
_Kim.png)
Role of PARP Inhibitors: A New Hope for Breast Cancer Therapy


Abstract

1. Introduction
2. The BRCA1 Gene
3. The BRCA2 Gene
4. The Interaction Between BRCA1 and BRCA2 Genes
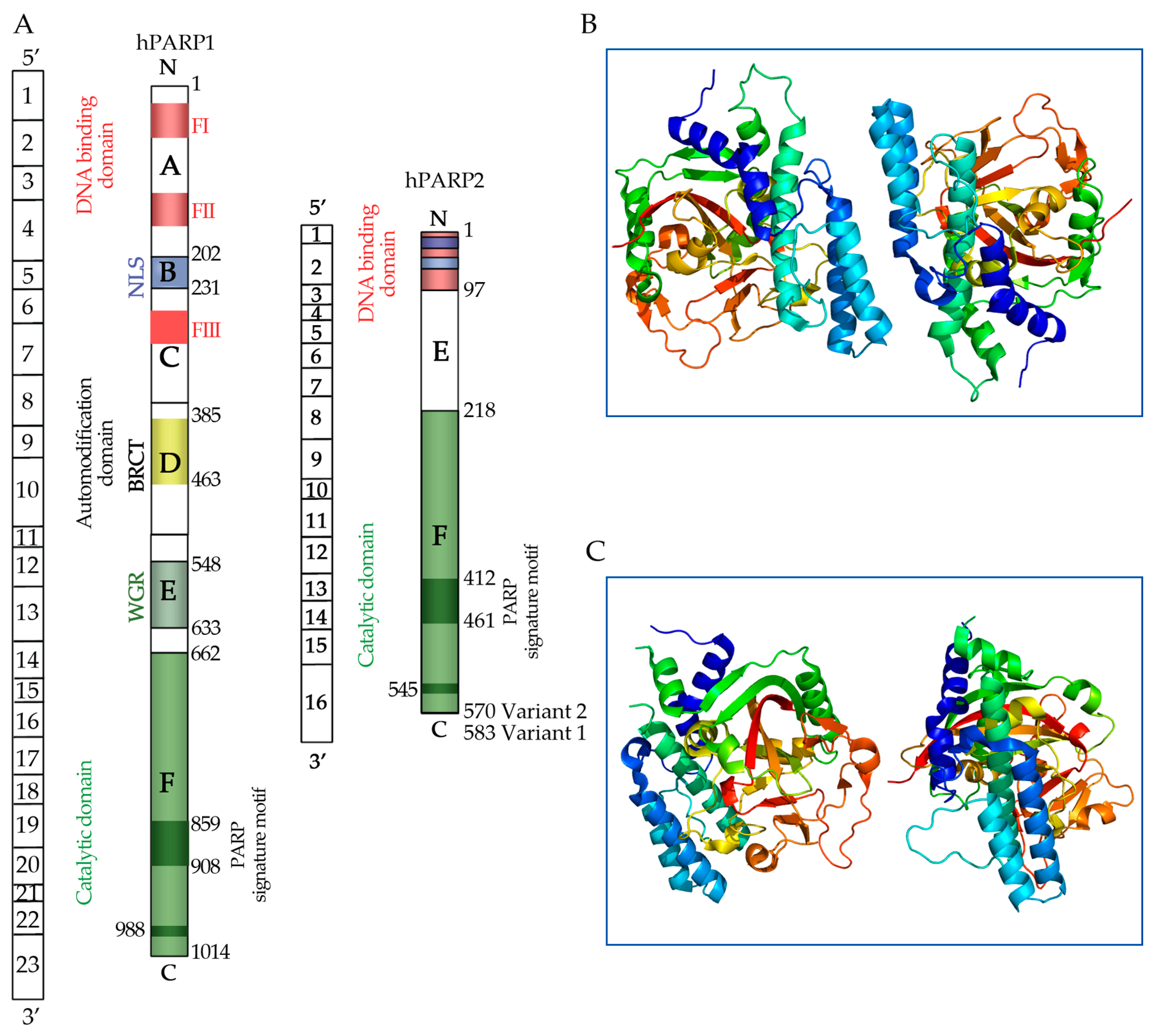
5. Poly (ADP-Ribose) Polymerase (PARP) Molecules
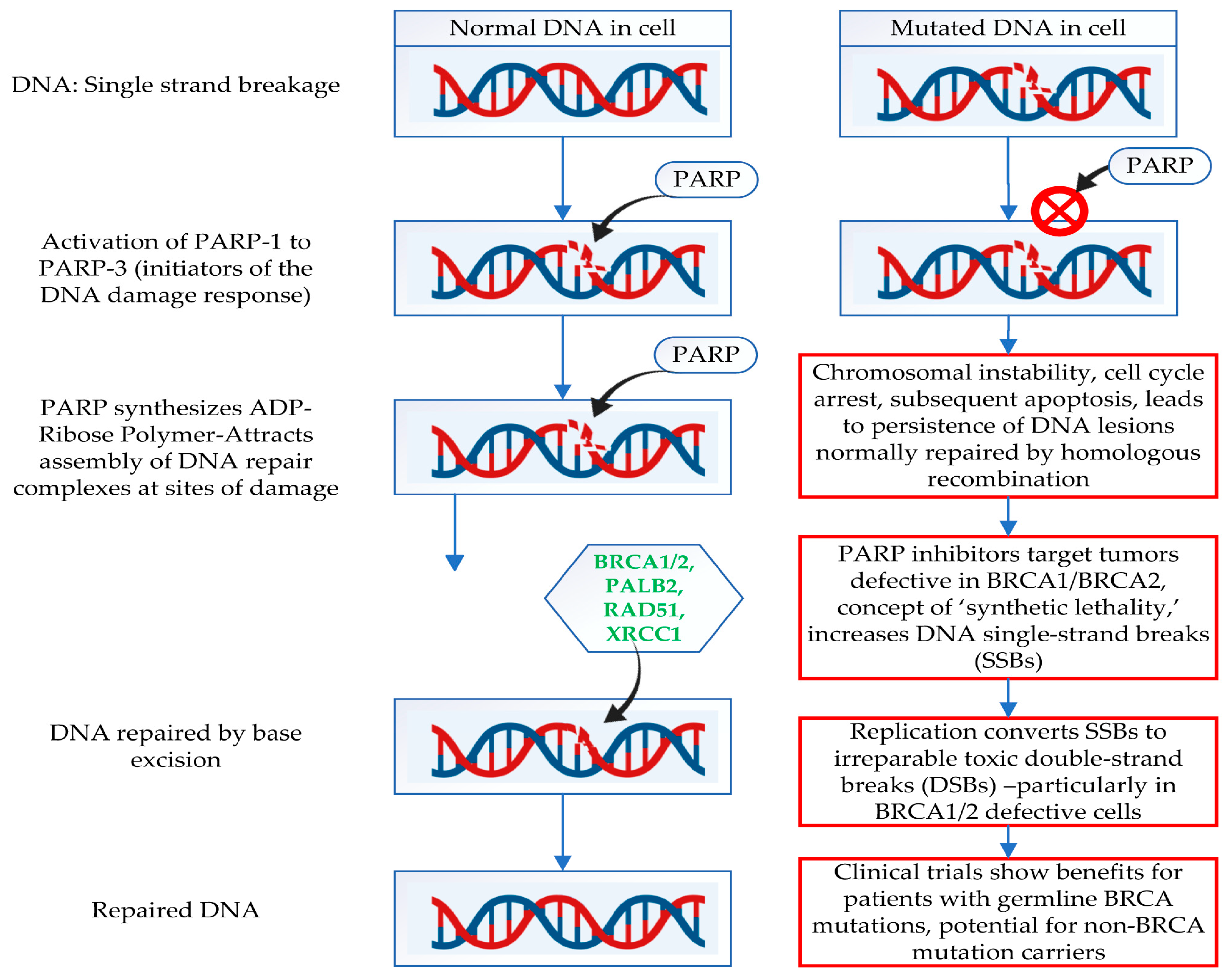
6. Mechanism of Action of PARP Inhibitors
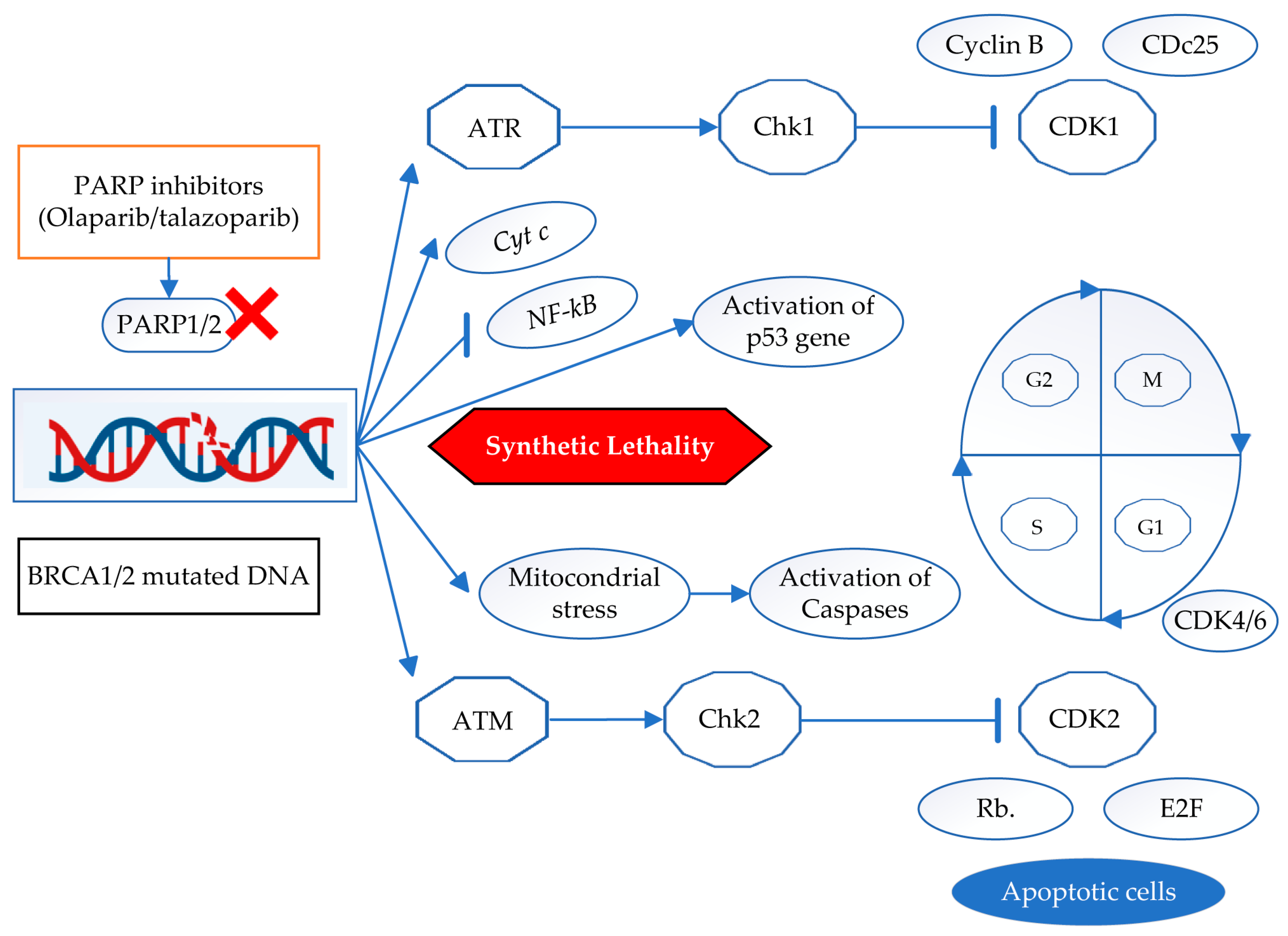
7. Cell Signaling Pathways of PARP Inhibitors
8. Clinical Implications
