

A First-in-Class Dual Degrader of Bcl-2/Bcl-xL Reverses HIV Latency and Minimizes Ex Vivo Reservoirs from Patients

 ,
, 
Abstract

1. Introduction
2. Results
2.1. Evaluation of BCL-2/BCL-XL Antagonists for Reactivating and Selective Killing of HIV from Latency
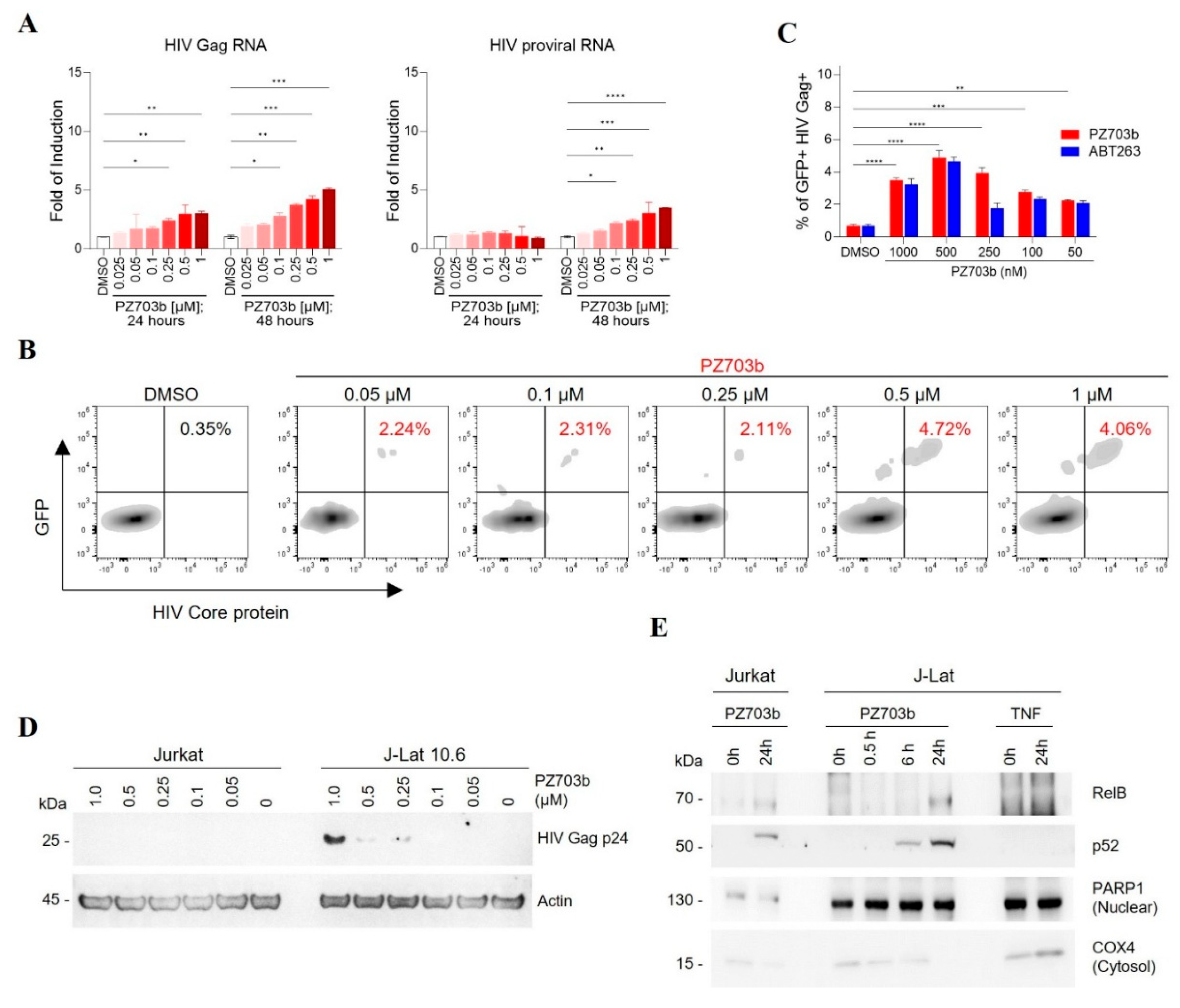
2.2. PZ703b Reactivates Latent HIV Through the Non-Canonical NF-kB Signaling Pathway
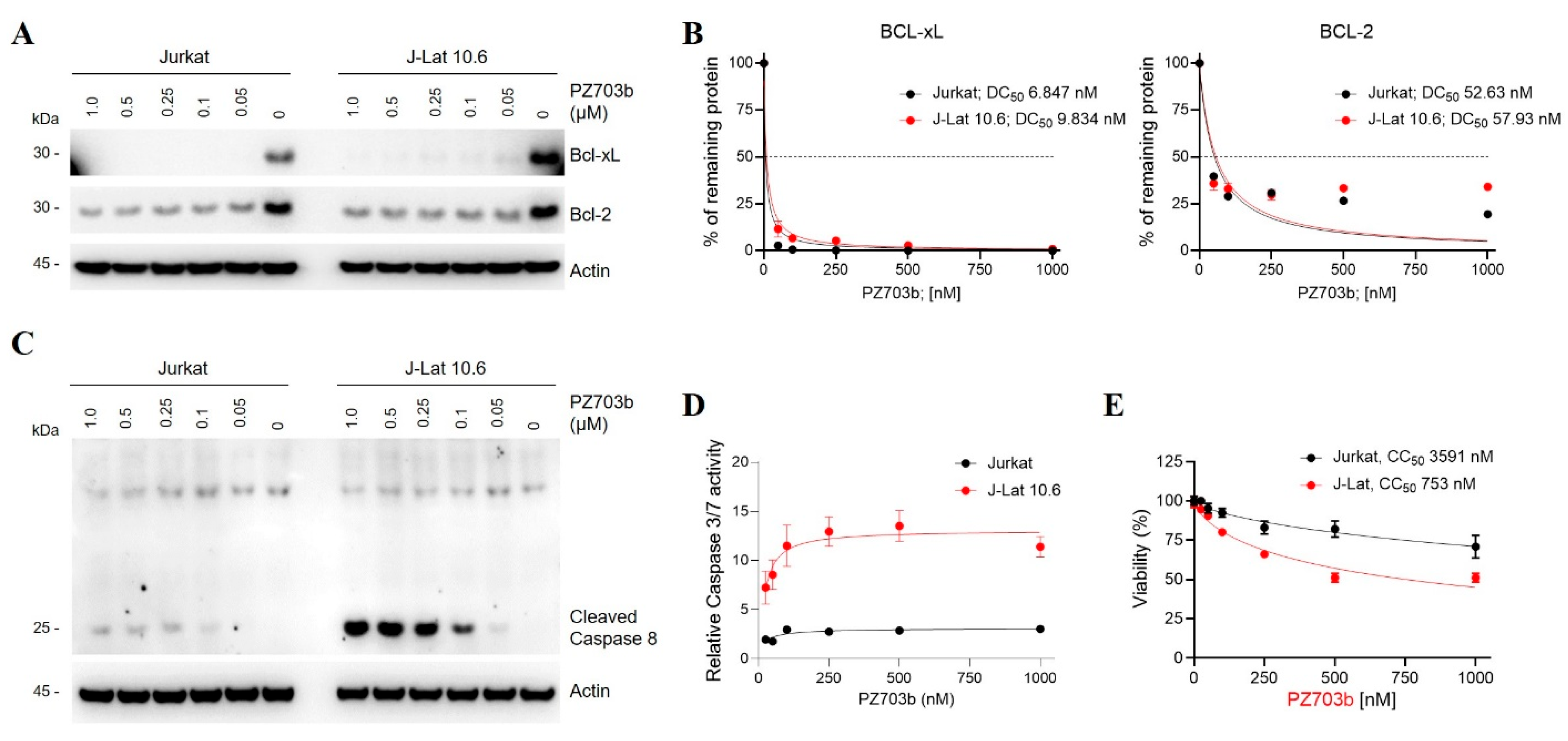
2.3. Mechanisms of Selective Killing of HIV-Latently Infected Cells by PZ703b
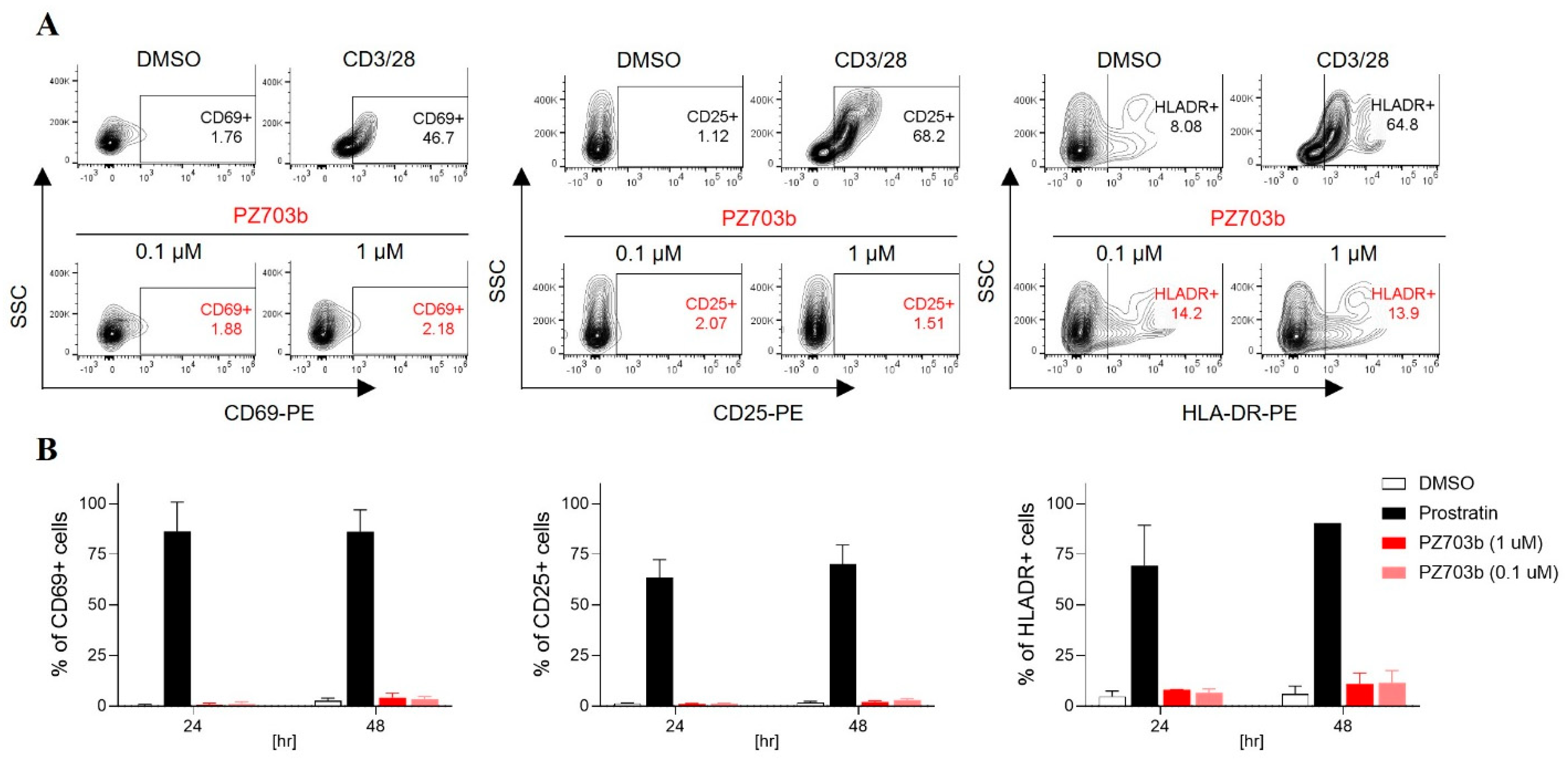
2.4. PZ703b Does Not Lead to Global Activation of Primary T Cells
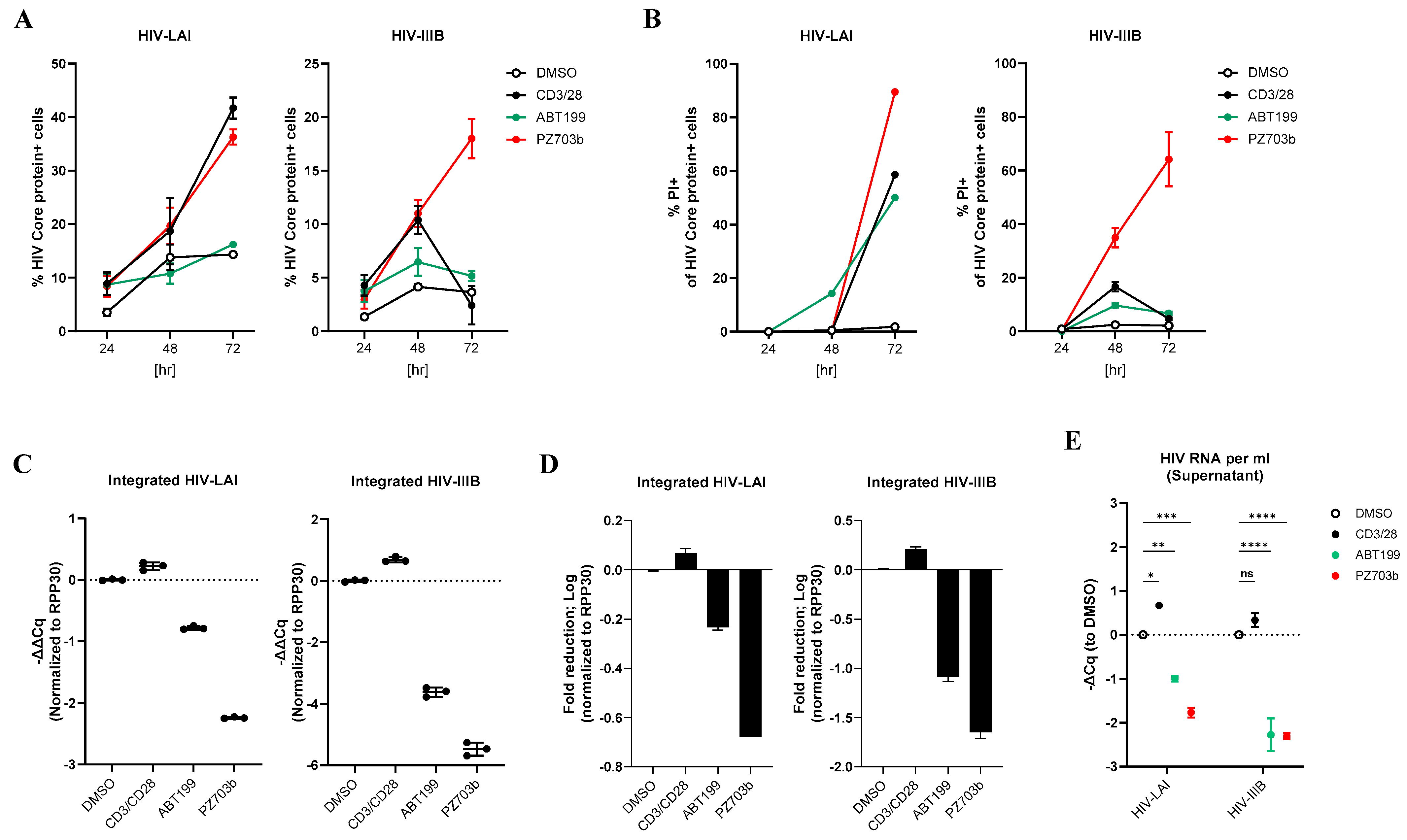
2.5. PZ703b Reverses Latency and Selectively Kills Reactivating Cells in the Central Memory T Cell (TCM) Model of HIV-1 Latency
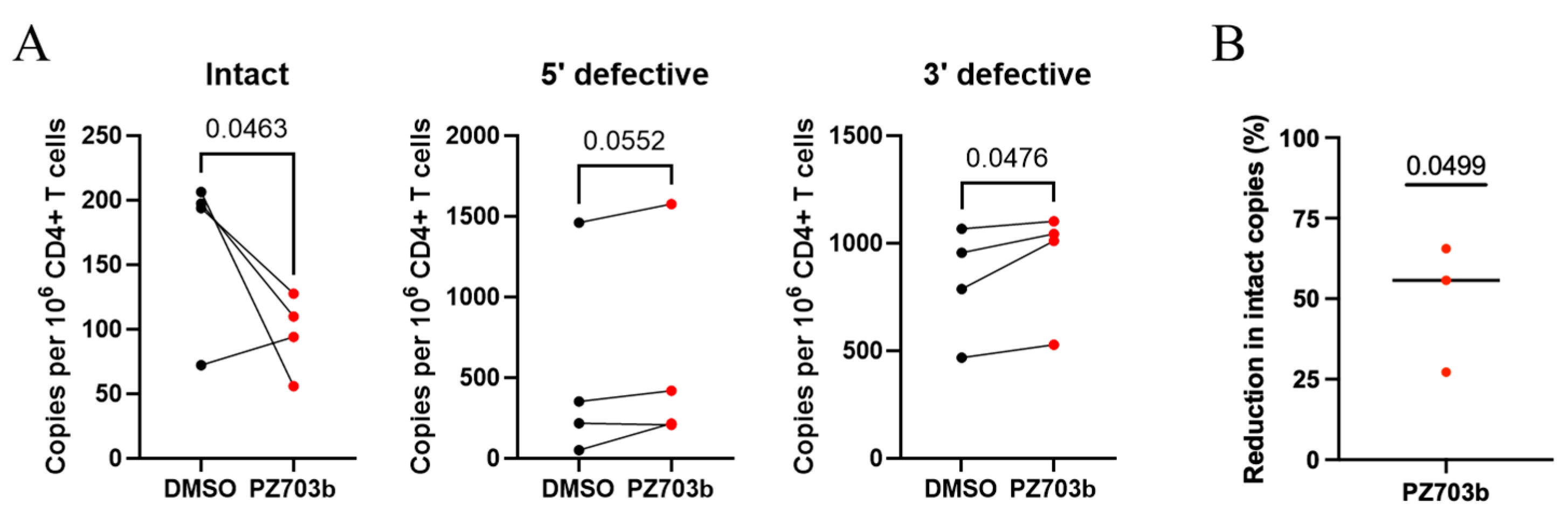
2.6. Ex Vivo Treatment with PZ703b Reduces HIV-1 Reservoir
3. Discussion
4. Materials and Methods
4.1. Cell Line Culture
4.2. Human Cell Isolation and Culture
4.3. In Vitro Cell Line Model for Screening BCL-2/BCL-XL Antagonists
4.4. Measurement of Supernatant HIV-RNA by TaqMan PCR
4.5. Protein Expression Analysis by Immunoblotting
4.6. Cytotoxicity MTS Assay
4.7. Measurement of Caspase Activity
4.8. Flow Cytometry
4.9. Generated and Cultured Central Memory CD4+ Primary T Cells
4.10. Infection of Central Memory CD4+ T Cells to Establish Latency
4.11. Analysis of Activation Markers in Human Primary T Cells
4.12. Measurement of Total Cell-Associated Integrated HIV Genome
4.13. Ex Vivo Elimination of Latent Reservoir
4.14. IPDA® Assay
4.15. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Board, N.L.; Moskovljevic, M.; Wu, F.; Siliciano, R.F.; Siliciano, J.D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 2022, 22, 499–512. [Google Scholar] [CrossRef]
