
Enhancing HCV NS3 Inhibitor Classification with Optimized Molecular Fingerprints Using Random Forest

Abstract
1. Introduction
Literature Review
2. Results and Discussion
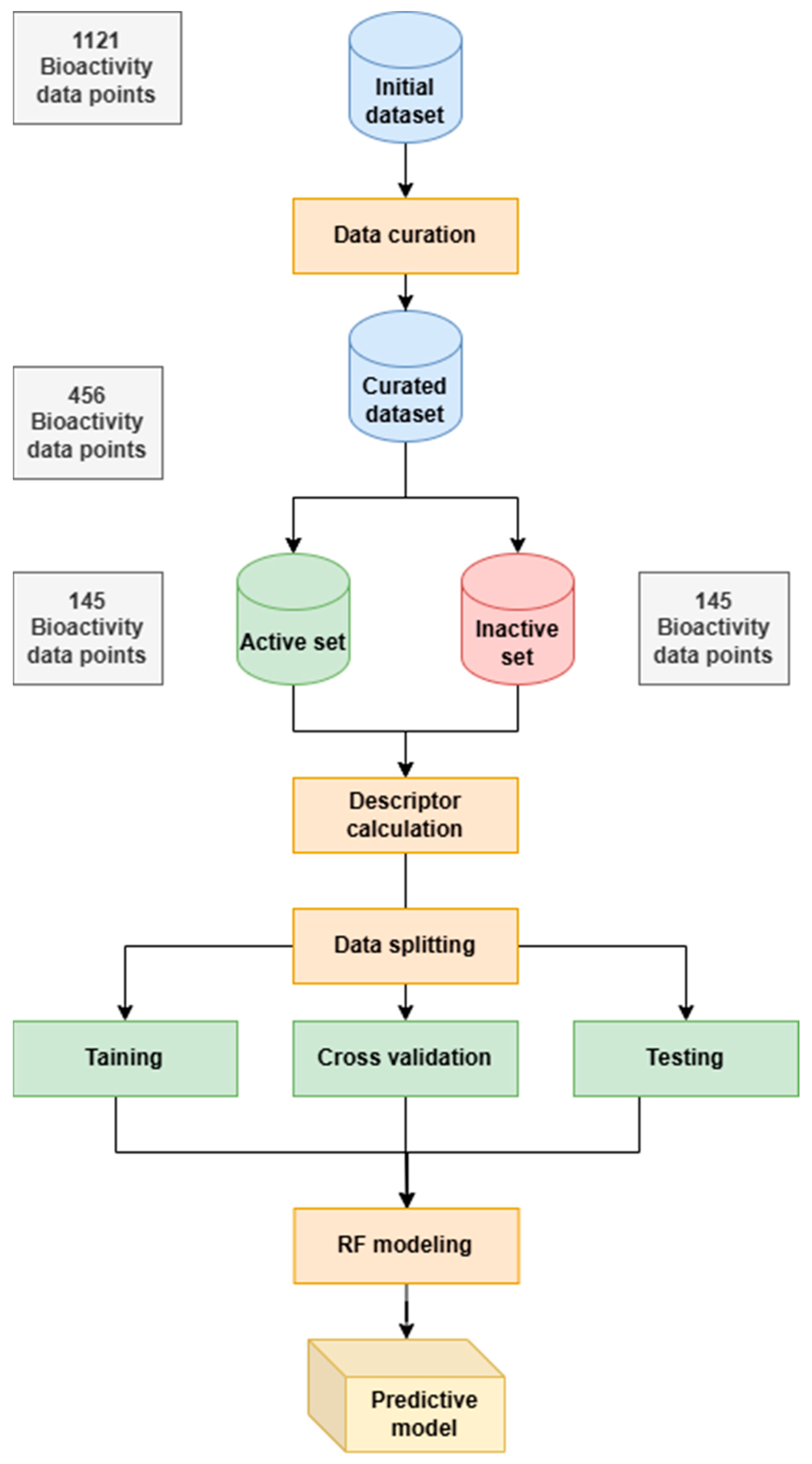
2.1. Experimental Setup
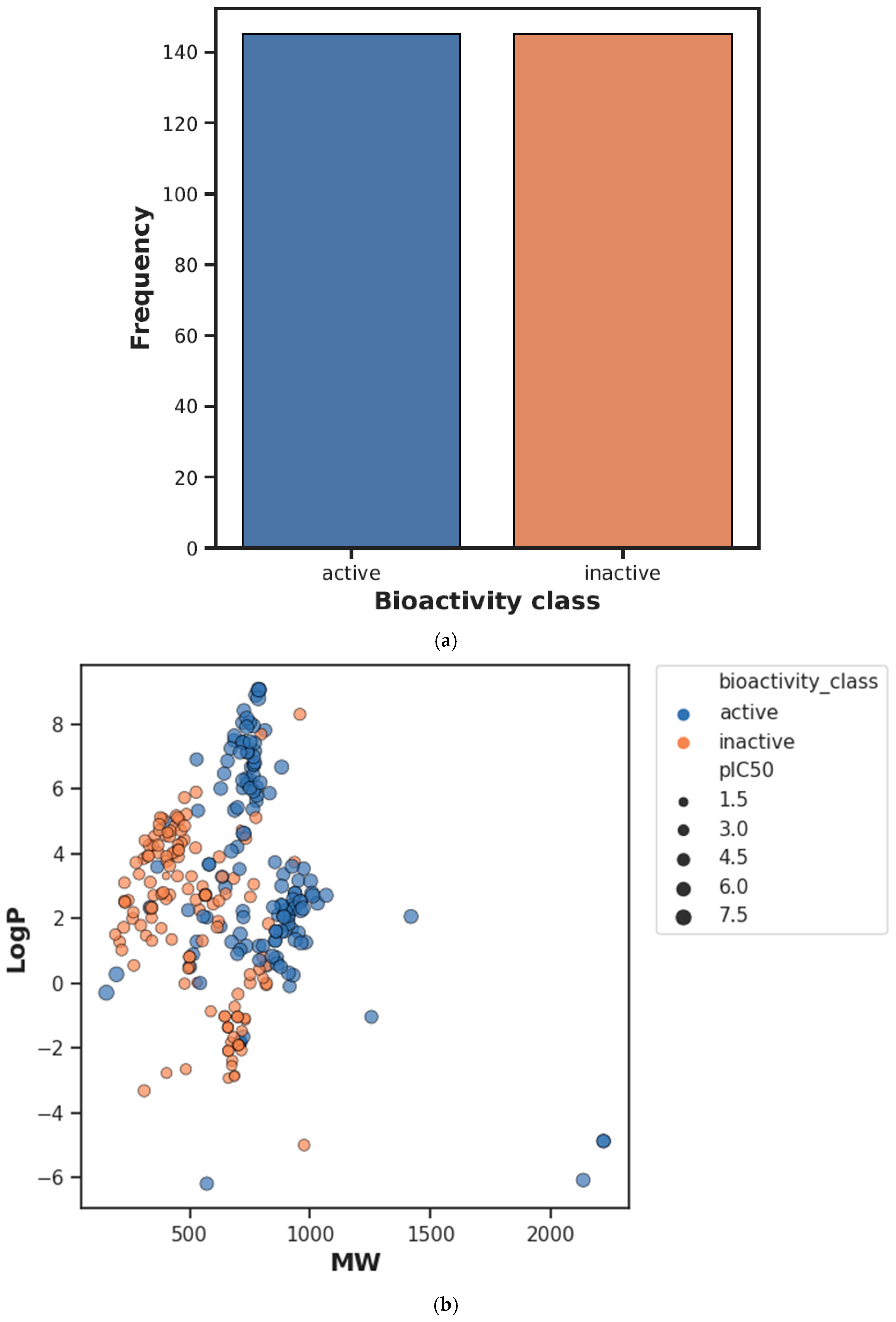
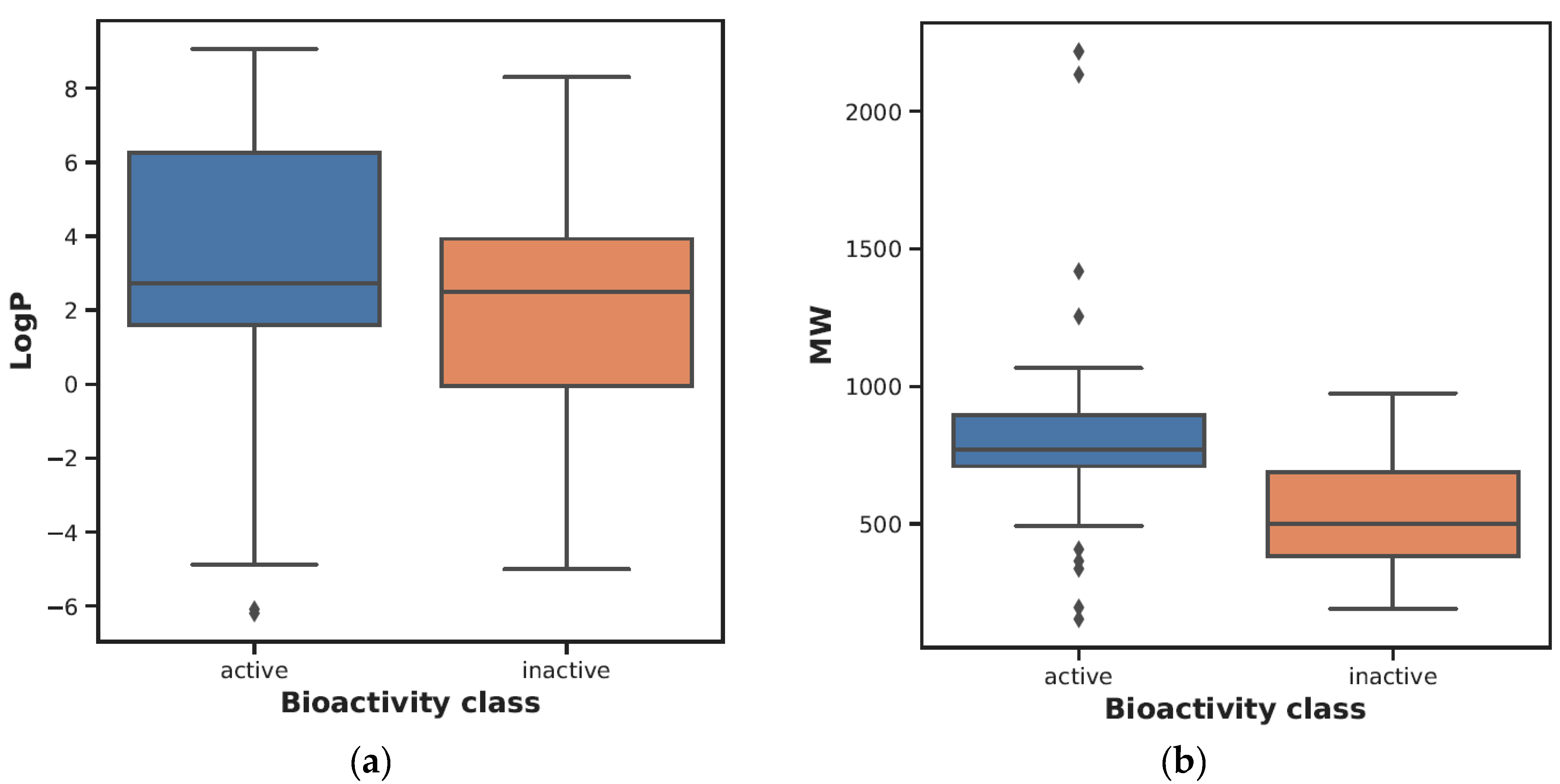
