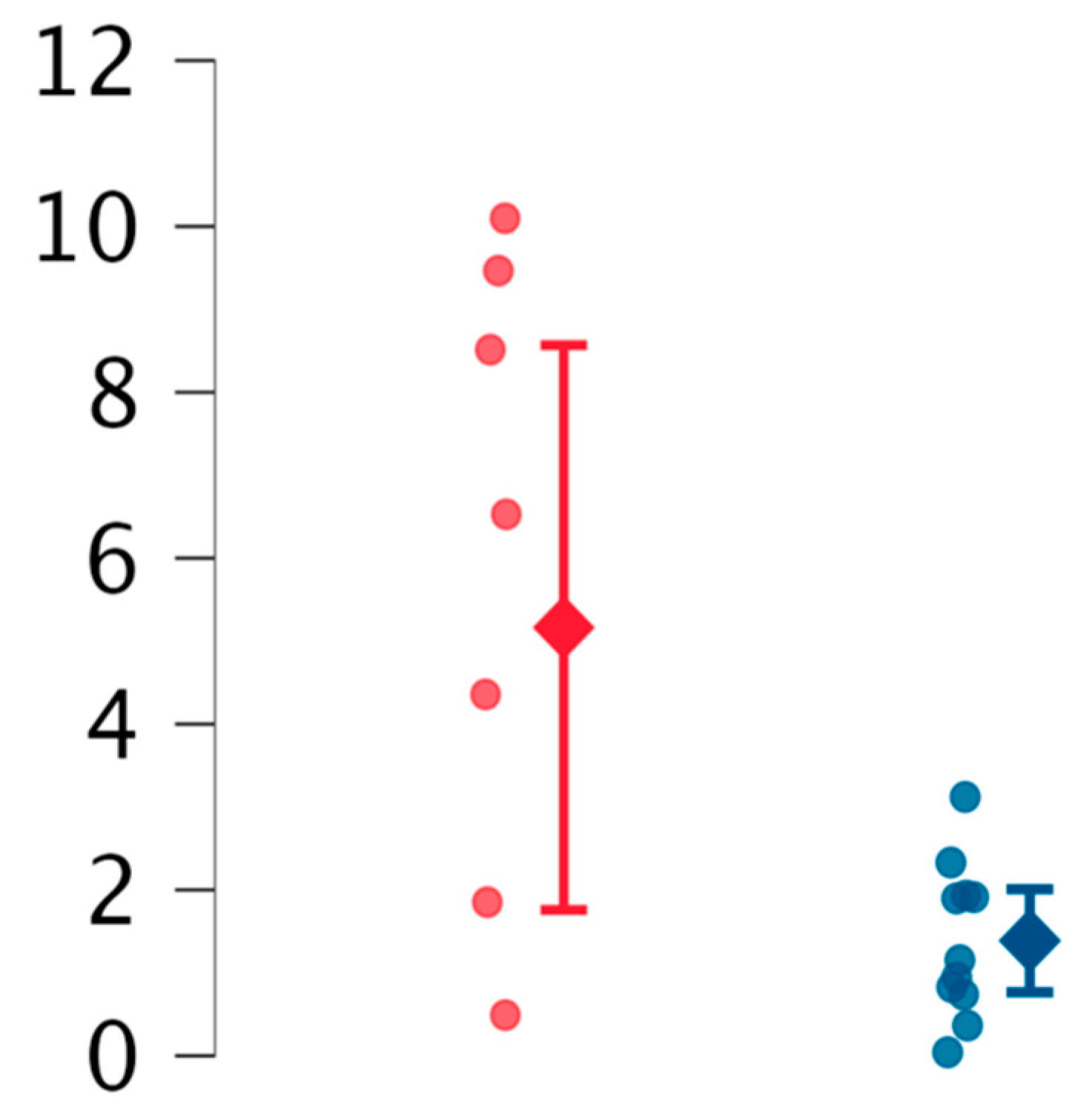
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Flow Cytometric Detection of EVs
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sammaritano, L.R. Antiphospholipid syndrome. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101463. [Google Scholar] [CrossRef]
- Linnemann, B. Antiphospholipid syndrome-an update. Vasa 2018, 47, 451–464. [Google Scholar] [CrossRef]
- Pires da Rosa, G.; Bettencourt, P.; Rodríguez-Pintó, I.; Cervera, R.; Espinosa, G. “Non-criteria” antiphospholipid syndrome: A nomenclature proposal. Autoimmun. Rev. 2020, 19, 102689. [Google Scholar] [CrossRef] [PubMed]
- Sacharidou, A.; Shaul, P.W.; Mineo, C. New Insights in the Pathophysiology of Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2018, 44, 475–482. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.G. The antiphospholipid syndrome finally fathomed? Blood 2018, 131, 2091–2092. [Google Scholar] [CrossRef]
- Giannakopoulos, B.; Krilis, S.A. The Pathogenesis of the Antiphospholipid Syndrome. N. Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Muller-Calleja, N.; Lackner, K.J. Mechanisms of Cellular Activation in the Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2018, 44, 483–492. [Google Scholar] [CrossRef]
- Bradacova, P.; Slavik, L.; Ulehlova, J.; Prochazkova, J.; Hlusi, A.; Manukyan, G.; Kriegova, E. Current Promising Biomarkers and Methods in the Diagnostics of Antiphospholipid Syndrome: A Review. Biomedicines 2021, 9, 166. [Google Scholar] [CrossRef]
- de Laat, B.; Wu, X.X.; van Lummel, M.; Derksen, R.H.W.M.; de Groot, P.G.; Rand, J.H. Correlation between antiphospholipid antibodies that recognize domain I of beta2-glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood 2007, 109, 1490–1494. [Google Scholar] [CrossRef]
- Radic, M.; Pattanaik, D. Cellular and Molecular Mechanisms of Anti-Phospholipid Syndrome. Front. Immunol. 2018, 9, 969. [Google Scholar] [CrossRef]
- Allen, K.L.; Fonseca, F.V.; Betapudi, V.; Willard, B.; Zhang, J.; McCrae, K.R. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2012, 119, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; De Groot, P.G.; Thorpe, P.E.; et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J. Clin. Investig. 2011, 121, 120–131. [Google Scholar] [CrossRef]
- Velasquez, M.; Rojas, M.; Abrahams, V.M.; Escudero, C.; Cadavid, A.P. Mechanisms of Endothelial Dysfunction in Antiphospholipid Syndrome: Association with Clinical Manifestations. Front. Physiol. 2018, 9, 1840. [Google Scholar] [CrossRef] [PubMed]