- Collins, D.R.; Gaiha, G.D.; Walker, B.D. CD8(+) T cells in HIV control, cure and prevention. Nat. Rev. Immunol. 2020, 20, 471–482. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Aillet, F.; Masutani, H.; Elbim, C.; Raoul, H.; Chene, L.; Nugeyre, M.T.; Paya, C.; Barre-Sinoussi, F.; Gougerot-Pocidalo, M.A.; Israel, N. Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4(+) T- or monocytic cell lines. J. Virol. 1998, 72, 9698–9705. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, S.H.; Macedo, A.B.; Ward, A.R.; Alberto, W.D.C.; Klevorn, T.; Leyre, L.; Copertino, D.C.; Mota, T.M.; Chan, D.; et al. Selective BCL-X(L) Antagonists Eliminate Infected Cells from a Primary-Cell Model of HIV Latency but Not from Ex Vivo Reservoirs. J. Virol. 2021, 95, e0242520. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, S.H.; Patel, S.; Alberto, W.D.C.; Magat, D.; Ahimovic, D.; Macedo, A.B.; Durga, R.; Chan, D.; Zale, E.; et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J. Clin. Investig. 2020, 130, 2542–2559. [Google Scholar] [CrossRef]
- Li, M.; Liu, W.; Bauch, T.; Graviss, E.A.; Arduino, R.C.; Kimata, J.T.; Chen, M.; Wang, J. Clearance of HIV infection by selective elimination of host cells capable of producing HIV. Nat. Commun. 2020, 11, 4051. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug. Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Thummuri, D.; Lv, D.; Liu, X.; Zhang, P.; Hu, W.; Poddar, S.K.; Hua, N.; Khan, S.; Yuan, Y.; et al. Discovery of a Novel BCL-X(L) PROTAC Degrader with Enhanced BCL-2 Inhibition. J. Med. Chem. 2021, 64, 14230–14246. [Google Scholar] [CrossRef]
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [PubMed]
