
Metabolic Pattern of Brain Death—NMR-Based Metabolomics of Cerebrospinal Fluid

 ,
,
Abstract
1. Introduction
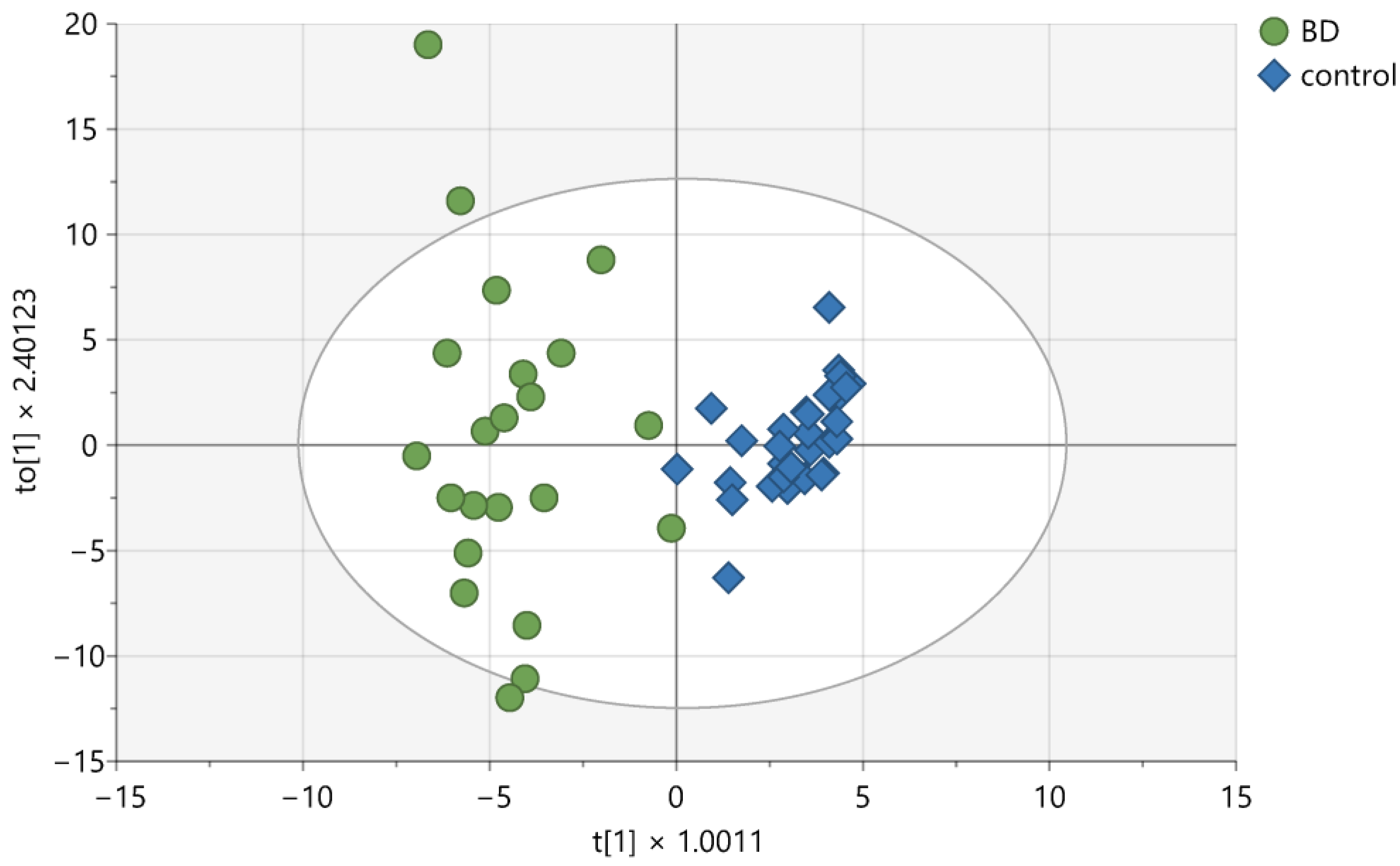
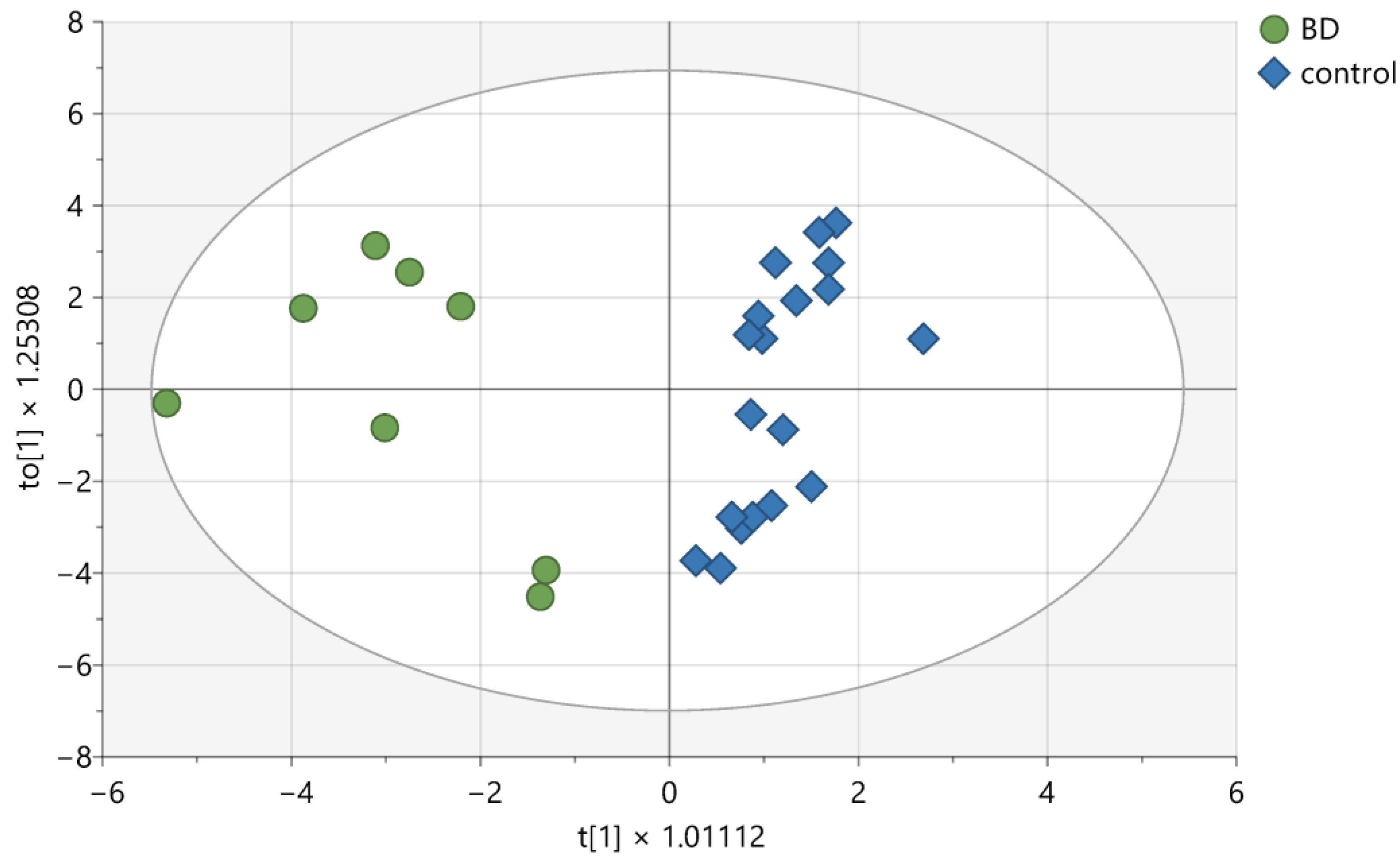
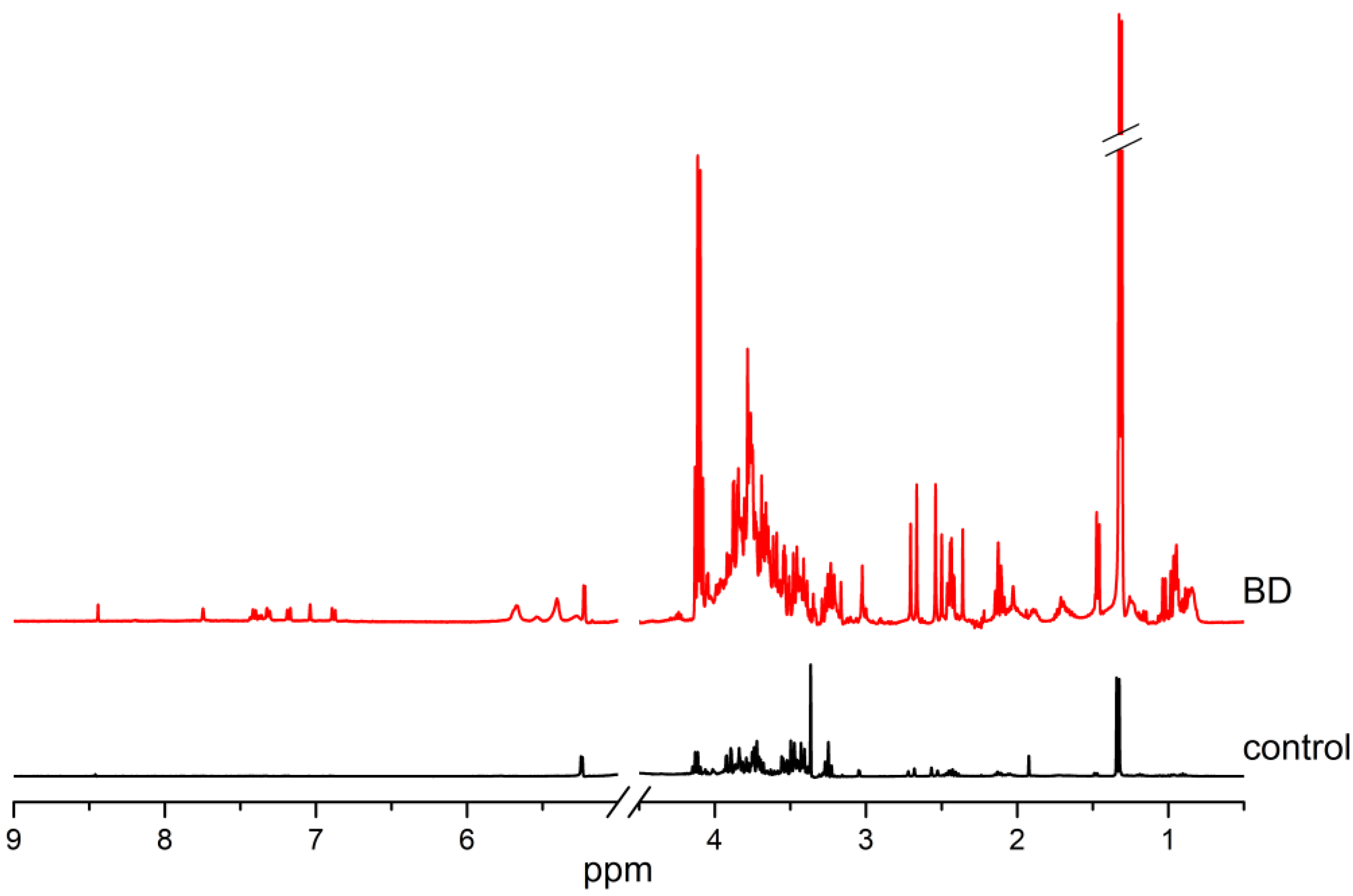
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients and Materials
4.2. Sample Preparation and Spectrum Acquisition
4.3. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dronavalli, V.B.; Ranasinghe, A.M.; Venkateswaran, R.J.; James, S.R.; McCabe, C.J.; Wilson, I.C.; Mascaro, J.G.; Bonser, R.S. N-terminal pro-brain-type natriuretic peptide: A biochemical surrogate of cardiac function in the potential heart donor. Eur. J. Cardio-Thorac. Surg. 2010, 38, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Floerchinger, B.; Oberhuber, R.; Tullius, S.G. Effects of brain death on organ quality and transplant outcome. Transplant. Rev. 2012, 26, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Greer, D.M.; Shemie, S.D.; Lewis, A.; Torrance, S.; Varelas, P.; Goldenberg, F.D.; Bernat, J.L.; Souter, M.; Topcuoglu, M.A.; Alexandrov, A.W.; et al. Determination of Brain Death/Death by Neurologic Criteria: The World Brain Death Project. JAMA 2020, 324, 1078–1097. [Google Scholar] [CrossRef]
- Greer, D.M.; Lewis, A.; Kirschen, M.P. New developments in guidelines for brain death/death by neurological criteria. Nat. Rev. Neurol. 2024, 20, 151–161. [Google Scholar] [CrossRef]
- Kirschen, M.P.; Lewis, A.; Rubin, M.; Kurtz, P.; Greer, D.M. New perspectives on brain death. J. Neurol. Neurosurg. Psychiatry 2021, 92, 255–262. [Google Scholar] [CrossRef]
- Chan, J.Y.; Tsai, C.Y.; Wu, C.H.; Li, F.C.; Dai, K.Y.; Sun, E.Y.; Chan, S.H.; Chang, A.Y. Sumoylation of hypoxia-inducible factor-1alpha ameliorates failure of brain stem cardiovascular regulation in experimental brain death. PLoS ONE 2011, 6, e17375. [Google Scholar] [CrossRef]
- Dai, K.Y.; Chan, S.H.; Chang, A.Y. Heme oxygenase-1 plays a pro-life role in experimental brain stem death via nitric oxide synthase I/protein kinase G signaling at rostral ventrolateral medulla. J. Biomed. Sci. 2010, 17, 72. [Google Scholar] [CrossRef]
- Wolff, B.; Machill, K.; Schumacher, D.; Schulzki, I.; Werner, D. Early achievement of mild therapeutic hypothermia and the neurologic outcome after cardiac arrest. Int. J. Cardiol. 2009, 133, 223–228. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Zielman, R.; Postma, R.; Verhoeven, A.; Bakels, F.; van Oosterhout, W.P.; Meissner, A.; van den Maagdenberg, A.M.; Terwindt, G.M.; Mayboroda, O.A.; Ferrari, M.D. Metabolomic changes in CSF of migraine patients measured with 1H-NMR spectroscopy. Mol. Biosyst. 2016, 12, 3674–3682. [Google Scholar] [CrossRef]