- Corban, M.T.; Duarte-Garcia, A.; McBane, R.D.; Matteson, E.L.; Lerman, L.O.; Lerman, A. Antiphospholipid Syndrome: Role of Vascular Endothelial Cells and Implications for Risk Stratification and Targeted Therapeutics. J. Am. Coll. Cardiol. 2017, 69, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Alves, J.D.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 2009, 302, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Sacharidou, A.; Chambliss, K.L.; Ulrich, V.; Salmon, J.E.; Shen, Y.-M.; Herz, J.; Hui, D.Y.; Terada, L.S.; Shaul, P.W.; Mineo, C. Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood 2018, 131, 2097–2110. [Google Scholar] [CrossRef]
- Tong, M.; Tsai, B.W.; Chamley, L.W. Antiphospholipid antibodies and extracellular vesicles in pregnancy. Am. J. Reprod. Immunol. 2021, 85, e13312. [Google Scholar] [CrossRef]
- Pericleous, C.; Giles, I.; Rahman, A. Are endothelial microparticles potential markers of vascular dysfunction in the antiphospholipid syndrome? Lupus 2009, 18, 671–675. [Google Scholar] [CrossRef]
- Argentino, G.; Olivieri, B.; Barbieri, A.; Beri, R.; Bason, C.; Friso, S.; Tinazzi, E. Exploring the Utility of Circulating Endothelial Cell-Derived Extracellular Vesicles as Markers of Health and Damage of Vasal Endothelium in Systemic Sclerosis Patients Treated with Iloprost. Biomedicines 2024, 12, 295. [Google Scholar] [CrossRef]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303. [Google Scholar] [CrossRef]
- Forastiero, R.R.; Martinuzzo, M.E.; de Larrañaga, G.F. Circulating levels of tissue factor and proinflammatory cytokines in patients with primary antiphospholipid syndrome or leprosy related antiphospholipid antibodies. Lupus 2005, 14, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Yuan, H.X.; Ou, Z.J.; Ou, J.S. Microparticles (Exosomes) and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Yang, S.H.; Kwon, I.; Lee, O.H.; Heo, J.H. Role of tumour necrosis factor receptor-1 and nuclear factor-κB in production of TNF-α-induced pro-inflammatory microparticles in endothelial cells. Thromb. Haemost. 2017, 112, 580–588. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Sciascia, S.; Amigo, M.C.; Roccatello, D.; Khamashta, M. Diagnosing antiphospholipid syndrome: “extra-criteria” manifestations and technical advances. Nat. Rev. Rheumatol. 2017, 13, 548–560. [Google Scholar] [CrossRef]
- Dignat-George, F.; Camoin-Jau, L.; Sabatier, F.; Anfosso, F.; Bardin, N.; Veit, V.; Combes, V.; Gentile, S.; Moal, V.; Sanmarco, M.; et al. Endothelial microparticles: A potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb. Haemost. 2004, 91, 667–673. [Google Scholar] [CrossRef]
- Štok, U.; Blokar, E.; Lenassi, M.; Frank-Bertoncelj, M.; Erman, A.; Resnik, N.; Sodin-Šemrl, S.; Čučnik, S.; Pirkmajer, K.P. Characterization of Plasma-Derived Small Extracellular Vesicles Indicates Ongoing Endothelial and Platelet Activation in Patients with Thrombotic Antiphospholipid Syndrome. Cells 2020, 9, 1211. [Google Scholar] [CrossRef]
- Álvarez, D.; Rúa, C.; Cadavid, J.Á.P. Microparticles: An Alternative Explanation to the Behavior of Vascular Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2021, 47, 787–799. [Google Scholar] [CrossRef]