- Sang, X.; Wang, J.; Zhou, J.; Xu, Y.; An, J.; Warshel, A.; Huang, Z. A Chemical Strategy for the Degradation of the Main Protease of SARS-CoV-2 in Cells. J. Am. Chem. Soc. 2023, 145, 27248–27253. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Chavez, R.M.; Salerno, A.; Liuzzi, A.; Ilari, A.; Milelli, A.; Uliassi, E.; Bolognesi, M.L. Targeted Protein Degradation for Infectious Diseases: From Basic Biology to Drug Discovery. ACS Bio Med. Chem. Au 2023, 3, 32–45. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, J.; Pang, X.; Liu, Z.; Li, Q.; Yi, D.; Zhang, Y.; Fang, X.; Zhang, T.; Zhou, R.; et al. An anti-influenza A virus microbial metabolite acts by degrading viral endonuclease PA. Nat. Commun. 2022, 13, 2079. [Google Scholar] [CrossRef]
- Zhao, N.; Ho, J.S.Y.; Meng, F.; Zheng, S.; Kurland, A.P.; Tian, L.; Rea-Moreno, M.; Song, X.; Seo, J.S.; Kaniskan, H.U.; et al. Generation of host-directed and virus-specific antivirals using targeted protein degradation promoted by small molecules and viral RNA mimics. Cell Host Microbe 2023, 31, 1154–1169. [Google Scholar] [CrossRef]
- Emert-Sedlak, L.A.; Tice, C.M.; Shi, H.; Alvarado, J.J.; Shu, S.T.; Reitz, A.B.; Smithgall, T.E. PROTAC-mediated degradation of HIV-1 Nef efficiently restores cell-surface CD4 and MHC-I expression and blocks HIV-1 replication. Cell Chem. Biol. 2024, 31, 658–668. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- Zhou, C.; Li, T.; Xia, M.; Wu, Z.; Zhong, X.; Li, A.; Rashid, H.K.; Ma, C.; Zhou, R.; Duan, H.; et al. Bcl-2 Antagonist Obatoclax Reactivates Latent HIV-1 via the NF-kappaB Pathway and Induces Latent Reservoir Cell Apoptosis in Latently Infected Cells. ACS Infect. Dis. 2023, 9, 2105–2118. [Google Scholar] [CrossRef]