9. Bioinformatic Analysis of Clinical Datasets
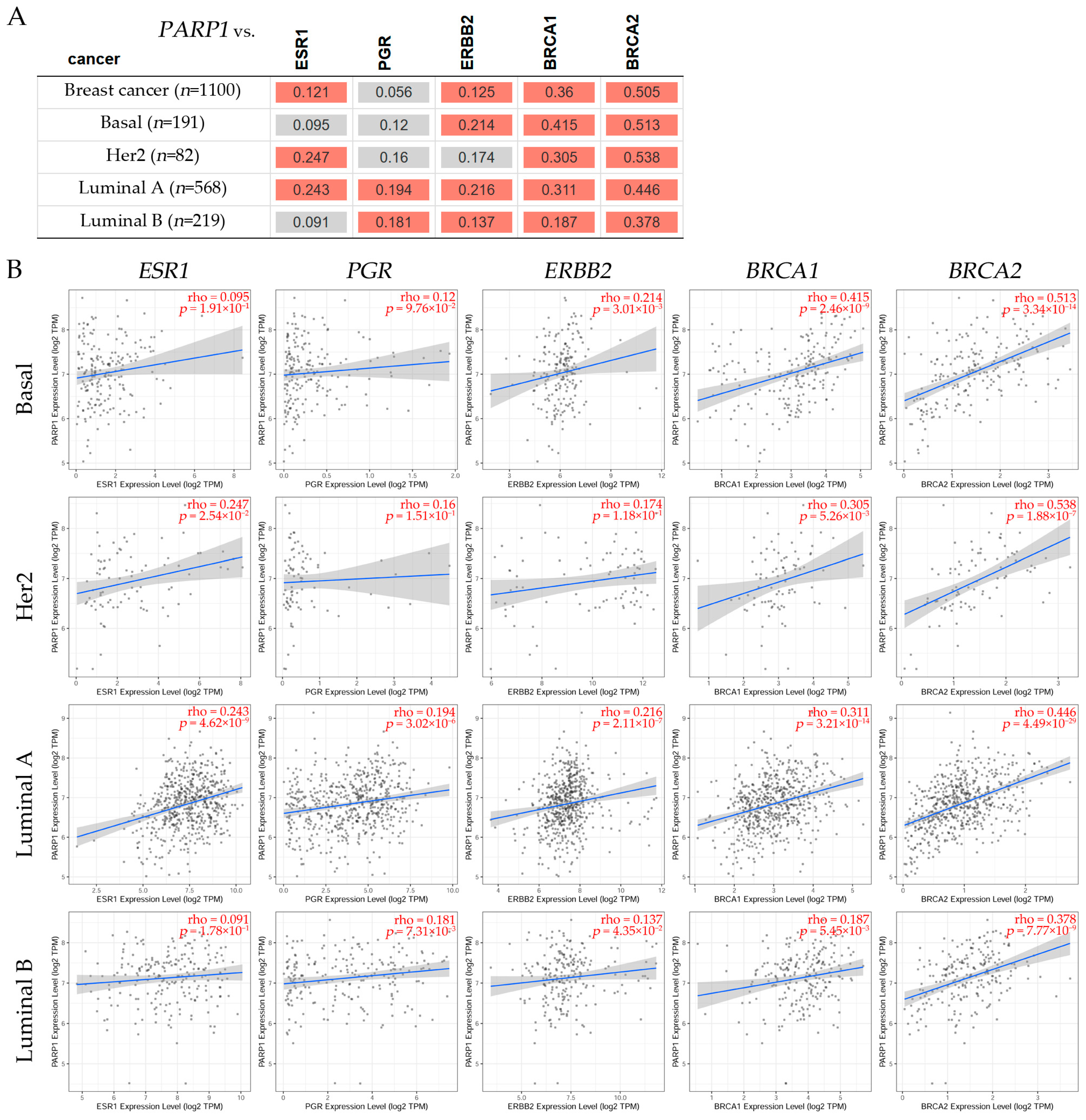
9.1. Correlation Analysis Between PARP1/2 and Genes Such as ESR1, PGR, ERBB2, BRCA1, and BRCA2 Expression Levels in Breast Cancer Subtypes
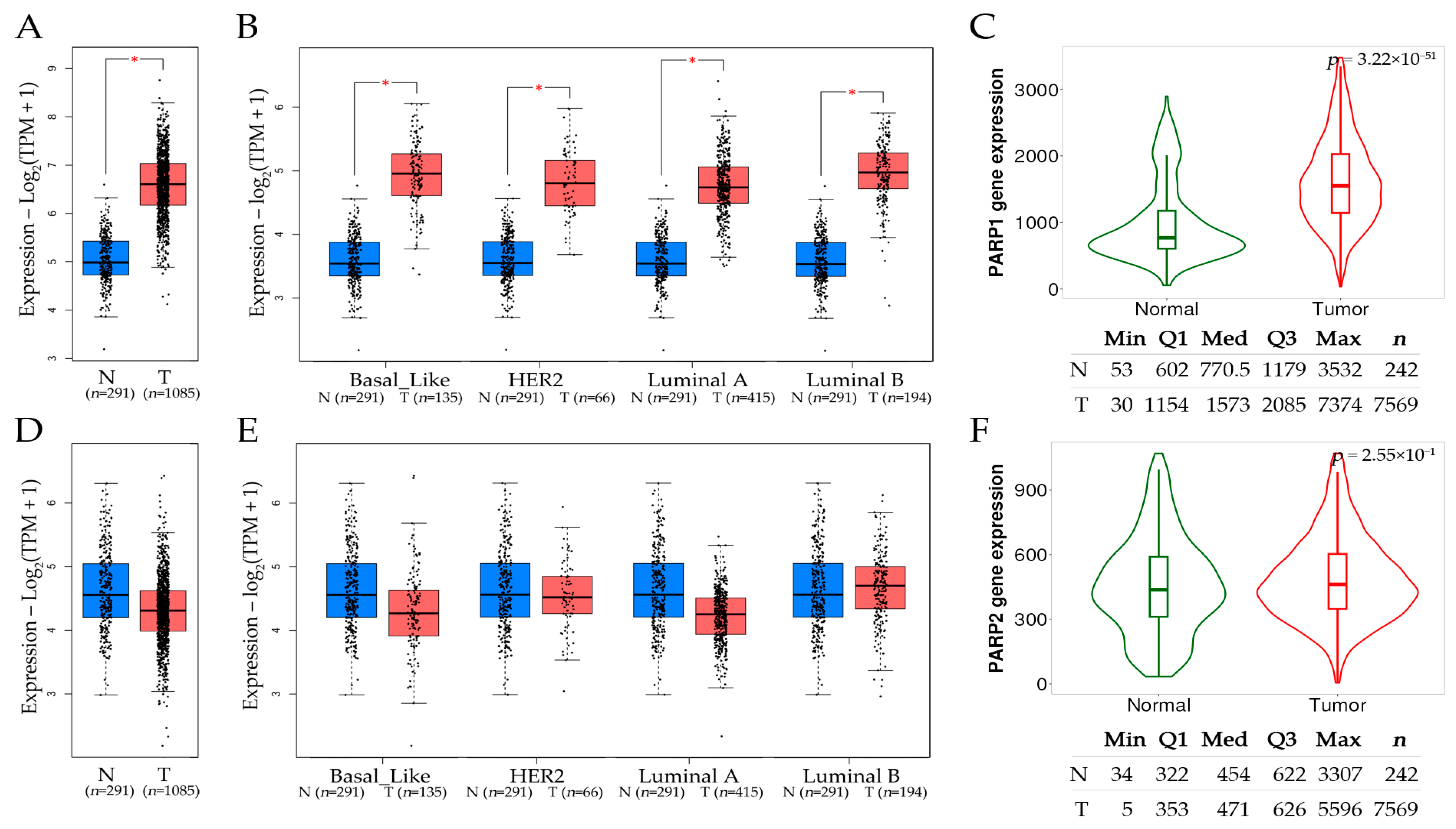
9.2. Comparative Assessment of PARP1 and PARP2 Expression Levels in Tumor and Normal Tissues in Breast Cancer Subtypes
9.3. PARP1 and PARP2 as Potential Markers in Chemotherapy-Treated Breast Cancer Patients
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ismail, T.; Alzneika, S.; Riguene, E.; Al-Maraghi, S.; Alabdulrazzak, A.; Al-Khal, N.; Fetais, S.; Thanassoulas, A.; AlFarsi, H.; Nomikos, M. BRCA1 and Its Vulnerable C-Terminal BRCT Domain: Structure, Function, Genetic Mutations and Links to Diagnosis and Treatment of Breast and Ovarian Cancer. Pharmaceuticals 2024, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- WHO. Breast Cancer. World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 12 November 2024).