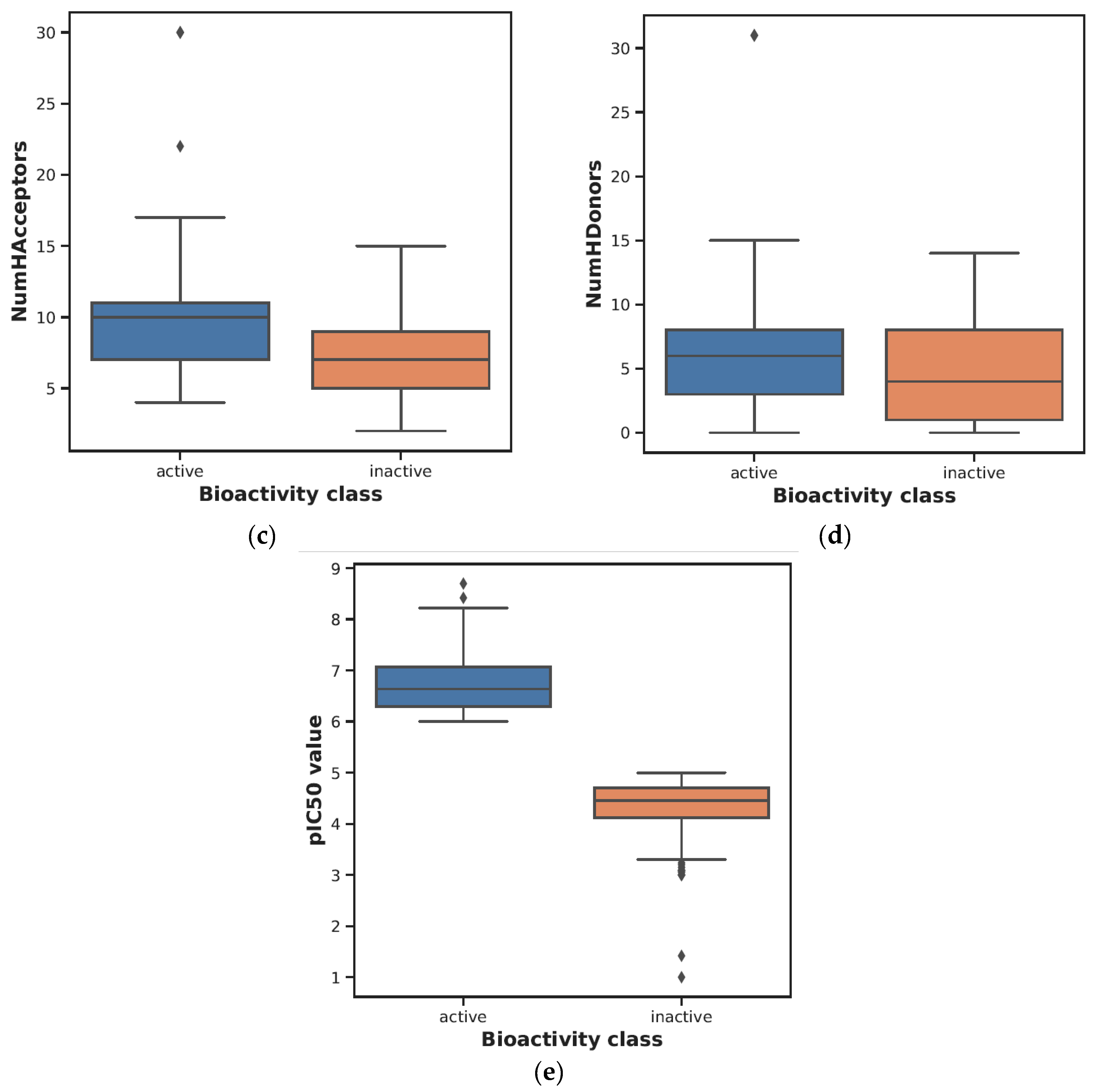
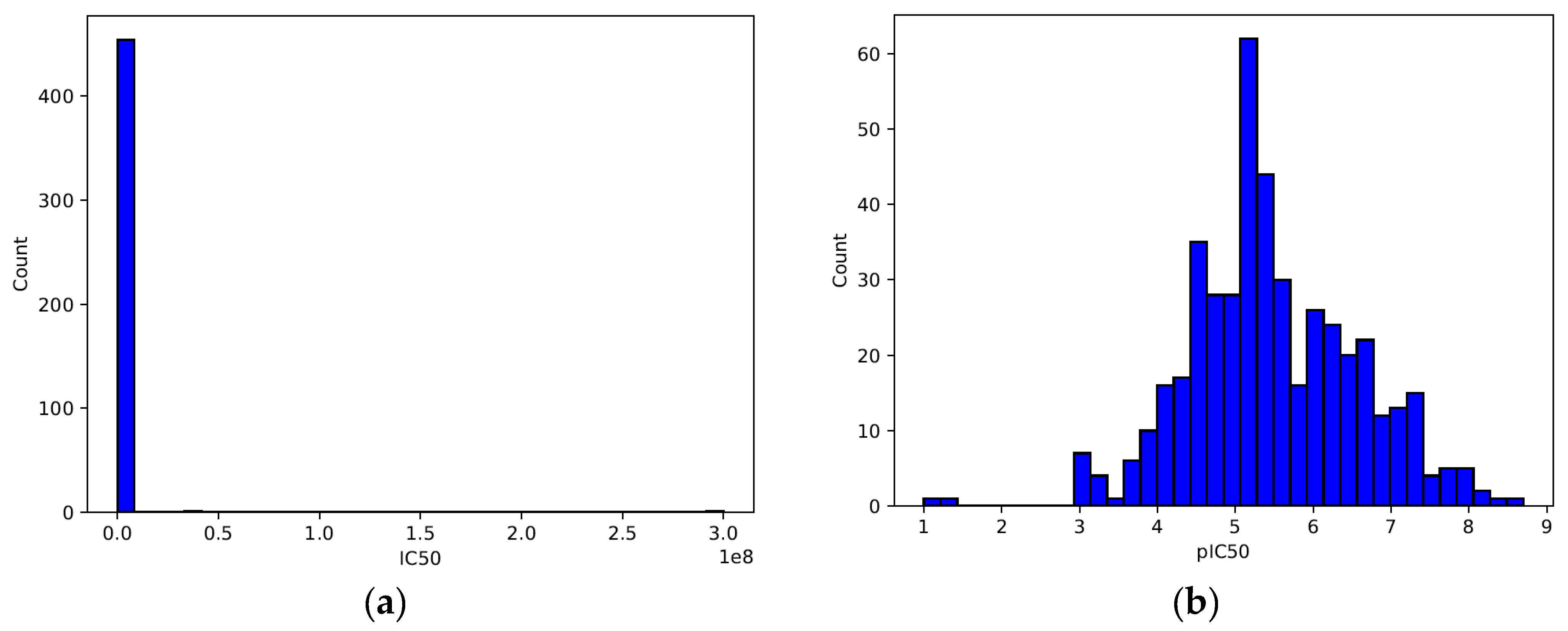
2.2. Chemical Space Analysis
2.3. QSAR Modeling and Bioactivity Class Analysis
2.4. Model Evaluation