- Badrichani, A.Z.; Stroka, D.M.; Bilbao, G.; Curiel, D.T.; Bach, F.H.; Ferran, C. Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kappaB. J. Clin. Investig. 1999, 103, 543–553. [Google Scholar] [CrossRef]
- Grimm, S.; Bauer, M.K.; Baeuerle, P.A.; Schulze-Osthoff, K. Bcl-2 down-regulates the activity of transcription factor NF-kappaB induced upon apoptosis. J. Cell Biol. 1996, 134, 13–23. [Google Scholar] [CrossRef]
- Rodari, A.; Darcis, G.; Van Lint, C.M. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu. Rev. Virol. 2021, 8, 491–514. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Koch, R.; Budamagunta, V.; Zhang, P.; Zhang, X.; Khan, S.; Thummuri, D.; Ortiz, Y.T.; Zhang, X.; Lv, D.; et al. DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 2020, 13, 95. [Google Scholar] [CrossRef]
- Nayak, D.; Lv, D.; Yuan, Y.; Zhang, P.; Hu, W.; Nayak, A.; Ruben, E.A.; Lv, Z.; Sung, P.; Hromas, R.; et al. Development and crystal structures of a potent second-generation dual degrader of BCL-2 and BCL-xL. Nat. Commun. 2024, 15, 2743. [Google Scholar] [CrossRef]
- Hashimoto, F.; Oyaizu, N.; Kalyanaraman, V.S.; Pahwa, S. Modulation of Bcl-2 protein by CD4 cross-linking: A possible mechanism for lymphocyte apoptosis in human immunodeficiency virus infection and for rescue of apoptosis by interleukin-2. Blood 1997, 90, 745–753. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. NF-kappaB and p53 are the dominant apoptosis-inducing transcription factors elicited by the HIV-1 envelope. J. Exp. Med. 2004, 199, 629–640. [Google Scholar] [CrossRef]
- Sainski, A.M.; Natesampillai, S.; Cummins, N.W.; Bren, G.D.; Taylor, J.; Saenz, D.T.; Poeschla, E.M.; Badley, A.D. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J. Virol. 2011, 85, 7965–7975. [Google Scholar] [CrossRef]