- Obeagu, E.I.; Obeagu, G.U. Breast cancer: A review of risk factors and diagnosis. Medicine 2024, 103, e36905. [Google Scholar] [CrossRef] [PubMed]
- Marino, P.; Mininni, M.; Deiana, G.; Marino, G.; Divella, R.; Bochicchio, I.; Giuliano, A.; Lapadula, S.; Lettini, A.R.; Sanseverino, F. Healthy Lifestyle and Cancer Risk: Modifiable Risk Factors to Prevent Cancer. Nutrients 2024, 16, 800. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhu, Y.; Ren, Y.; Chen, L.; Dai, X.; Wang, Y.; Huang, Y.; Wang, H. Global burden and attributable risk factors of breast cancer in young women: Historical trends from 1990 to 2019 and forecasts to 2030 by sociodemographic index regions and countries. J. Glob. Health 2024, 14, 04142. [Google Scholar] [CrossRef]
- Grauman, A.; Sundell, E.; Johansson, J.V.; Cavalli-Bjorkman, N.; Fahlquist, J.N.; Hedstrom, M. Perceptions of lifestyle-related risk communication in patients with breast and colorectal cancer: A qualitative interview study in Sweden. Arch. Public Health 2024, 82, 154. [Google Scholar] [CrossRef]
- Gao, M.; Wik, S.L.; Yu, Q.; Xue, F.; Chan, S.C.; Chow, S.H.; Adebisi, Y.A.; Zhong, C.C.; Lucero-Prisno, D.E., III; Wong, M.C.; et al. Disease Burden, Risk Factors, and Temporal Trends in Breast Cancer in Low- and Middle-Income Countries: A Global Study. Public Health Chall. 2024, 3, e223. [Google Scholar] [CrossRef]
- Conte, L.; Lupo, R.; Lezzi, A.; Sciolti, S.; Rubbi, I.; Carvello, M.; Calabro, A.; Botti, S.; Fanizzi, A.; Massafra, R.; et al. Breast Cancer Prevention Practices and Knowledge in Italian and Chinese Women in Italy: Clinical Checkups, Free NHS Screening Adherence, and Breast Self-Examination (BSE). J. Cancer Educ. 2024, 40, 30–43. [Google Scholar] [CrossRef]
- Chou, C.Y.; Shen, T.T.; Wang, W.C.; Wu, M.P. Favorable breast cancer mortality-to-incidence ratios of countries with good human development index rankings and high health expenditures. Taiwan J. Obstet. Gynecol. 2024, 63, 527–531. [Google Scholar] [CrossRef]
- Goh, S.P.; Ong, S.C.; Chan, J.E. Economic evaluation of germline genetic testing for breast cancer in low- and middle-income countries: A systematic review. BMC Cancer 2024, 24, 316. [Google Scholar] [CrossRef]
- Khorasani, A.B.S.; Hafezi, N.; Sanaei, M.J.; Jafari-Raddani, F.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/AKT/mTOR signaling pathway in breast cancer: Review of clinical trials and latest advances. Cell Biochem. Funct. 2024, 42, e3998. [Google Scholar] [CrossRef]
- Mustafa, M.; Abbas, K.; Alam, M.; Ahmad, W.; Moinuddin; Usmani, N.; Siddiqui, S.A.; Habib, S. Molecular pathways and therapeutic targets linked to triple-negative breast cancer (TNBC). Mol. Cell Biochem. 2024, 479, 895–913. [Google Scholar] [CrossRef]
- Varzaru, V.B.; Vlad, T.; Popescu, R.; Vlad, C.S.; Moatar, A.E.; Cobec, I.M. Triple-Negative Breast Cancer: Molecular Particularities Still a Challenge. Diagnostics 2024, 14, 1875. [Google Scholar] [CrossRef]
- Hernandez, A.E.; Westrick, A.C.; Stoler, J.; Kesmodel, S.B.; Pinheiro, P.S.; Figueroa, M.; Kobetz, E.N.; Rebbeck, T.; Goel, N. Associations Between Neighborhood-Level Income and Triple-Negative Breast Cancer in a Majority-Minority Population. Ann. Surg. Oncol. 2024, 31, 988–996. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Wu, S.; Ge, A.; Deng, X.; Liu, L.; Wang, Y. Evolving immunotherapeutic solutions for triple-negative breast carcinoma. Cancer Treat. Rev. 2024, 130, 102817. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Gusterson, B. Do ‘basal-like’ breast cancers really exist? Nat. Rev. Cancer 2009, 9, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Impact of basal-like breast carcinoma determination for a more specific therapy. Pathobiology 2008, 75, 95–103. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Reis-Filho, J.; Ellis, I.O. Patho-biological aspects of basal-like breast cancer. Breast Cancer Res. Treat. 2009, 113, 411–422. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef]
- Fulford, L.G.; Easton, D.F.; Reis-Filho, J.S.; Sofronis, A.; Gillett, C.E.; Lakhani, S.R.; Hanby, A. Specific morphological features predictive for the basal phenotype in grade 3 invasive ductal carcinoma of breast. Histopathology 2006, 49, 22–34. [Google Scholar] [CrossRef]
- Banerjee, S.; Reis-Filho, J.S.; Ashley, S.; Steele, D.; Ashworth, A.; Lakhani, S.R.; Smith, I.E. Basal-like breast carcinomas: Clinical outcome and response to chemotherapy. J. Clin. Pathol. 2006, 59, 729–735. [Google Scholar] [CrossRef]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef]
- Rakha, E.A.; Ellis, I.O.; Reis-Filho, J.S. Immunohistochemical heterogeneity of breast carcinomas negative for estrogen receptors, progesterone receptors and Her2/neu (basal-like breast carcinomas). Mod. Pathol. 2008, 21, 1060–1061; author reply 1061–1062. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Thike, A.A.; Cheok, P.Y.; Jara-Lazaro, A.R.; Tan, B.; Tan, P.; Tan, P.H. Triple-negative breast cancer: Clinicopathological characteristics and relationship with basal-like breast cancer. Mod. Pathol. 2010, 23, 123–133. [Google Scholar] [CrossRef]
- Thike, A.A.; Iqbal, J.; Cheok, P.Y.; Chong, A.P.; Tse, G.M.; Tan, B.; Tan, P.; Wong, N.S.; Tan, P.H. Triple negative breast cancer: Outcome correlation with immunohistochemical detection of basal markers. Am. J. Surg. Pathol. 2010, 34, 956–964. [Google Scholar] [CrossRef]
- Fulford, L.G.; Reis-Filho, J.S.; Ryder, K.; Jones, C.; Gillett, C.E.; Hanby, A.; Easton, D.; Lakhani, S.R. Basal-like grade III invasive ductal carcinoma of the breast: Patterns of metastasis and long-term survival. Breast Cancer Res. 2007, 9, R4. [Google Scholar] [CrossRef]
- Arnes, J.B.; Brunet, J.S.; Stefansson, I.; Begin, L.R.; Wong, N.; Chappuis, P.O.; Akslen, L.A.; Foulkes, W.D. Placental cadherin and the basal epithelial phenotype of BRCA1-related breast cancer. Clin. Cancer Res. 2005, 11, 4003–4011. [Google Scholar] [CrossRef]
- Pinilla, S.M.; Honrado, E.; Hardisson, D.; Benitez, J.; Palacios, J. Caveolin-1 expression is associated with a basal-like phenotype in sporadic and hereditary breast cancer. Breast Cancer Res. Treat. 2006, 99, 85–90. [Google Scholar] [CrossRef]