- Mussap, M.; Antonucci, R.; Noto, A.; Fanos, V. The role of metabolomics in neonatal and pediatric laboratory medicine. Clin. Chim. Acta 2013, 426, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Schnackenberg, L.K. Global metabolic profiling and its role in systems biology to advance personalized medicine in the 21st century. Expert Rev. Mol. Diagn. 2007, 7, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Fonville, J.M.; Maher, A.D.; Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Evaluation of full-resolution J-resolved 1H NMR projections of biofluids for metabonomics information retrieval and biomarker identification. Anal. Chem. 2010, 82, 1811–1821. [Google Scholar] [CrossRef]
- Wishart, D.S. Quantitative metabolomics using NMR. Trac-Trend Anal. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Wang, Y. Mass spectrometry: From proteomics to metabolomics and lipidomics. Chem. Soc. Rev. 2009, 38, 1882–1896. [Google Scholar] [CrossRef]
- Maeda, H.; Zhu, B.L.; Ishikawa, T.; Quan, L.; Michiue, T. Significance of postmortem biochemistry in determining the cause of death. Leg. Med. 2009, 11 (Suppl. S1), S46–S49. [Google Scholar] [CrossRef]
- Mandal, R.; Guo, A.C.; Chaudhary, K.K.; Liu, P.; Yallou, F.S.; Dong, E.; Aziat, F.; Wishart, D.S. Multi-platform characterization of the human cerebrospinal fluid metabolome: A comprehensive and quantitative update. Genome Med. 2012, 4, 38. [Google Scholar] [CrossRef]
- Pesko, B.K.; Weidt, S.; McLaughlin, M.; Wescott, D.J.; Torrance, H.; Burgess, K.; Burchmore, R. Postmortomics: The Potential of Untargeted Metabolomics to Highlight Markers for Time Since Death. OMICS 2020, 24, 649–659. [Google Scholar] [CrossRef]
- Mora-Ortiz, M.; Trichard, M.; Oregioni, A.; Claus, S.P. Thanatometabolomics: Introducing NMR-based metabolomics to identify metabolic biomarkers of the time of death. Metabolomics 2019, 15, 37. [Google Scholar] [CrossRef]
- Dawiskiba, T.; Wojtowicz, W.; Qasem, B.; Lukaszewski, M.; Mielko, K.A.; Dawiskiba, A.; Banasik, M.; Skora, J.P.; Janczak, D.; Mlynarz, P. Brain-dead and coma patients exhibit different serum metabolic profiles: Preliminary investigation of a novel diagnostic approach in neurocritical care. Sci. Rep. 2021, 11, 15519. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aguilera, M.E.; de San Miguel, E.R.; Cruz-Perez, J.; Aguirre-Cruz, L.; Ramirez-Alfaro, C.M.; Esturau-Escofet, N. NMR-based metabolomics of human cerebrospinal fluid identifies signature of brain death. Metabolomics 2021, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.D.; Nayak, M.K.; Ghatge, M.; Chauhan, A.K. Metabolic targeting of platelets to combat thrombosis: Dawn of a new paradigm? Cardiovasc. Res. 2023, 119, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: Its importance in health and disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Atila, A.; Alay, H.; Yaman, M.E.; Akman, T.C.; Cadirci, E.; Bayrak, B.; Celik, S.; Atila, N.E.; Yaganoglu, A.M.; Kadioglu, Y.; et al. The serum amino acid profile in COVID-19. Amino Acids 2021, 53, 1569–1588. [Google Scholar] [CrossRef]