- Jy, W.; Tiede, M.; Bidot, C.J.; Horstman, L.L.; Jimenez, J.J.; Chirinos, J.; Ahn, Y.S. Platelet activation rather than endothelial injury identifies risk of thrombosis in subjects positive for antiphospholipid antibodies. Thromb. Res. 2007, 121, 319–325. [Google Scholar] [CrossRef]
- Vikerfors, A.; Mobarrez, F.; Bremme, K.; Holmström, M.; Ågren, A.; Eelde, A.; Bruzelius, M.; Antovic, A.; Wallén, H.; Svenungsson, E. Studies of microparticles in patients with the antiphospholipid syndrome (APS). Lupus 2012, 21, 802–805. [Google Scholar] [CrossRef]
- Combes, V.; Simon, A.C.; Grau, G.E.; Arnoux, D.; Camoin, L.; Sabatier, F.; Mutin, M.; Sanmarco, M.; Sampol, J.; Dignat-George, F. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J. Clin. Investig. 1999, 104, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Alluri, R.; McCrae, K.R. Extracellular Vesicles in the Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2018, 44, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Horstman, L.L.; Ahn, Y.S. Platelet microparticles: A wide-angle perspective. Crit. Rev. Oncol. Hematol. 1999, 30, 111–142. [Google Scholar]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Noulsri, E. Effects of Cell-Derived Microparticles on Immune Cells and Potential Implications in Clinical Medicine. Lab. Med. 2021, 52, 122–135. [Google Scholar] [CrossRef]
- Martinez, M.C.; Andriantsitohaina, R. Microparticles in angiogenesis: Therapeutic potential. Circ. Res. 2011, 109, 110–119. [Google Scholar] [CrossRef]
- Kim, H.K.; Song, K.S.; Chung, J.H.; Lee, K.R.; Lee, S.N. Platelet microparticles induce angiogenesis in vitro. Br. J. Haematol. 2004, 124, 376–384. [Google Scholar] [CrossRef]
- Brill, A.; Dashevsky, O.; Rivo, J.; Gozal, Y.; Varon, D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc. Res. 2005, 67, 30–38. [Google Scholar] [CrossRef]
- Urbanus, R.T.; Pennings, M.T.T.; Derksen, R.H.W.M.; de Groot, P.G. Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J. Thromb. Haemost. 2008, 6, 1405–1412. [Google Scholar] [CrossRef]
- Gasa, N.; Meiring, M. Microparticles: A link to increased thrombin generation. Blood Coagul. Fibrinolysis 2021, 32, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Breen, K.A.; Sanchez, K.; Kirkman, N.; Seed, P.; Parmar, K.; Moore, G.; Hunt, B. Endothelial and platelet microparticles in patients with antiphospholipid antibodies. Thromb. Res. 2015, 135, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Nomura, S.; Kanazawa, S.; Ozaki, Y.; Kagawa, H.; Fukuhara, S. Significance of anti-oxidized LDL antibody and monocyte-derived microparticles in anti-phospholipid antibody syndrome. Autoimmunity 2003, 36, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Radu, C.M.; Campello, E.; Spiezia, L.; Dhima, S.; Visentin, S.; Gavasso, S.; Woodhams, B.; Cosmi, E.; Simioni, P. Origin and levels of circulating microparticles in normal pregnancy: A longitudinal observation in healthy women. Scand. J. Clin. Lab. Investig. 2015, 75, 487–495. [Google Scholar] [CrossRef]