- Natesampillai, S.; Cummins, N.W.; Nie, Z.; Sampath, R.; Baker, J.V.; Henry, K.; Pinzone, M.; O’Doherty, U.; Polley, E.C.; Bren, G.D.; et al. HIV Protease-Generated Casp8p41, When Bound and Inactivated by Bcl2, Is Degraded by the Proteasome. J. Virol. 2018, 92, e00037-18. [Google Scholar] [CrossRef]
- Balestrieri, E.; Grelli, S.; Matteucci, C.; Minutolo, A.; d’Ettorre, G.; Di Sora, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptosis-associated gene expression in HIV-infected patients in response to successful antiretroviral therapy. J. Med. Virol. 2007, 79, 111–117. [Google Scholar] [CrossRef]
- Akari, H.; Bour, S.; Kao, S.; Adachi, A.; Strebel, K. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J. Exp. Med. 2001, 194, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.J.; Bonczkowski, P.; Spivak, A.M.; De Spiegelaere, W.; Novis, C.L.; DePaula-Silva, A.B.; Malatinkova, E.; Trypsteen, W.; Bosque, A.; Vanderkerckhove, L.; et al. Modeling HIV-1 Latency in Primary T Cells Using a Replication-Competent Virus. AIDS Res. Hum. Retroviruses 2016, 32, 187–193. [Google Scholar] [CrossRef]
- Vandergeeten, C.; Fromentin, R.; Merlini, E.; Lawani, M.B.; DaFonseca, S.; Bakeman, W.; McNulty, A.; Ramgopal, M.; Michael, N.; Kim, J.H.; et al. Cross-clade ultrasensitive PCR-based assays to measure HIV persistence in large-cohort studies. J. Virol. 2014, 88, 12385–12396. [Google Scholar] [CrossRef]
- Konvalinka, J.; Krausslich, H.G.; Muller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Kuang, X.T.; Brockman, M.A. Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses 2018, 10, 677. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses 2017, 9, 237. [Google Scholar] [CrossRef]