- Savage, K.; Leung, S.; Todd, S.K.; Brown, L.A.; Jones, R.L.; Robertson, D.; James, M.; Parry, S.; Rodrigues Pinilla, S.M.; Huntsman, D.; et al. Distribution and significance of caveolin 2 expression in normal breast and invasive breast cancer: An immunofluorescence and immunohistochemical analysis. Breast Cancer Res. Treat. 2008, 110, 245–256. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pinheiro, C.; Lambros, M.B.; Milanezi, F.; Carvalho, S.; Savage, K.; Simpson, P.T.; Jones, C.; Swift, S.; Mackay, A.; et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J. Pathol. 2006, 209, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Goetz, M.P.; Reynolds, C.A.; Ingle, J.N.; Giordano, K.F.; Suman, V.J.; Blair, H.E.; Jenkins, R.B.; Lingle, W.L.; Reinholz, M.M.; et al. Molecular analysis of metaplastic breast carcinoma: High EGFR copy number via aneusomy. Mol. Cancer Ther. 2008, 7, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.; Ellis, I.; Reis-Filho, J. Are triple-negative and basal-like breast cancer synonymous? Clin. Cancer Res. 2008, 14, 618. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Savage, K.; Lambros, M.B.; James, M.; Steele, D.; Jones, R.L.; Dowsett, M. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: An immunohistochemical and chromogenic in situ hybridisation analysis. Mod. Pathol. 2006, 19, 999–1009. [Google Scholar] [CrossRef]
- Subhawong, A.P.; Subhawong, T.; Nassar, H.; Kouprina, N.; Begum, S.; Vang, R.; Westra, W.H.; Argani, P. Most basal-like breast carcinomas demonstrate the same Rb-/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am. J. Surg. Pathol. 2009, 33, 163–175. [Google Scholar] [CrossRef]
- Katz, E.; Dubois-Marshall, S.; Sims, A.H.; Gautier, P.; Caldwell, H.; Meehan, R.R.; Harrison, D.J. An in vitro model that recapitulates the epithelial to mesenchymal transition (EMT) in human breast cancer. PLoS ONE 2011, 6, e17083. [Google Scholar] [CrossRef]
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumours: A critical review. Histopathology 2008, 52, 108–118. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Lakhani, S.R. Breast cancer special types: Why bother? J. Pathol. 2008, 216, 394–398. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Brunet, J.S.; Begin, L.R.; Huntsman, D.G.; Cheang, M.C.; Akslen, L.A.; Nielsen, T.O.; Foulkes, W.D. Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC Cancer 2007, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.N.; Broadwater, G.; Lin, N.U.; Miron, A.; Schnitt, S.J.; Cowan, D.; Lara, J.; Bleiweiss, I.; Berry, D.; Ellis, M.; et al. Molecular subtypes of breast cancer in relation to paclitaxel response and outcomes in women with metastatic disease: Results from CALGB 9342. Breast Cancer Res. 2006, 8, R66. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-negative breast cancer: Risk factors to potential targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 2007, 131, 18–43. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Wolff, A.C.; Mangu, P.B.; Temin, S. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Oncol. Pract. 2010, 6, 195–197. [Google Scholar] [CrossRef]
- Rakha, E.A.; Tan, D.S.; Foulkes, W.D.; Ellis, I.O.; Tutt, A.; Nielsen, T.O.; Reis-Filho, J.S. Are triple-negative tumours and basal-like breast cancer synonymous? Breast Cancer Res. 2007, 9, 404. [Google Scholar] [CrossRef]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Calza, S.; Hall, P.; Auer, G.; Bjohle, J.; Klaar, S.; Kronenwett, U.; Liu, E.T.; Miller, L.; Ploner, A.; Smeds, J.; et al. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006, 8, R34. [Google Scholar] [CrossRef] [PubMed]
- Jumppanen, M.; Gruvberger-Saal, S.; Kauraniemi, P.; Tanner, M.; Bendahl, P.O.; Lundin, M.; Krogh, M.; Kataja, P.; Borg, A.; Ferno, M.; et al. Basal-like phenotype is not associated with patient survival in estrogen-receptor-negative breast cancers. Breast Cancer Res. 2007, 9, R16. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef]
- de Ronde, J.J.; Hannemann, J.; Halfwerk, H.; Mulder, L.; Straver, M.E.; Vrancken Peeters, M.J.; Wesseling, J.; van de Vijver, M.; Wessels, L.F.; Rodenhuis, S. Concordance of clinical and molecular breast cancer subtyping in the context of preoperative chemotherapy response. Breast Cancer Res. Treat. 2010, 119, 119–126. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006, 25, 5846–5853. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Brunet, J.S.; Stefansson, I.M.; Straume, O.; Chappuis, P.O.; Begin, L.R.; Hamel, N.; Goffin, J.R.; Wong, N.; Trudel, M.; et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004, 64, 830–835. [Google Scholar] [CrossRef]
- Vaziri, S.A.; Krumroy, L.M.; Elson, P.; Budd, G.T.; Darlington, G.; Myles, J.; Tubbs, R.R.; Casey, G. Breast tumor immunophenotype of BRCA1-mutation carriers is influenced by age at diagnosis. Clin. Cancer Res. 2001, 7, 1937–1945. [Google Scholar]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed]
- Palacios, J.; Honrado, E.; Osorio, A.; Cazorla, A.; Sarrio, D.; Barroso, A.; Rodriguez, S.; Cigudosa, J.C.; Diez, O.; Alonso, C.; et al. Phenotypic characterization of BRCA1 and BRCA2 tumors based in a tissue microarray study with 37 immunohistochemical markers. Breast Cancer Res. Treat. 2005, 90, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Ford, E.; Gillett, C.; Ryder, K.; Merrett, S.; Reis-Filho, J.S.; Fulford, L.G.; Hanby, A.; Lakhani, S.R. Molecular cytogenetic identification of subgroups of grade III invasive ductal breast carcinomas with different clinical outcomes. Clin. Cancer Res. 2004, 10, 5988–5997. [Google Scholar] [CrossRef] [PubMed]
- van Beers, E.H.; van Welsem, T.; Wessels, L.F.; Li, Y.; Oldenburg, R.A.; Devilee, P.; Cornelisse, C.J.; Verhoef, S.; Hogervorst, F.B.; van’t Veer, L.J.; et al. Comparative genomic hybridization profiles in human BRCA1 and BRCA2 breast tumors highlight differential sets of genomic aberrations. Cancer Res. 2005, 65, 822–827. [Google Scholar] [CrossRef]
- Wessels, L.F.; van Welsem, T.; Hart, A.A.; van’t Veer, L.J.; Reinders, M.J.; Nederlof, P.M. Molecular classification of breast carcinomas by comparative genomic hybridization: A specific somatic genetic profile for BRCA1 tumors. Cancer Res. 2002, 62, 7110–7117. [Google Scholar]