- Ardawi, M.S.; Newsholme, E.A. Glutamine metabolism in lymphocytes of the rat. Biochem. J. 1983, 212, 835–842. [Google Scholar] [CrossRef]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife 2017, 6, e21330. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Cornille, E.; Abou-Hamdan, M.; Khrestchatisky, M.; Nieoullon, A.; de Reggi, M.; Gharib, B. Enhancement of L-3-hydroxybutyryl-CoA dehydrogenase activity and circulating ketone body levels by pantethine. Relevance to dopaminergic injury. BMC Neurosci. 2010, 11, 51. [Google Scholar] [CrossRef]
- Fedorovich, S.V.; Voronina, P.P.; Waseem, T.V. Ketogenic diet versus ketoacidosis: What determines the influence of ketone bodies on neurons? Neural Regen. Res. 2018, 13, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, C.D.; Mukhtarov, M.; Malkov, A.E.; Popova, I.Y.; Bregestovski, P.; Zilberter, Y. Energy substrate availability as a determinant of neuronal resting potential, GABA signaling and spontaneous network activity in the neonatal cortex in vitro. J. Neurochem. 2010, 112, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Voronina, P.P.; Adamovich, K.V.; Adamovich, T.V.; Dubouskaya, T.G.; Hrynevich, S.V.; Waseem, T.V.; Fedorovich, S.V. High Concentration of Ketone Body beta-Hydroxybutyrate Modifies Synaptic Vesicle Cycle and Depolarizes Plasma Membrane of Rat Brain Synaptosomes. J. Mol. Neurosci. 2020, 70, 112–119. [Google Scholar] [CrossRef]
- Mayor, D.; Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 2018, 134 Pt B, 178–188. [Google Scholar] [CrossRef]
- Kriat, M.; Nicoli, F.; Vion-Dury, J.; Confort-Gouny, S.; Dano, P.; Grisoli, F.; Gastaut, J.L.; Cozzone, P.J. High resolution NMR spectroscopy of CSF: Methodological issues and perspective clinical applications. Ann. Biol. Clin. 1991, 49, 461–467. [Google Scholar]
- Brinton, R.D. Estrogen regulation of glucose metabolism and mitochondrial function: Therapeutic implications for prevention of Alzheimer’s disease. Adv. Drug Deliv. Rev. 2008, 60, 1504–1511. [Google Scholar] [CrossRef]
- Rettberg, J.R.; Yao, J.; Brinton, R.D. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 2014, 35, 8–30. [Google Scholar] [CrossRef]
- Balthazart, J.; Ball, G.F. Is brain estradiol a hormone or a neurotransmitter? Trends Neurosci. 2006, 29, 241–249. [Google Scholar] [CrossRef]
- Garcia-Ovejero, D.; Azcoitia, I.; Doncarlos, L.L.; Melcangi, R.C.; Garcia-Segura, L.M. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res. Brain Res. Rev. 2005, 48, 273–286. [Google Scholar] [CrossRef]
- Mitterling, K.L.; Spencer, J.L.; Dziedzic, N.; Shenoy, S.; McCarthy, K.; Waters, E.M.; McEwen, B.S.; Milner, T.A. Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J. Comp. Neurol. 2010, 518, 2729–2743. [Google Scholar] [CrossRef]