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK2. Retrovirology 2015, 12, 74. [Google Scholar] [CrossRef]
- Impens, F.; Timmerman, E.; Staes, A.; Moens, K.; Arien, K.K.; Verhasselt, B.; Vandekerckhove, J.; Gevaert, K. A catalogue of putative HIV-1 protease host cell substrates. Biol. Chem. 2012, 393, 915–931. [Google Scholar] [CrossRef]
- Rumlova, M.; Krizova, I.; Keprova, A.; Hadravova, R.; Dolezal, M.; Strohalmova, K.; Pichova, I.; Hajek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Centazzo, M.; Manganaro, L.; Alvisi, G. Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses 2023, 15, 712. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.; Richman, D.; Siliciano, R.F.; Nussenzweig, M.C.; Howell, B.J.; Martinez-Picado, J.; Chomont, N.; Bar, K.J.; Yu, X.G.; Lichterfeld, M.; et al. Recommendations for measuring HIV reservoir size in cure-directed clinical trials. Nat. Med. 2020, 26, 1339–1350. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Board, N.L.; Yuan, Z.; Wu, F.; Moskovljevic, M.; Ravi, M.; Sengupta, S.; Mun, S.S.; Simonetti, F.R.; Lai, J.; Tebas, P.; et al. Bispecific antibodies promote natural killer cell-mediated elimination of HIV-1 reservoir cells. Nat. Immunol. 2024, 25, 462–470. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Lopez-Huertas, M.R.; Palladino, C.; Garrido-Arquero, M.; Esteban-Cartelle, B.; Sanchez-Carrillo, M.; Martinez-Roman, P.; Martin-Carbonero, L.; Ryan, P.; Dominguez-Dominguez, L.; Santos, I.L.; et al. HCV-coinfection is related to an increased HIV-1 reservoir size in cART-treated HIV patients: A cross-sectional study. Sci. Rep. 2019, 9, 5606. [Google Scholar] [CrossRef]