- Melchor, L.; Honrado, E.; Garcia, M.J.; Alvarez, S.; Palacios, J.; Osorio, A.; Nathanson, K.L.; Benitez, J. Distinct genomic aberration patterns are found in familial breast cancer associated with different immunohistochemical subtypes. Oncogene 2008, 27, 3165–3175. [Google Scholar] [CrossRef]
- Valenza, C.; Marsicano, R.M.; Trapani, D.; Curigliano, G. PARP inhibitor resistant BRCA-mutated advanced breast cancer: Current landscape and emerging treatments. Curr. Opin. Oncol. 2024, 36, 474–479. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; Gillett, C.E.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007, 26, 2126–2132. [Google Scholar] [CrossRef]
- Zhu, S.; Meng, L.; Wei, P.; Gu, G.; Duan, K. Sinensetin suppresses breast cancer cell progression via Wnt/beta-catenin pathway inhibition. Transl. Cancer Res. 2024, 13, 348–362. [Google Scholar] [CrossRef]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. Wnt/beta-catenin-driven EMT regulation in human cancers. Cell Mol. Life Sci. 2024, 81, 79. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Awaji, A.A.; Maigoro, A.Y.; Aborode, A.T.; Akintola, A.A.; Fatoba, D.O.; Idris, E.B.; Idris, A.B.; Jafri, S.; Shoaib, E.; Onifade, I.A.; et al. Identification of key molecular pathways and genes in BRCA1 and BRCA2-mutant ovarian cancer: Evidence from bioinformatics analysis. Genome Instab. Dis. 2024, 5, 164–182. [Google Scholar] [CrossRef]
- Young, A.L.; Butow, P.N.; Rhodes, P.; Tucker, K.M.; Williams, R.; Healey, E.; Wakefield, C.E. Talking across generations: Family communication about BRCA1 and BRCA2 genetic cancer risk. J. Genet. Couns. 2019, 28, 516–532. [Google Scholar] [CrossRef]
- Daniele, A.; Divella, R.; Pilato, B.; Tommasi, S.; Pasanisi, P.; Patruno, M.; Digennaro, M.; Minoia, C.; Dellino, M.; Pisconti, S.; et al. Can harmful lifestyle, obesity and weight changes increase the risk of breast cancer in BRCA 1 and BRCA 2 mutation carriers? A Mini review. Hered. Cancer Clin. Pract. 2021, 19, 45. [Google Scholar] [CrossRef]
- Kufel-Grabowska, J.; Wasag, B. Diagnosis and treatment of patients with breast cancer and mutation in the BRCA1/2 genes. Oncol. Clin. Pract. 2024, 20, 222–228. [Google Scholar] [CrossRef]
- Yu, S.; Qiu, X.; Wang, Z.; Xiao, J.; Ji, H.; Shan, H.; Shao, Q.; Xia, H.; Cao, F.; Li, J.; et al. Breast cancer risk associated with BRCA1 and BRCA2 pathogenic variants in the Eastern Chinese population. Cancer Pathog. Ther. 2024, 2, e01–e026. [Google Scholar] [CrossRef]
- Minello, A.; Carreira, A. BRCA1/2 Haploinsufficiency: Exploring the Impact of Losing one Allele. J. Mol. Biol. 2024, 436, 168277. [Google Scholar] [CrossRef]
- Barili, V.; Ambrosini, E.; Bortesi, B.; Minari, R.; De Sensi, E.; Cannizzaro, I.R.; Taiani, A.; Michiara, M.; Sikokis, A.; Boggiani, D.; et al. Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing. Genes 2024, 15, 219. [Google Scholar] [CrossRef]
- Szentmartoni, G.; Muhl, D.; Csanda, R.; Szasz, A.M.; Herold, Z.; Dank, M. Predictive Value and Therapeutic Significance of Somatic BRCA Mutation in Solid Tumors. Biomedicines 2024, 12, 593. [Google Scholar] [CrossRef]
- Yi, H.; Trivedi, M.S.; Crew, K.D.; Schechter, I.; Appelbaum, P.; Chung, W.K.; Allegrante, J.P.; Kukafka, R. Understanding Social, Cultural, and Religious Factors Influencing Medical Decision-Making on BRCA1/2 Genetic Testing in the Orthodox Jewish Community. Public Health Genom. 2024, 27, 57–67. [Google Scholar] [CrossRef]
- Ren, P.; Zhang, J.; Vijg, J. Somatic mutations in aging and disease. Geroscience 2024, 46, 5171–5189. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, A.P.S.; Tanvir; Singh, K.; Singh, S.; Antaal, H.; Luthra, S.; Singla, A.; Nijjar, G.S.; Aulakh, S.K.; Kaur, Y. Decoding Cancer Risk: Understanding Gene-Environment Interactions in Cancer Development. Cureus 2024, 16, e64936. [Google Scholar] [CrossRef] [PubMed]
- Agboola, O.O.; Emmanuel, A.O.; Sabina, C.E.; Anejo-Okopi, J.; Naomi, A.I.; Ene, U.I.; Olowoyo, J.O. Occurrence of Carcinogens and their Potential Effects on Human Health—A Review. UMYU Sci. 2024, 3, 129–143. [Google Scholar] [CrossRef]
- Yuan, P.; Ma, N.; Xu, B. Poly (adenosine diphosphate-ribose) polymerase inhibitors in the treatment of triple-negative breast cancer with homologous repair deficiency. Med. Res. Rev. 2024, 44, 2774–2792. [Google Scholar] [CrossRef]
- Previtali, V.; Bagnolini, G.; Ciamarone, A.; Ferrandi, G.; Rinaldi, F.; Myers, S.H.; Roberti, M.; Cavalli, A. New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives. J. Med. Chem. 2024, 67, 11488–11521. [Google Scholar] [CrossRef]
- Subhan, M.A.; Parveen, F.; Shah, H.; Yalamarty, S.S.K.; Ataide, J.A.; Torchilin, V.P. Recent Advances with Precision Medicine Treatment for Breast Cancer including Triple-Negative Sub-Type. Cancers 2023, 15, 2204. [Google Scholar] [CrossRef]
- Miresmaeili, M.; Nabi-Afjadi, M.; Lagzian, A.; Fathi, Z.; Yazdanpour, M.; Zalpoor, H.; Yaghoubzad-Maleki, M.; Moeini, A.M.; Arman, I. Identification of Novel Functional Single Nucleotide Polymorphisms in the BRCA1 Gene of Breast Cancer Patients. Iran. J. Sci. 2024, 48, 821–833. [Google Scholar] [CrossRef]
- Aubuchon, L.N.; Verma, P. Endogenous base damage as a driver of genomic instability in homologous recombination-deficient cancers. DNA Repair. 2024, 141, 103736. [Google Scholar] [CrossRef]
- Khamidullina, A.I.; Abramenko, Y.E.; Bruter, A.V.; Tatarskiy, V.V. Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets. Int. J. Mol. Sci. 2024, 25, 1263. [Google Scholar] [CrossRef]
- Mubarak, M.H.; Kharisma, M. Impact of Estrogen Therapy on BRCA1-Associated Breast Cancer Progression in Transgender Women. Int. J. Health Med. 2024, 1, 134–155. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xiao, D.; Li, D.; Peng, M.; Peng, W.; Duan, H.; Yang, X. Combined strategies with PARP inhibitors for the treatment of BRCA wide type cancer. Front. Oncol. 2024, 14, 1441222. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, L.; Calheiros, J.; Saraiva, L. Exploiting DNA Damage Repair in Precision Cancer Therapy: BRCA1 as a Prime Therapeutic Target. Cancers 2021, 13, 3438. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ravendranathan, N.; Frisbee, J.C.; Singh, K.K. Complex Interplay between DNA Damage and Autophagy in Disease and Therapy. Biomolecules 2024, 14, 922. [Google Scholar] [CrossRef]