- Nunez, J.L.; McCarthy, M.M. Androgens predispose males to GABAA-mediated excitotoxicity in the developing hippocampus. Exp. Neurol. 2008, 210, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, C.; Fernell, E.; Kocovska, E.; Minnis, H.; Bourgeron, T.; Thompson, L.; Allely, C.S. The role of cholesterol metabolism and various steroid abnormalities in autism spectrum disorders: A hypothesis paper. Autism Res. 2017, 10, 1022–1044. [Google Scholar] [CrossRef] [PubMed]
- Madeira, C.; Vargas-Lopes, C.; Brandao, C.O.; Reis, T.; Laks, J.; Panizzutti, R.; Ferreira, S.T. Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients With Probable Alzheimer’s Disease and Depression. Front. Psychiatry 2018, 9, 561. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Jourdain, P.; Allaman, I.; Rothenfusser, K.; Fiumelli, H.; Marquet, P.; Magistretti, P.J. L-Lactate protects neurons against excitotoxicity: Implication of an ATP-mediated signaling cascade. Sci. Rep. 2016, 6, 21250. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Tabas, I. Cholesterol in health and disease. J. Clin. Investig. 2002, 110, 583–590. [Google Scholar] [CrossRef]
- Gill, S.; Chow, R.; Brown, A.J. Sterol regulators of cholesterol homeostasis and beyond: The oxysterol hypothesis revisited and revised. Prog. Lipid Res. 2008, 47, 391–404. [Google Scholar] [CrossRef]
- Du, X.; Pham, Y.H.; Brown, A.J. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: Implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 47010–47016. [Google Scholar] [CrossRef]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.; Shah, R.; Kotti, T. Cholesterol 24-hydroxylase: An enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef]
- Meljon, A.; Theofilopoulos, S.; Shackleton, C.H.; Watson, G.L.; Javitt, N.B.; Knolker, H.J.; Saini, R.; Arenas, E.; Wang, Y.; Griffiths, W.J. Analysis of bioactive oxysterols in newborn mouse brain by LC/MS. J. Lipid Res. 2012, 53, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Urano, Y.; Takabe, W.; Saito, Y.; Noguchi, N. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA: Cholesterol acyltransferase 1. Cell Death Dis. 2014, 5, e990. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Doherty, J.J.; Robichaud, A.J.; Belfort, G.M.; Chow, B.Y.; Hammond, R.S.; Crawford, D.C.; Linsenbardt, A.J.; Shu, H.J.; Izumi, Y.; et al. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci. 2013, 33, 17290–17300. [Google Scholar] [CrossRef]
- Taylor, A.M.; Bus, T.; Sprengel, R.; Seeburg, P.H.; Rawlins, J.N.; Bannerman, D.M. Hippocampal NMDA receptors are important for behavioural inhibition but not for encoding associative spatial memories. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130149. [Google Scholar] [CrossRef]
- Sun, M.Y.; Taylor, A.; Zorumski, C.F.; Mennerick, S. 24S-hydroxycholesterol and 25-hydroxycholesterol differentially impact hippocampal neuronal survival following oxygen-glucose deprivation. PLoS ONE 2017, 12, e0174416. [Google Scholar] [CrossRef]