- Korner, C.; Kramer, B.; Schulte, D.; Coenen, M.; Mauss, S.; Fatkenheuer, G.; Oldenburg, J.; Nattermann, J.; Rockstroh, J.K.; Spengler, U. Effects of HCV co-infection on apoptosis of CD4+ T-cells in HIV-positive patients. Clin. Sci. 2009, 116, 861–870. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Cillo, A.R.; Sobolewski, M.D.; Bosch, R.J.; Fyne, E.; Piatak, M., Jr.; Coffin, J.M.; Mellors, J.W. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 7078–7083. [Google Scholar] [CrossRef]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef]
- Rao, S.; Lungu, C.; Crespo, R.; Steijaert, T.H.; Gorska, A.; Palstra, R.J.; Prins, H.A.B.; van Ijcken, W.; Mueller, Y.M.; van Kampen, J.J.A.; et al. Selective cell death in HIV-1-infected cells by DDX3 inhibitors leads to depletion of the inducible reservoir. Nat. Commun. 2021, 12, 2475. [Google Scholar] [CrossRef]
- Yin, M.T.; Zhang, C.A.; McMahon, D.J.; Ferris, D.C.; Irani, D.; Colon, I.; Cremers, S.; Shane, E. Higher rates of bone loss in postmenopausal HIV-infected women: A longitudinal study. J. Clin. Endocrinol. Metab. 2012, 97, 554–562. [Google Scholar] [CrossRef]
- Yin, M.T.; Bucovsky, M.; Williams, J.; Brunjes, D.; RoyChoudhury, A.; Colon, I.; Ferris, D.C.; Olender, S.; Schulze, P.C.; Sharma, A.; et al. Effect of vitamin D(3) and calcium carbonate supplementation on muscle strength in postmenopausal women living with HIV. Antivir. Ther. 2020, 25, 411–418. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age | Sex | HCV | Height (cm) | Weight (kg) | HIV Regimen | CD4 T Cells | Viral Load |
|---|---|---|---|---|---|---|---|---|
| 1 | 49 | Female | + | 167.9 | 105 | Abacavir, lamivudine, darunavir/ritonavir | 953 | <50 |