- Pourmasoumi, P.; Moradi, A.; Bayat, M. BRCA1/2 Mutations and Breast/Ovarian Cancer Risk: A New Insights Review. Reprod. Sci. 2024, 31, 3624–3634. [Google Scholar] [CrossRef]
- Bedrosian, I.; Somerfield, M.R.; Achatz, M.I.; Boughey, J.C.; Curigliano, G.; Friedman, S.; Kohlmann, W.K.; Kurian, A.W.; Laronga, C.; Lynce, F.; et al. Germline Testing in Patients With Breast Cancer: ASCO-Society of Surgical Oncology Guideline. J. Clin. Oncol. 2024, 42, 584–604. [Google Scholar] [CrossRef]
- Andrian, S.M.; Benny, A.; Hemalatha, N. Oncogenomic analysis to find the role of BRCA1 and BRCA2 in male. In Information Technology & Bioinformatics International Conference On Advance It, Engineering and Management SACAIM 2023; Hemalatha, N., Shetty, K.A., Rakesh Kumar, B., Eds.; Redshine Publication: Mumbai, Maharashtra, India, 2023; Volume 2, p. 26. [Google Scholar]
- Ramirez-Otero, M.A.; Costanzo, V. “Bridging the DNA divide”: Understanding the interplay between replication- gaps and homologous recombination proteins RAD51 and BRCA1/2. DNA Repair. 2024, 141, 103738. [Google Scholar] [CrossRef]
- Patel, V.; Casimiro, S.; Abreu, C.; Barroso, T.; de Sousa, R.T.; Torres, S.; Ribeiro, L.A.; Nogueira-Costa, G.; Pais, H.L.; Pinto, C.; et al. DNA damage targeted therapy for advanced breast cancer. Explor. Target. Antitumor Ther. 2024, 5, 678–698. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Polak, P. Probing the relevance of BRCA1 and BRCA2 germline pathogenic variants beyond breast and ovarian cancer. J. Natl. Cancer Inst. 2024, 116, 1871–1874. [Google Scholar] [CrossRef]
- Chahat; Nainwal, N.; Murti, Y.; Yadav, S.; Rawat, P.; Dhiman, S.; Kumar, B. Chahat; Nainwal, N.; Murti, Y.; Yadav, S.; Rawat, P.; Dhiman, S.; Kumar, B. Advancements in targeting tumor suppressor genes (p53 and BRCA 1/2) in breast cancer therapy. Mol. Divers. 2024. [Google Scholar] [CrossRef]
- Reddy, P.P.; Phale, A.; Das, R. Structural analysis of genetic variants of the human tumor suppressor Palb2 coiled-coil domain. BioRxvid 2024, 3, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Munyembaraga, V.; Cyuzuzo, D.; Dao, T.N.P.; Oyinloye, B.E.; Onikanni, S.A.; Chang, H.H. Modulatory Potential of Poly (ADP-Ribose) Polymerase 1 (PARP1) in BRCA-Mutated Tumors. Eur. J. Cancer Care 2024, 2024, e1–e12. [Google Scholar] [CrossRef]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar]
- Wikipedia. PARP1. Available online: https://en.wikipedia.org/wiki/PARP1 (accessed on 3 February 2025).
- Wikipedia. PARP2. Available online: https://en.wikipedia.org/wiki/PARP2 (accessed on 3 February 2025).
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Deeksha, W.; Rajakumara, E. Regulatory apoptotic fragment of PARP1 complements catalytic fragment for PAR and DNA-dependent activity but inhibits DNA-induced catalytic stimulation of PARP2. DNA Repair. 2024, 133, 103593. [Google Scholar] [CrossRef]
- Bastos, I.M.; Rebelo, S.; Silva, V.L.M. A review of poly(ADP-ribose)polymerase-1 (PARP1) role and its inhibitors bearing pyrazole or indazole core for cancer therapy. Biochem. Pharmacol. 2024, 221, 116045. [Google Scholar] [CrossRef]
- van Beek, L.; McClay, E.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef]
- Al-Rahahleh, R.Q.; Sobol, R.W. Poly-ADP-ribosylation dynamics, signaling, and analysis. Environ. Mol. Mutagen. 2024, 65, 315–337. [Google Scholar] [CrossRef]
- Nosella, M.L.; Kim, T.H.; Huang, S.K.; Harkness, R.W.; Goncalves, M.; Pan, A.; Tereshchenko, M.; Vahidi, S.; Rubinstein, J.L.; Lee, H.O.; et al. Poly(ADP-ribosyl)ation enhances nucleosome dynamics and organizes DNA damage repair components within biomolecular condensates. Mol. Cell 2024, 84, 429–446.e17. [Google Scholar] [CrossRef]
- Marti, J.M.; Fernandez-Cortes, M.; Serrano-Saenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef]
- Ramesh, S.; Almeida, S.D.; Hammigi, S.; Radhakrishna, G.K.; Sireesha, G.; Panneerselvam, T.; Vellingiri, S.; Kunjiappan, S.; Ammunje, D.N.; Pavadai, P. A Review of PARP-1 Inhibitors: Assessing Emerging Prospects and Tailoring Therapeutic Strategies. Drug Res. 2023, 73, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, X.; Liu, W.; Chen, Z.; Xiao, X.; Deng, G. Supplementation with NAD+ and its precursors: A rescue of female reproductive diseases. Biochem. Biophys. Rep. 2024, 38, 101715. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A.; Stravodimou, A. Homologous Recombination Defects and Mutations in DNA Damage Response (DDR) Genes Besides BRCA1 and BRCA2 as Breast Cancer Biomarkers for PARP Inhibitors and Other DDR Targeting Therapies. Anticancer. Res. 2023, 43, 967–981. [Google Scholar] [CrossRef]
- Che, H.; Wang, L.W.; Ye, X.Y.; He, X. Olaparib research update: Mechanism, structure and clinical trials. Med. Chem. Res. 2025, 34, 535–548. [Google Scholar] [CrossRef]
- Dibitetto, D.; Widmer, C.A.; Rottenberg, S. PARPi, BRCA, and gaps: Controversies and future research. Trends Cancer 2024, 10, 857–869. [Google Scholar] [CrossRef]
- Li, X.; Zou, L. BRCAness, DNA gaps, and gain and loss of PARP inhibitor-induced synthetic lethality. J. Clin. Investig. 2024, 134, e181062. [Google Scholar] [CrossRef]
- Malamos, P.; Papanikolaou, C.; Gavriatopoulou, M.; Dimopoulos, M.A.; Terpos, E.; Souliotis, V.L. The Interplay between the DNA Damage Response (DDR) Network and the Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway in Multiple Myeloma. Int. J. Mol. Sci. 2024, 25, 6991. [Google Scholar] [CrossRef]
- Tan, J.; Sun, X.; Zhao, H.; Guan, H.; Gao, S.; Zhou, P.K. Double-strand DNA break repair: Molecular mechanisms and therapeutic targets. MedComm (2020) 2023, 4, e388. [Google Scholar] [CrossRef]
- Ndlovu, H.; Lawal, I.O.; Mdanda, S.; Kgatle, M.M.; Mokoala, K.M.G.; Al-Ibraheem, A.; Sathekge, M.M. [(18)F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology. J. Clin. Med. 2024, 13, 3426. [Google Scholar] [CrossRef]