- Bhatti, U.F.; Karnovsky, A.; Dennahy, I.S.; Kachman, M.; Williams, A.M.; Nikolian, V.C.; Biesterveld, B.E.; Siddiqui, A.; O’Connell, R.L.; Liu, B.; et al. Pharmacologic modulation of brain metabolism by valproic acid can induce a neuroprotective environment. J. Trauma. Acute Care Surg. 2021, 90, 507–514. [Google Scholar] [CrossRef]
- Kamat, D.V.; Chakravorty, B.P. Comparative values of CSF-cholesterol and CSF-triglycerides along with other biochemical parameters in neurological disorders. Indian J. Med. Sci. 1996, 50, 280–284. [Google Scholar]
- Almaguel, F.G.; Liu, J.W.; Pacheco, F.J.; De Leon, D.; Casiano, C.A.; De Leon, M. Lipotoxicity-mediated cell dysfunction and death involve lysosomal membrane permeabilization and cathepsin L activity. Brain Res. 2010, 1318, 133–143. [Google Scholar] [CrossRef]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Ghauri, F.Y.; Nicholson, J.K.; Sweatman, B.C.; Wood, J.; Beddell, C.R.; Lindon, J.C.; Cairns, N.J. NMR spectroscopy of human post mortem cerebrospinal fluid: Distinction of Alzheimer’s disease from control using pattern recognition and statistics. NMR Biomed. 1993, 6, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kalloniatis, M. Amino acids in neurotransmission and disease. J. Am. Optom. Assoc. 1995, 66, 750–757. [Google Scholar] [PubMed]
- Sadasivudu, B.; Rao, T.I. Studies on functional and metabolic role of urea cycle intermediates in brain. J. Neurochem. 1976, 27, 785–794. [Google Scholar] [CrossRef]
- McAdoo, D.J.; Hughes, M.G.; Nie, L.; Shah, B.; Clifton, C.; Fullwood, S.; Hulsebosch, C.E. The effect of glutamate receptor blockers on glutamate release following spinal cord injury. Lack of evidence for an ongoing feedback cascade of damage → glutamate release → damage → glutamate release → etc. Brain Res. 2005, 1038, 92–99. [Google Scholar] [CrossRef]
- Wagner, A.K.; Bayir, H.; Ren, D.; Puccio, A.; Zafonte, R.D.; Kochanek, P.M. Relationships between cerebrospinal fluid markers of excitotoxicity, ischemia, and oxidative damage after severe TBI: The impact of gender, age, and hypothermia. J. Neurotrauma 2004, 21, 125–136. [Google Scholar] [CrossRef]
- Kim, H.H.; Jeong, I.H.; Hyun, J.S.; Kong, B.S.; Kim, H.J.; Park, S.J. Metabolomic profiling of CSF in multiple sclerosis and neuromyelitis optica spectrum disorder by nuclear magnetic resonance. PLoS ONE 2017, 12, e0181758. [Google Scholar] [CrossRef]
- Leoni, V.; Strittmatter, L.; Zorzi, G.; Zibordi, F.; Dusi, S.; Garavaglia, B.; Venco, P.; Caccia, C.; Souza, A.L.; Deik, A.; et al. Metabolic consequences of mitochondrial coenzyme A deficiency in patients with PANK2 mutations. Mol. Genet. Metab. 2012, 105, 463–471. [Google Scholar] [CrossRef]