| 2 | 57 | Female | + | 157.5 | 79.4 | Tenofovir disoproxil fumarate, nelfinavir | 794 | <50 |
| 3 | 65 | Female | + | 147.6 | 56.5 | Tenofovir disoproxil fumarate, lamivudine, lopinavir/ritonavir saquinavir | 1144 | <50 |
| 4 | 57 | Female | + | 157.5 | 79.4 | Tenofovir, lamivudine, nelfinavir | 794 | <50 |
| 5 | 47 | Female | - | 171.7 | 87.5 | darunavir/ritonavir | 750 | <50 |
| 6 | 48 | Female | - | 168.15 | 75 | darunavir/ritonavir | 731 | <50 |
| 7 | 64 | Female | - | 152.4 | 52.6 | lopinavir/ritonavir | 746 | <50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, L.-C.; Yin, M.T.; Laird, G.M.; Ritter, K.D.; Shah, J.G.; Debnath, A.K. A First-in-Class Dual Degrader of Bcl-2/Bcl-xL Reverses HIV Latency and Minimizes Ex Vivo Reservoirs from Patients. Int. J. Mol. Sci. 2025, 26, 2772. https://doi.org/10.3390/ijms26062772
Chang L-C, Yin MT, Laird GM, Ritter KD, Shah JG, Debnath AK. A First-in-Class Dual Degrader of Bcl-2/Bcl-xL Reverses HIV Latency and Minimizes Ex Vivo Reservoirs from Patients. International Journal of Molecular Sciences. 2025; 26(6):2772. https://doi.org/10.3390/ijms26062772
Chicago/Turabian StyleChang, Lin-Chun, Michael T. Yin, Gregory M. Laird, Kristen D. Ritter, Jayesh G. Shah, and Asim K. Debnath. 2025. "A First-in-Class Dual Degrader of Bcl-2/Bcl-xL Reverses HIV Latency and Minimizes Ex Vivo Reservoirs from Patients" International Journal of Molecular Sciences 26, no. 6: 2772. https://doi.org/10.3390/ijms26062772
APA StyleChang, L.-C., Yin, M. T., Laird, G. M., Ritter, K. D., Shah, J. G., & Debnath, A. K. (2025). A First-in-Class Dual Degrader of Bcl-2/Bcl-xL Reverses HIV Latency and Minimizes Ex Vivo Reservoirs from Patients. International Journal of Molecular Sciences, 26(6), 2772. https://doi.org/10.3390/ijms26062772