- Xu, L.; Xu, P.; Wang, J.; Ji, H.; Zhang, L.; Tang, Z. Advancements in clinical research and emerging therapies for triple-negative breast cancer treatment. Eur. J. Pharmacol. 2025, 988, 177202. [Google Scholar] [CrossRef]
- Huang, L.; Shao, J.; Lai, W.; Gu, H.; Yang, J.; Shi, S.; Wufoyrwoth, S.; Song, Z.; Zou, Y.; Xu, Y.; et al. Discovery of the first ataxia telangiectasia and Rad3-related (ATR) degraders for cancer treatment. Eur. J. Med. Chem. 2024, 267, 116159. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Gallyas, F., Jr.; Sumegi, B. Mitochondrial Protection by PARP Inhibition. Int. J. Mol. Sci. 2020, 21, 2767. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Leung, H.Y. Evasion of cell death: A contributory factor in prostate cancer development and treatment resistance. Cancer Lett. 2021, 520, 213–221. [Google Scholar] [CrossRef]
- Klapp, V.; Alvarez-Abril, B.; Leuzzi, G.; Kroemer, G.; Ciccia, A.; Galluzzi, L. The DNA Damage Response and Inflammation in Cancer. Cancer Discov. 2023, 13, 1521–1545. [Google Scholar] [CrossRef]
- Jin, W.; Yang, Q.; Zhang, Z.; Li, J. Olaparib-associated toxicity in cancer patients: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2025, 81, 65–81. [Google Scholar] [CrossRef]
- Pacaud, R.; Thomas, S.; Chaudhuri, S.; Lazar, A.; Timmerman, L.A.; Munster, P.N. Low dose DNA methyltransferase inhibitors potentiate PARP inhibitors in homologous recombination repair deficient tumors. Breast Cancer Res. 2025, 27, 8. [Google Scholar] [CrossRef]
- Narang, A.; Hage Chehade, C.; Ozay, Z.I.; Nordblad, B.; Swami, U.; Agarwal, N. Talazoparib for the treatment of prostate cancer. Expert. Opin. Pharmacother. 2024, 25, 1717–1727. [Google Scholar] [CrossRef]
- Sun, K.; Mikule, K.; Wang, Z.; Poon, G.; Vaidyanathan, A.; Smith, G.; Zhang, Z.Y.; Hanke, J.; Ramaswamy, S.; Wang, J. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018, 9, 37080–37096. [Google Scholar] [CrossRef]
- Longoria, T.C.; Tewari, K.S. Pharmacokinetic drug evaluation of niraparib for the treatment of ovarian cancer. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 543–550. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int. J. Mol. Sci. 2021, 22, 4203. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wang, L.; Xing, Z.; Wang, F.; Cheng, X. Update on Combination Strategies of PARP Inhibitors. Cancer Control 2024, 31, 10732748241298329. [Google Scholar] [CrossRef] [PubMed]
- Pearre, D.C.; Tewari, K.S. Targeted treatment of advanced ovarian cancer: Spotlight on rucaparib. Ther. Clin. Risk Manag. 2018, 14, 2189–2201. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R. D 2020, 20, 55–73. [Google Scholar] [CrossRef]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell Mol. Life Sci. 2020, 77, 677–703. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmana, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- McCann, K.E. Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: Talazoparib. Future Oncol. 2019, 15, 1707–1715. [Google Scholar] [CrossRef]
- Kowalik, A.; Chudecka-Głaz, A.; Kufel-Grabowska, J.; Skoneczna, I.; Kubiatowski, T. Diagnostics and treatment of BRCA-associated cancers with olaparib—expert position statement. Oncol. Clin. Pract. 2024, 20, 229–244. [Google Scholar] [CrossRef]
- Jones, P.; Wilcoxen, K.; Rowley, M.; Toniatti, C. Niraparib: A Poly(ADP-ribose) Polymerase (PARP) Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J. Med. Chem. 2015, 58, 3302–3314. [Google Scholar] [CrossRef] [PubMed]
- Musella, A.; Bardhi, E.; Marchetti, C.; Vertechy, L.; Santangelo, G.; Sassu, C.; Tomao, F.; Rech, F.; D’Amelio, R.; Monti, M.; et al. Rucaparib: An emerging parp inhibitor for treatment of recurrent ovarian cancer. Cancer Treat. Rev. 2018, 66, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Bartha, A.; Gyorffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Met-astatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Fekete, J.T.; Gyorffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SL. No | Name of PARP Inhibitors | Stage of Application | References |
|---|---|---|---|
| 1 | Olaparib | Olaparib, the first PARP inhibitor to be approved, is used in advanced breast cancer treatment after chemotherapy and provides clinically effective results. It is especially useful in patients with BRCA mutations and HER2-negative metastatic breast cancer (mBC). | (Tutt et al., 2021) [152] |
| 2 | Talazoparib | Talazoparib is a stronger PARP inhibitor than olaparib, more effective against BRCA-mutant breast cancer, and longer-acting because of its strong affinity for PARP. | (McCann, 2019; Kowalik et al., 2024) [153,154] |
| 3 | Niraparib | Developed originally for ovarian cancer, it is being studied for breast cancer. Regardless of a patient’s BRCA status, niraparib is a strong inhibitor that can be used because it may affect other DNA repair pathways. | (Jones et al., 2015) [155] |
| 4 | Rucaparib | Like olaparib and talazoparib, rucaparib works well in cancers with BRCA mutations. It is used after patients have undergone prior treatment and has demonstrated efficacy in treating both breast and ovarian cancers. | (Musella et al., 2018) [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De, K.; Jana, M.; Chowdhury, B.; Calaf, G.M.; Roy, D. Role of PARP Inhibitors: A New Hope for Breast Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 2773. https://doi.org/10.3390/ijms26062773
De K, Jana M, Chowdhury B, Calaf GM, Roy D. Role of PARP Inhibitors: A New Hope for Breast Cancer Therapy. International Journal of Molecular Sciences. 2025; 26(6):2773. https://doi.org/10.3390/ijms26062773
Chicago/Turabian StyleDe, Kamalendu, Malabendu Jana, Bhabadeb Chowdhury, Gloria M. Calaf, and Debasish Roy. 2025. "Role of PARP Inhibitors: A New Hope for Breast Cancer Therapy" International Journal of Molecular Sciences 26, no. 6: 2773. https://doi.org/10.3390/ijms26062773
APA StyleDe, K., Jana, M., Chowdhury, B., Calaf, G. M., & Roy, D. (2025). Role of PARP Inhibitors: A New Hope for Breast Cancer Therapy. International Journal of Molecular Sciences, 26(6), 2773. https://doi.org/10.3390/ijms26062773