- Toczylowska, B.; Chalimoniuk, M.; Wodowska, M.; Mayzner-Zawadzk, E. Changes in concentration of cerebrospinal fluid components in patients with traumatic brain injury. Brain Res. 2006, 1104, 183–189. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Simpkins, J.W.; Alkon, D.L.; Smith, K.E.; Chen, Y.W.; Tan, Z.; Huber, J.D.; Rosen, C.L. Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimers Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef]
- Wuttke, A.; Yu, Q.; Tengholm, A. Autocrine Signaling Underlies Fast Repetitive Plasma Membrane Translocation of Conventional and Novel Protein Kinase C Isoforms in beta Cells. J. Biol. Chem. 2016, 291, 14986–14995. [Google Scholar] [CrossRef] [PubMed]
- Stejskalova, C.; Arrigoni, F.; Albanesi, R.; Bertini, L.; Mollica, L.; Coscia, F. A conserved acidic residue drives thyroxine synthesis within thyroglobulin and other protein precursors. J. Biol. Chem. 2024, 301, 108026. [Google Scholar] [CrossRef] [PubMed]
- Schuurs, T.A.; Morariu, A.M.; Ottens, P.J.; Hart, N.T.; Popma, S.H.; Leuvenink, H.G.; Ploeg, R.J. Time-dependent changes in donor brain death related processes. Am. J. Transplant. 2006, 6, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Belzberg, H.; Shoemaker, W.C.; Wo, C.C.; Nicholls, T.P.; Dang, A.B.; Zelman, V.; Gruen, J.P.; Berne, T.V.; Demetriades, D. Hemodynamic and oxygen transport patterns after head trauma and brain death: Implications for management of the organ donor. J. Trauma 2007, 63, 1032–1042. [Google Scholar] [CrossRef]
- Chen, E.P.; Bittner, H.B.; Kendall, S.W.; Van Trigt, P. Hormonal and hemodynamic changes in a validated animal model of brain death. Crit. Care Med. 1996, 24, 1352–1359. [Google Scholar] [CrossRef]
- Herijgers, P.; Leunens, V.; Tjandra-Maga, T.B.; Mubagwa, K.; Flameng, W. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation 1996, 62, 330–335. [Google Scholar] [CrossRef]
- Novitzky, D.; Cooper, D.K.; Morrell, D.; Isaacs, S. Change from aerobic to anaerobic metabolism after brain death, and reversal following triiodothyronine therapy. Transplantation 1988, 45, 32–36. [Google Scholar] [CrossRef]
- Smith, M. Physiologic changes during brain stem death—Lessons for management of the organ donor. J. Heart Lung Transplant. 2004, 23 (Suppl. S9), S217–S222. [Google Scholar] [CrossRef]
- Podlecka-Pietowska, A.; Kacka, A.; Zakrzewska-Pniewska, B.; Nojszewska, M.; Zieminska, E.; Chalimoniuk, M.; Toczylowska, B. Altered Cerebrospinal Fluid Concentrations of Hydrophobic and Hydrophilic Compounds in Early Stages of Multiple Sclerosis-Metabolic Profile Analyses. J. Mol. Neurosci. 2019, 69, 94–105. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Li, X.; Yang, S.B.; Qiu, Y.P.; Zhao, T.; Chen, T.L.; Su, M.M.; Chu, L.X.; Lv, A.P.; Liu, P.; Jia, W. Urinary metabolomics as a potentially novel diagnostic and stratification tool for knee osteoarthritis. Metabolomics 2010, 6, 109–118. [Google Scholar] [CrossRef]