3. Materials and Methods
3.1. Dataset
3.2. Data Preprocessing
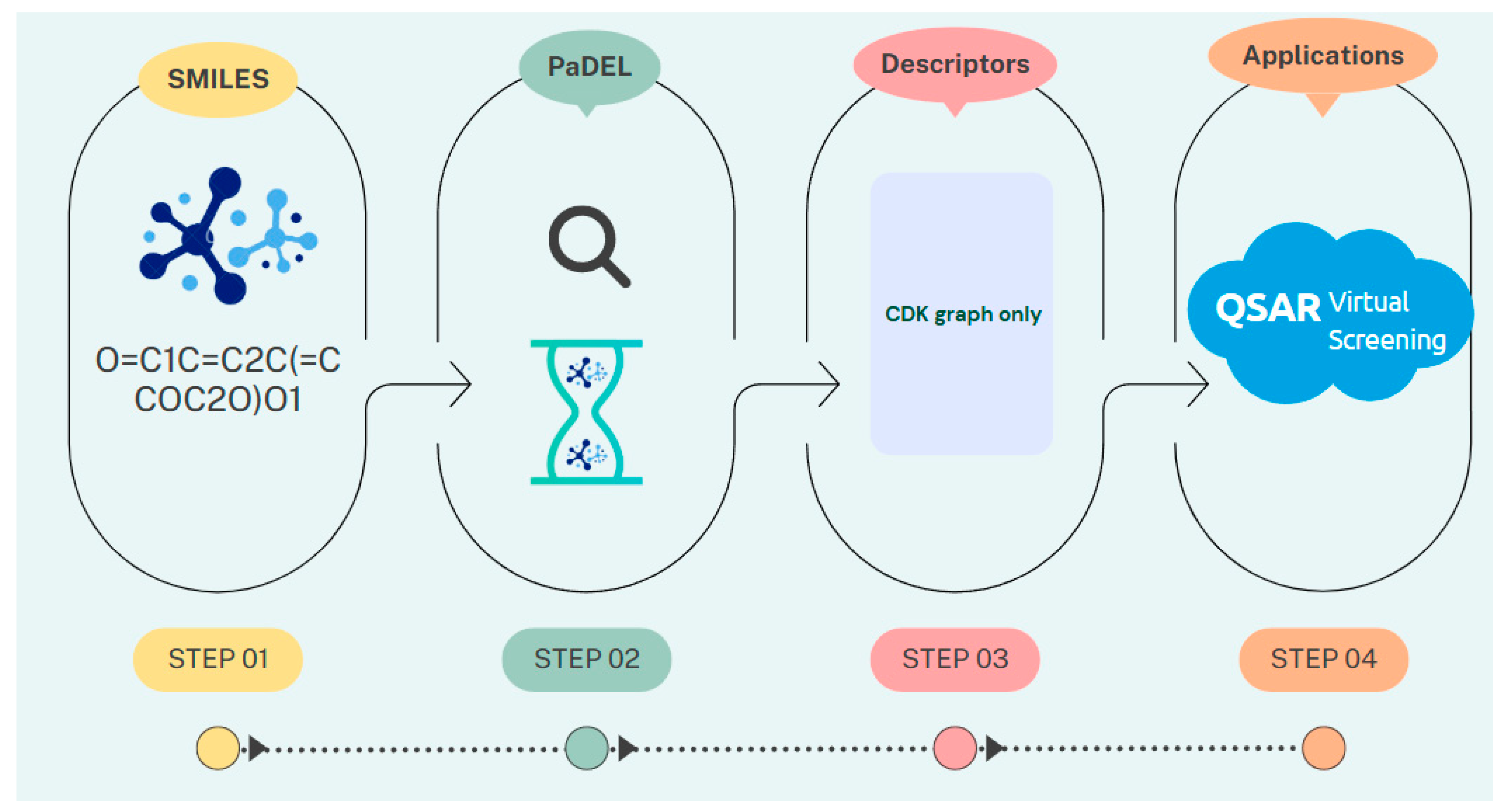
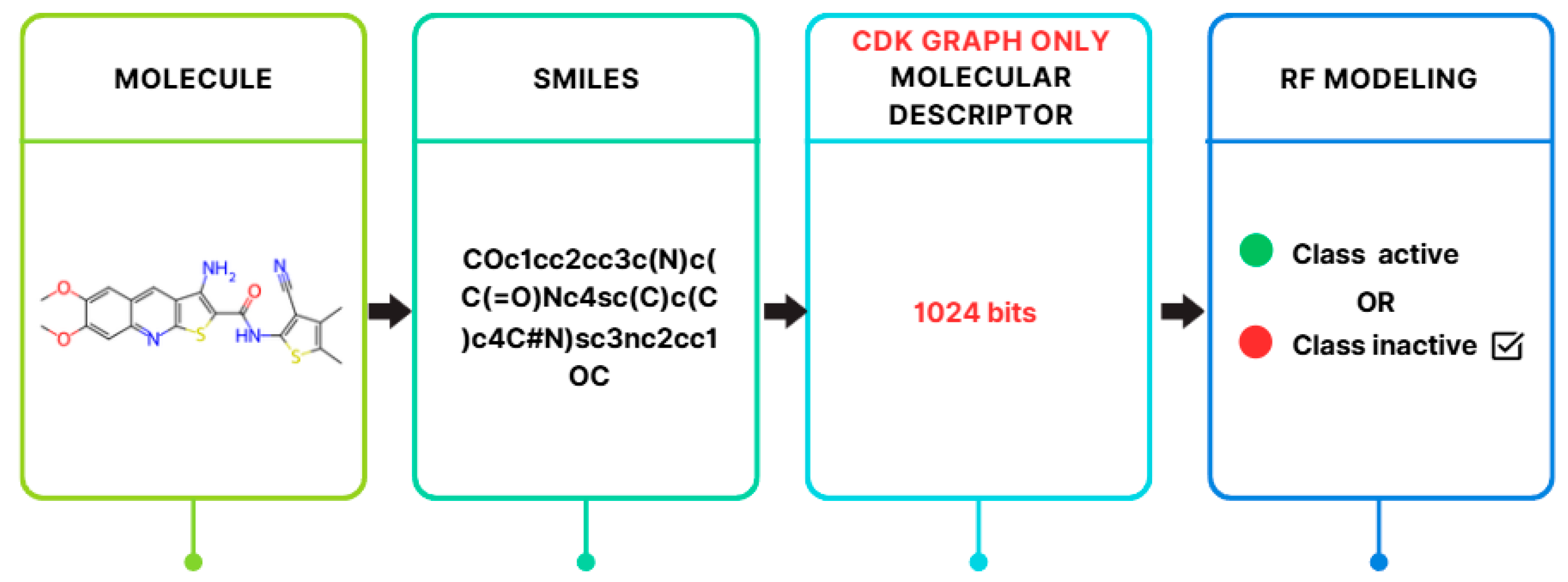
3.3. Molecular Fingerprint
3.4. QSAR Modeling
3.5. Exploratory Data Analysis (EDA)
3.6. Assessment of Model Performance
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 20 December 2024).