- Barbhaiya, M.; Zuily, S.; Naden, R.; Hendry, A.; Manneville, F.; Amigo, M.; Amoura, Z.; Andrade, D.; Andreoli, L.; Artim-Esen, B.; et al. The 2023 ACR/EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. 2023, 75, 1687–1702. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Cockrell, E.; Espinola, R.G.; Hsi, L.; Fulton, S.; Khan, M.; Li, L.; Fonseca, F.; Kundu, S.; McCrae, K.R. Circulating microparticles in patients with antiphospholipid antibodies: Characterization and associations. Thromb. Res. 2015, 135, 102–108. [Google Scholar] [CrossRef]
- Iversen, L.; Østergaard, O.; Ullman, S.; Nielsen, C.; Halberg, P.; Karlsmark, T.; Heegaard, N.; Jacobsen, S. Circulating microparticles and plasma levels of soluble E- and P-selectins in patients with systemic sclerosis. Scand. J. Rheumatol. 2013, 42, 473–482. [Google Scholar] [CrossRef]
- Rodríguez-Carrio, J.; Alperi-López, M.; López, P.; Alonso-Castro, S.; Carro-Esteban, S.R.; Ballina-Garcia, F.J.; Suárez, A. Altered profile of circulating microparticles in rheumatoid arthritis patients. Clin. Sci. 2015, 128, 437–448. [Google Scholar] [CrossRef]
- Sellam, J.; Proulle, V.; Jüngel, A.; Ittah, M.; Richard, C.M.; Gottenberg, J.-E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.-M.; et al. Increased levels of circulating microparticles in primary Sjögren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 2009, 11, R156. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013, 330, 150–162. [Google Scholar] [CrossRef]
- Deng, F.; Wang, S.; Zhang, L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of circulatory hypoxia-related diseases: A literature review. J. Cell. Mol. Med. 2017, 21, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Baffour, S.; Adjei, J.; Aryeh, C.; Kyeremeh, R.; Kyei, F.; Seidu, M.A. Understanding the biosynthesis of platelets-derived extracellular vesicles. Immun. Inflamm. Dis. 2015, 3, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chyrchel, B.; Drozdz, A.; Dlugosz, D.; Stepien, E.L.; Surdacki, A. Platelet Reactivity And Circulating Platelet-Derived Microvesicles Are Differently Affected By P2Y12 Receptor Antagonists. Int. J. Med. Sci. 2019, 16, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087288. [Google Scholar] [CrossRef]
- Edvardsson, M.; Oweling, M.; Järemo, P. Small procoagulant platelets in diabetes type 2. Thromb. Res. 2020, 195, 1–7. [Google Scholar] [CrossRef]
- Lentz, B.R. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 2003, 42, 423–438. [Google Scholar]
- JASP Team. JASP, Version 0.17.3; JASP Team: 2023 (Amsterdam, The Netherlands). Available online: https://jasp-stats.org/ (accessed on 1 February 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EVs Type | Subgroup | Median EVs | IQR | p Value |