- Lutz, N.W.; Maillet, S.; Nicoli, F.; Viout, P.; Cozzone, P.J. Further assignment of resonances in 1H NMR spectra of cerebrospinal fluid (CSF). FEBS Lett. 1998, 425, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, A.; Rosel, P.; Marron, T. Cerebrospinal fluid: Postmortem biochemical study. J. Clin. Forensic Med. 2005, 12, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification. J. Chemom. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BD vs. Controls [%] | p Value | VIP Value |
|---|---|---|---|
| Formate | >1000 | 0.003 | |
| Nicotinamide N-oxide | >1000 | 0.011 | |
| Guanine | >1000 | 0.021 | |
| Phenylalanine | >1000 | 0.001 | 1.18 |
| Tyrosine | >1000 | 0.002 | |
| Histidine | >1000 | 0.001 | 1.21 |
| Urea | >1000 | 0.019 | |
| α-D glucose | >1000 | 0.020 | |
| β-D-glucose | >1000 | 0.088 | |
| Threonine | >1000 | 0.001 | 1.19 |
| Myo-inositol | >1000 | 0.001 | |
| Guanidinoacetate | >1000 | 0.002 | 1.07 |
| Glycine | >1000 | 0.009 | |
| Glycerol | >1000 | 0.001 | |
| Scyllo-inositol | 224 | 0.001 | |
| Taurine | >1000 | 0.001 | |
| Glycerophosphocholine | >1000 | 0.001 | 1.07 |
| Choline | >1000 | 0.001 | 1.26 |
| Citrulline | >1000 | 0.003 | |
| Creatine/Creatinine | >1000 | 0.002 | |
| Sarcosine/2-Ketobutyric acid | >1000 | 0.105 | |
| Citrate | >1000 | 0.001 | 1.03 |
| Glutamine | >1000 | 0.001 | 1.01 |
| Oxoglutarate/Succinate | >1000 | 0.001 | 1.01 |
| Pyruvate | >1000 | 0.001 | 1.16 |
| N-Acetylaspartylglutamate | >1000 | 0.066 | |
| Acetoacetate | >1000 | 0.001 | |
| Acetone | >1000 | 0.003 | |
| N-Acetyl-L-Aspartate | >1000 | 0.001 | |
| Acetate | >1000 | 0.212 | |
| Lysine | >1000 | 0.001 | 1.11 |
| Alanine | >1000 | 0.001 | 1.16 |
| Lactate | >1000 | 0.001 | 1.08 |
| 3-Hydroxybutyrate | >1000 | 0.009 | |
| α-Ketoisovaleric acid | >1000 | 0.001 | 1.40 |
| Isobutyric acid | >1000 | 0.001 | 1.01 |
| Valine | >1000 | 0.001 | 1.21 |
| Isoleucine | >1000 | 0.001 | 1.17 |
| Leucine | >1000 | 0.001 | 1.15 |
| Valeric acid | >1000 | 0.007 | |
| Lipids | >1000 | 0.001 | 1.46 |
| Compound/Functional Group | BD vs. Controls [%] | p Value | VIP Value |
|---|---|---|---|
| Estriol | 16 | 0.018 | 1.10 |
| Sphingomyelin | 44 | 0.621 | |
| Phosphatidylcholine Phosphatidylethanoloamine Sphingomyelin | 27 | 0.750 | |
| PUFA and MUFA | 28 | 0.519 | |
| Trigliceride | 211 | 0.259 | |
| 1,2 Diacylgliceride | >1000 | 0.001 | 1.02 |
| Phosphocholine | 322 | 0.111 | |
| Phosphocholine/Sphingomyelin | 85 | 0.065 | |
| Phosphoetanolamine | >1000 | 0.040 | 1.10 |
| Arachidonic, alfa-linolenic | 267 | 0.255 | |
| Linoleic acid | 194 | 0.241 | |
| Lauric, myristic/palmitic, arachidonic, alfa-linolenic/oleic | 896 | 0.624 | |
| Arachidonic acid | 19 | 0.397 | |
| Pelargonic, arachidonic, oleic and palmitoleic acids | 98 | 0.727 | |