- Salam, K.A.; Akimitsu, N. Hepatitis C Virus NS3 Inhibitors: Current and Future Perspectives. Biomed. Res. Int. 2013, 2013, 467869. [Google Scholar] [CrossRef]
- Bunally, S.B.; Luscombe, C.N.; Young, R.J. Using Physicochemical Measurements to Influence Better Compound Design. SLAS Discov. Adv. Life Sci. R&D 2019, 24, 791–801. [Google Scholar]
- Malik, A.A.; Phanus-umporn, C.; Schaduangrat, N.; Shoombuatong, W.; Isarankura-Na-Ayudhya, C.; Nantasenamat, C. HCVpred: A Web Server for Predicting the Bioactivity of Hepatitis C Virus NS5B Inhibitors. J. Comput. Chem. 2020, 41, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Atasever, S. In Silico Drug Discovery: A Machine Learning-Driven Systematic Review. Med. Chem. Res. 2024, 33, 1465–1490. [Google Scholar] [CrossRef]
- Bajorath, J. Computer-Aided Drug Discovery. F1000Research 2015, 4, 630. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in Drug Design—A Review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Ponzoni, I.; Sebastián-Pérez, V.; Martínez, M.J.; Roca, C.; la Cruz Pérez, C.; Cravero, F.; Vazquez, G.E.; Páez, J.A.; Díaz, M.F.; Campillo, N.E. QSAR Classification Models for Predicting the Activity of Inhibitors of Beta-Secretase (BACE1) Associated with Alzheimer’s Disease. Sci. Rep. 2019, 9, 9102. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Pérez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools against the DUD Data Set Reveals Limitations of Current 3D Methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance Evaluation of 2D Fingerprint and 3D Shape Similarity Methods in Virtual Screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef]
- Jaganathan, K.; Tayara, H.; Chong, K.T. Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. Int. J. Mol. Sci. 2021, 22, 8073. [Google Scholar] [CrossRef]
- Musmuca, I.; Caroli, A.; Mai, A.; Kaushik-Basu, N.; Arora, P.; Ragno, R. Combining 3-D Quantitative Structure- Activity Relationship with Ligand Based and Structure Based Alignment Procedures for in Silico Screening of New Hepatitis C Virus NS5B Polymerase Inhibitors. J. Chem. Inf. Model. 2010, 50, 662–676. [Google Scholar] [CrossRef]
- Wang, M.; Xuan, S.; Yan, A.; Yu, C. Classification Models of HCV NS3 Protease Inhibitors Based on Support Vector Machine (SVM). Comb. Chem. High Throughput Screen. 2015, 18, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Meewan, I.; Zhang, X.; Roy, S.; Ballatore, C.; O’Donoghue, A.J.; Schooley, R.T.; Abagyan, R. Discovery of New Inhibitors of Hepatitis C Virus NS3/4A Protease and Its D168A Mutant. ACS Omega 2019, 4, 16999–17008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Y.; Gao, Y.; Hu, W.; Qu, Y.; Lou, N.; Zhu, Y.; Zhang, X.; Yang, H. Hepatitis C Virus NS3 Protein Enhances Hepatocellular Carcinoma Cell Invasion by Promoting PPM1A Ubiquitination and Degradation. J. Exp. Clin. Cancer Res. 2017, 36, 42. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Takegami, T.; Shiozaki, T.; Miyazaki, T. Hepatitis C Virus NS3 Protein Can Activate the Notch-Signaling Pathway through Binding to a Transcription Factor, SRCAP. PLoS ONE 2011, 6, e20718. [Google Scholar] [CrossRef]
- Kamboj, S.; Rajput, A.; Rastogi, A.; Thakur, A.; Kumar, M. Targeting Non-Structural Proteins of Hepatitis C Virus for Predicting Repurposed Drugs Using QSAR and Machine Learning Approaches. Comput. Struct. Biotechnol. J. 2022, 20, 3422–3438. [Google Scholar] [CrossRef]
- Hentabli, H.; Saeed, F.; Abdo, A.; Salim, N. A New Graph-Based Molecular Descriptor Using the Canonical Representation of the Molecule. Sci. World J. 2014, 2014, 286974. [Google Scholar] [CrossRef]
- Gong, Y.; Teng, D.; Wang, Y.; Gu, Y.; Wu, Z.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Potential Drug-Induced Nephrotoxicity with Machine Learning Methods. J. Appl. Toxicol. 2022, 42, 1639–1650. [Google Scholar] [CrossRef]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo-and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- PubChem Substructure Fingerprint Description. Available online: https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt (accessed on 25 July 2024).
- Laggner, C. SMARTS Patterns for Functional Group Classification; Inte: Ligand Software-Entwicklungs und Consulting GmbH: Wien, Austria, 2005. [Google Scholar]
- Klekota, J.; Roth, F.P. Chemical Substructures That Enrich for Biological Activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Phanus-Umporn, C.; Shoombuatong, W.; Prachayasittikul, V.; Anuwongcharoen, N.; Nantasenamat, C. Privileged Substructures for Anti-Sickling Activity via Cheminformatic Analysis. RSC Adv. 2018, 8, 5920–5935. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Aqeel, I.; Bilal, M.; Majid, A.; Majid, T. Hybrid Approach to Identifying Druglikeness Leading Compounds Against COVID-19 3CL Protease. Pharmaceuticals 2022, 15, 1333. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.A.; Chotpatiwetchkul, W.; Phanus-Umporn, C.; Nantasenamat, C.; Charoenkwan, P.; Shoombuatong, W. StackHCV: A Web-Based Integrative Machine-Learning Framework for Large-Scale Identification of Hepatitis C Virus NS5B Inhibitors. J. Comput. Aided Mol. Des. 2021, 35, 1037–1053. [Google Scholar] [CrossRef]
- Molecular Descriptors. Available online: https://chem.libretexts.org/Courses/Intercollegiate_Courses/Cheminformatics/06%3A_Molecular_Similarity/6.01%3A_Molecular_Descriptors (accessed on 25 July 2024).