|---|---|---|---|---|
| reEVs | Healthy | 0.44 | 0.77 | Mann–Whitney |
| Patients | 1.88 | 3.46 | 0.01 | |
| aeEVs | Healthy | 0.00 | 0.00 | Mann–Whitney |
| Patients | 0.30 | 1.67 | <0.01 | |
| opEVs | Healthy | 12.75 | 9.21 | Mann–Whitney |
| Patients | 22.70 | 16.63 | <0.01 | |
| ppEVs | Healthy | 1.09 | 1.54 | Mann–Whitney |
| Patients | 1.50 | 1.43 | 0.156 |
| EVs Type | Subgroup | Median EVs | IQR | p Value |
|---|---|---|---|---|
| reEVs | Neurological | 1.50 | 1.72 | Ns |
| Non neurological | 3.14 | 5.72 | ||
| aeEVs | Neurological | 0.39 | 0.65 | Ns |
| Non neurological | 0.29 | 0.48 | ||
| opEVs | Neurological | 32.77 (mean) | 18.68 (SD) | Welch 0.03 |
| Non neurological | 17.89 (mean) | 8.01 (SD) | ||
| ppEVs | Neurological | 1.61 | 1.52 | Ns |
| Non neurological | 1.24 | 2.25 |
| EVs Type | Subgroup | Median EVs | IQR | p Value |
|---|---|---|---|---|
| reEVs | Obstetric | 0.66 | 0.71 | Mann–Whitney |
| Non obstetric | 2.73 | 5.67 | 0.03 | |
| aeEVs | Obstetric | 0.04 | 0.23 | Ns |
| Non obstetric | 0.54 | 0.79 | ||
| opEVs | Obstetric | 25.50 | 10.18 | Ns |
| Non obstetric | 27.25 | 26.75 | ||
| ppEVs | Obstetric | 0.94 | 0.69 | Ns |
| Non obstetric | 1.87 | 1.75 |
| EVs Type | Subgroup | Median EVs | IQR | p Value |
|---|---|---|---|---|
| reEVs | ECD suspected | 1.39 (mean) | 0.93 (SD) | Welch |
| EC unaffected | 5.16 (mean) | 4.07 (SD) | 0.03 | |
| aeEVs | ECD suspected | 0.39 | 0.65 | Ns |
| EC unaffected | 0.30 | 0.39 | ||
| opEVs | ECD suspected | 30.49 (mean) | 21.51 (SD) | Ns |
| EC unaffected | 21.16 (mean) | 6.48 (SD) | ||
| ppEVs | ECD suspected | 1.52 | 1.35 | Ns |
| EC unaffected | 1.40 | 1.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonisoli, G.L.; Argentino, G.; Friso, S.; Tinazzi, E. Extracellular Vesicles Analysis as Possible Signatures of Antiphospholipid Syndrome Clinical Features. Int. J. Mol. Sci. 2025, 26, 2834. https://doi.org/10.3390/ijms26072834
Bonisoli GL, Argentino G, Friso S, Tinazzi E. Extracellular Vesicles Analysis as Possible Signatures of Antiphospholipid Syndrome Clinical Features. International Journal of Molecular Sciences. 2025; 26(7):2834. https://doi.org/10.3390/ijms26072834
Chicago/Turabian StyleBonisoli, Giulio Luigi, Giuseppe Argentino, Simonetta Friso, and Elisa Tinazzi. 2025. "Extracellular Vesicles Analysis as Possible Signatures of Antiphospholipid Syndrome Clinical Features" International Journal of Molecular Sciences 26, no. 7: 2834. https://doi.org/10.3390/ijms26072834
APA StyleBonisoli, G. L., Argentino, G., Friso, S., & Tinazzi, E. (2025). Extracellular Vesicles Analysis as Possible Signatures of Antiphospholipid Syndrome Clinical Features. International Journal of Molecular Sciences, 26(7), 2834. https://doi.org/10.3390/ijms26072834