| Lauric/palmitic | 2 | 0.671 | |
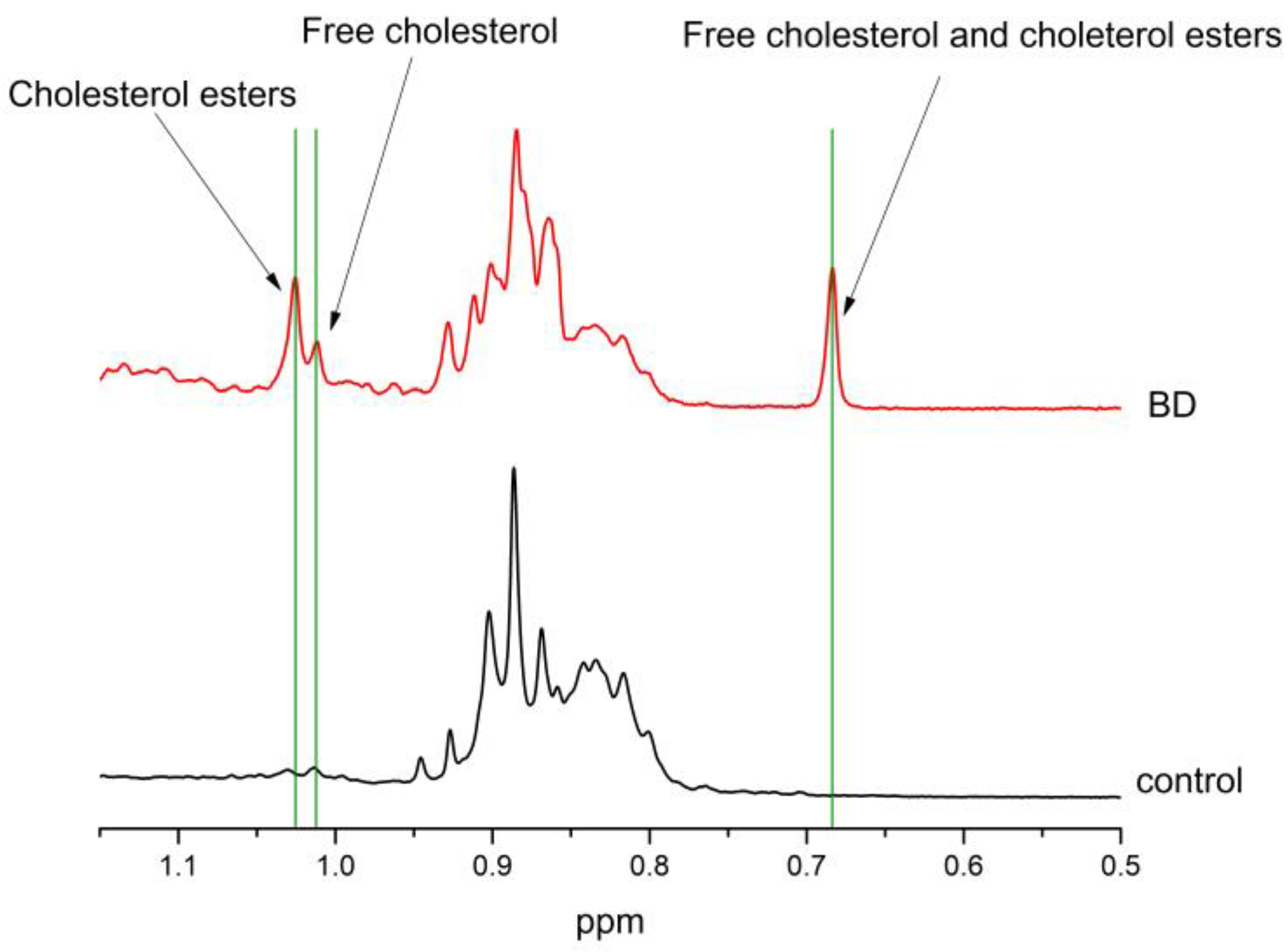
| Cholesterol esters | >1000 | 0.001 | 1.80 |
| Free cholesterol | >1000 | 0.001 | 1.62 |
| Dodecanonic, palmitic/ arachidonic/palmitoleic, oleic, | 73 | 0.873 | |
| Pelargonic acid | 41 | 0.671 | |
| Free cholesterol and cholesterol esters | >1000 | 0.001 | 1.68 |
| Metabolites | Metabolic Pathway | Most Likely Pathomechanism |
|---|---|---|
| Estriol | Steroid hormone biosynthesis | Disturbances in energy production and neurotransmission |
| 1,2 Diacylglyceride | Lipids metabolism | Disturbances in energy production and cell membrane function |
| Cholesterol Cholesterol ester | Cholesterol metabolism | Disturbances in neurotransmission |
| Oxoglutarate/Succinate Citrate Guanidinoacetate Threonine | TCA cycle Glycine, serine, and threonine metabolism | Oxidative stress and disturbances in energy production |
| Histidine Glutamine | Histidine metabolism Alanine, aspartate, and glutamate metabolism | Disturbances in neurotransmission |
| Choline | Glycerophospholipid metabolism | Disturbances in neurotransmission |
| Pyruvate Alanine Lactate | Taurine, alanine, and pyruvate metabolism, TCA cycle | Disturbances in neurotransmission and oxidative stress Disturbances in energy production |
| Isobutyric acid | Butanoate metabolism | Disturbances in neurotransmission |
| Lysine | Lysine biosynthesis or degradation | Disturbances in energy production and neurotransmission |
| α-ketoisovaleric acid Leucine Valine Isoleucine | Valine, leucine, and isoleucine biosynthesis or degradation | Disturbances in neurotransmission and oxidative stress |
| Phenylalanine | Phenylalanine metabolism | Disturbances in neurotransmission |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toczylowska, B.; Kalinowski, P.; Kacka-Piotrowska, A.; Duda, P.; Grąt, M.; Zieminska, E. Metabolic Pattern of Brain Death—NMR-Based Metabolomics of Cerebrospinal Fluid. Int. J. Mol. Sci. 2025, 26, 2719. https://doi.org/10.3390/ijms26062719
Toczylowska B, Kalinowski P, Kacka-Piotrowska A, Duda P, Grąt M, Zieminska E. Metabolic Pattern of Brain Death—NMR-Based Metabolomics of Cerebrospinal Fluid. International Journal of Molecular Sciences. 2025; 26(6):2719. https://doi.org/10.3390/ijms26062719
Chicago/Turabian StyleToczylowska, Beata, Piotr Kalinowski, Agata Kacka-Piotrowska, Paulina Duda, Michał Grąt, and Elzbieta Zieminska. 2025. "Metabolic Pattern of Brain Death—NMR-Based Metabolomics of Cerebrospinal Fluid" International Journal of Molecular Sciences 26, no. 6: 2719. https://doi.org/10.3390/ijms26062719
APA StyleToczylowska, B., Kalinowski, P., Kacka-Piotrowska, A., Duda, P., Grąt, M., & Zieminska, E. (2025). Metabolic Pattern of Brain Death—NMR-Based Metabolomics of Cerebrospinal Fluid. International Journal of Molecular Sciences, 26(6), 2719. https://doi.org/10.3390/ijms26062719