- Liu, Y.; Bi, M.; Zhang, X.; Zhang, N.; Sun, G.; Zhou, Y.; Zhao, L.; Zhong, R. Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors. Processes 2021, 9, 2074. [Google Scholar] [CrossRef]
- Amrehn, M.; Mualla, F.; Angelopoulou, E.; Steidl, S.; Maier, A. The Random Forest Classifier in WEKA: Discussion and New Developments for Imbalanced Data. arXiv 2018, arXiv:1812.08102. [Google Scholar]
- Osmanli, S.; Akansu, S.O.; Azginoglu, N.; Akansu, Y.E.; Develi, I. Investigation of S1046 Profile Bladed Vertical Axis Wind Turbine and Artificial Intelligence-Based Performance Evaluation. Energy Sources Part A Recovery Util. Environ. Eff. 2023, 45, 8771–8790. [Google Scholar] [CrossRef]
- Voršilák, M.; Kolář, M.; Čmelo, I.; Svozil, D. SYBA: Bayesian Estimation of Synthetic Accessibility of Organic Compounds. J. Cheminform. 2020, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.; Williams, J.; Nguyễn, Ð.-T.; Neyra, J.; Southall, N.; Mathé, E.; Xu, X.; Shah, P. Validating ADME QSAR Models Using Marketed Drugs. SLAS Discov. Sci. Drug Discov. 2021, 26, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A Review on Machine Learning Approaches and Trends in Drug Discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef] [PubMed]
- Prihandoko, P.; Bertalya, B.; Setyowati, L. City Health Prediction Model Using Random Forest Classification Method. In Proceedings of the 2020 Fifth International Conference on Informatics and Computing (ICIC), Gorontalo, Indonesia, 3–4 November 2020; pp. 1–5. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Statistics | p | Alpha | Interpretation |
|---|---|---|---|---|
| LogP | 13,400.5 | 0.000053 | 0.05 | Different distribution (reject H0) |
| MW | 17,897.5 | 4.550260 × 10−25 | 0.05 | Different distribution (reject H0) |
| NumHAcceptors | 14,950.5 | 3.587758 × 10−10 | 0.05 | Different distribution (reject H0) |
| NumHDonors | 13,257.0 | 0.000111 | 0.05 | Different distribution (reject H0) |
| pIC50 | 21,025.0 | 4.634027 × 10−49 | 0.05 | Different distribution (reject H0) |
| Fingerprint | #Features | Description | References |
|---|---|---|---|
| CDK | 1024 | Fingerprint of length 1024 and search depth of 8 | [20] |
| CDK extended | 1024 | Extends the fingerprint with additional bits describing ring features | [20] |
| CDK graph only | 1024 | A special version that considers only the connectivity and not bond order | [20] |
| E-state | 79 | Electrotopological state atom types | [21] |
| MACCS | 166 | Binary representation of chemical features defined by MACCS keys | [22] |
| PubChem | 881 | Binary representation of substructures defined by PubChem | [23] |
| Substructure | 307 | Presence of SMARTS patterns for functional groups | [24] |
| Substructure count | 307 | Count of SMARTS patterns for functional groups | [24] |
| Klekota–Roth | 4860 | Presence of chemical substructures | [25] |
| Klekota–Roth count | 4860 | Count of chemical substructures | [25] |
| 2D atom pairs | 780 | Presence of atom pairs at various topological distances | [26] |
| 2D atom pairs count | 780 | Count of atom pairs at various topological distances | [26] |
| Training Set | Ten-Fold CV Set | Test Set | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Descriptor Class | Method | A * | AccTrain (%) | MAE | RMSE | MCC | AccCv (%) | MAE | RMSE | MCC | AccTest (%) | MAE | RMSE | MCC |
| CDK | RF | 1022 | 97.9310 | 0.0870 | 0.1469 | 0.9790 | 88.2759 | 0.2201 | 0.3232 | 0.7660 | 86.2069 | 0.2263 | 0.3490 | 0.7320 |
| CDK | IBk | 1022 | 97.9310 | 0.0254 | 0.1059 | 0.9590 | 86.2069 | 0.1461 | 0.3711 | 0.7240 | 86.2069 | 0.1466 | 0.3726 | 0.7320 |
| CDK | LR | 1022 | 97.9310 | 0.0224 | 0.1059 | 0.9590 | 74.4828 | 0.2558 | 0.4983 | 0.4930 | 79.3103 | 0.2094 | 0.4460 | 0.6130 |
| CDK | AdaBoost | 1022 | 88.6207 | 0.1805 | 0.2950 | 0.7750 | 82.4138 | 0.2170 | 0.3584 | 0.6510 | 86.2069 | 0.1974 | 0.3451 | 0.7320 |
| CDK | OneR | 1022 | 78.2759 | 0.2172 | 0.4661 | 0.5720 | 74.4828 | 0.2552 | 0.5051 | 0.4950 | 70.6897 | 0.2931 | 0.5414 | 0.4410 |
| CDK extended | RF | 1007 | 97.9310 | 0.0845 | 0.1440 | 0.9590 | 87.5862 | 0.2172 | 0.3238 | 0.7520 | 86.2069 | 0.2170 | 0.3456 | 0.7260 |
| CDK extended | IBk | 1007 | 97.931 | 0.0254 | 0.1059 | 0.9590 | 86.5517 | 0.1427 | 0.3665 | 0.7310 | 86.2069 | 0.1466 | 0.3726 | 0.7320 |
| CDK extended | LR | 1007 | 97.9310 | 0.0224 | 0.1059 | 0.9590 | 75.1724 | 0.2506 | 0.4935 | 0.5060 | 77.5862 | 0.2330 | 0.4700 | 0.5560 |
| CDK extended | OneR | 1007 | 75.8621 | 0.2414 | 0.4913 | 0.5260 | 73.1034 | 0.2690 | 0.5186 | 0.4660 | 63.7931 | 0.3621 | 0.6017 | 0.2890 |
| CDK extended | AdaBoost | 1007 | 88.2759 | 0.1857 | 0.2896 | 0.7660 | 80.0000 | 0.2402 | 0.3756 | 0.6020 | 75.8621 | 0.2706 | 0.4054 | 0.5730 |
| Estate | RF | 34 | 94.8276 | 0.1095 | 0.2004 | 0.8980 | 87.9310 | 0.1996 | 0.3223 | 0.7600 | 82.7586 | 0.2366 | 0.3781 | 0.6630 |
| Estate | IBk | 34 | 94.8276 | 0.0697 | 0.1842 | 0.8990 | 87.2414 | 0.1555 | 0.3345 | 0.7480 | 82.7586 | 0.2127 | 0.4128 | 0.6720 |
| Estate | LR | 34 | 88.6207 | 0.1620 | 0.2856 | 0.7730 | 80.3448 | 0.2293 | 0.4017 | 0.6110 | 75.8621 | 0.2515 | 0.4364 | 0.5420 |
| Estate | AdaBoost | 34 | 81.0345 | 0.3266 | 0.3827 | 0.6320 | 78.6207 | 0.3253 | 0.3926 | 0.5790 | 79.3103 | 0.3309 | 0.3997 | 0.6280 |
| Estate | OneR | 34 | 69.6552 | 0.3034 | 0.5509 | 0.4110 | 67.2414 | 0.3276 | 0.5724 | 0.3500 | 63.7931 | 0.3621 | 0.6017 | 0.4140 |
| CDK graph only | RF | 979 | 97.5862 | 0.0864 | 0.1582 | 0.9520 | 88.9655 | 0.1994 | 0.3181 | 0.7790 | 89.6552 | 0.2114 | 0.3304 | 0.7950 |
| CDK graph only | IBk | 979 | 97.5862 | 0.0346 | 0.1264 | 0.9520 | 86.2069 | 0.1541 | 0.3694 | 0.7240 | 84.4828 | 0.1661 | 0.3659 | 0.7020 |
| CDK graph only | LR | 979 | 97.5862 | 0.0320 | 0.1264 | 0.9520 | 73.7931 | 0.2690 | 0.5040 | 0.4790 | 74.1379 | 0.2383 | 0.4695 | 0.4830 |
| CDK graph only | AdaBoost | 979 | 89.3103 | 0.2038 | 0.2959 | 0.7880 | 83.7931 | 0.2540 | 0.3558 | 0.6760 | 86.2069 | 0.1955 | 0.3094 | 0.7260 |
| CDK graph only | OneR | 979 | 72.7586 | 0.2724 | 0.5219 | 0.4610 | 66.5517 | 0.3345 | 0.5783 | 0.3350 | 67.2414 | 0.3276 | 0.5724 | 0.3800 |
| MACCS | RF | 145 | 98.2759 | 0.0769 | 0.1393 | 0.9660 | 87.2414 | 0.1946 | 0.3092 | 0.7450 | 87.9310 | 0.1990 | 0.3104 | 0.7630 |
| MACCS | IBk | 145 | 98.2759 | 0.0229 | 0.1003 | 0.9660 | 86.8966 | 0.1383 | 0.3585 | 0.7390 | 86.2069 | 0.1373 | 0.3548 | 0.7260 |
| MACCS | LR | 145 | 97.2414 | 0.0329 | 0.1286 | 0.9450 | 74.1379 | 0.2619 | 0.5026 | 0.4830 | 82.7586 | 0.1880 | 0.4218 | 0.6630 |
| MACCS | AdaBoost | 145 | 83.7931 | 0.2241 | 0.3337 | 0.6810 | 82.7586 | 0.2456 | 0.3600 | 0.6570 | 84.4828 | 0.2356 | 0.3568 | 0.7020 |
| MACCS | OneR | 145 | 75.1724 | 0.2483 | 0.4983 | 0.5060 | 75.1724 | 0.2483 | 0.4983 | 0.5060 | 70.6897 | 0.2931 | 0.5414 | 0.4130 |
| PubChem | RF | 561 | 98.2759 | 0.0806 | 0.1429 | 0.9660 | 87.2414 | 0.1927 | 0.3101 | 0.7450 | 87.9310 | 0.2118 | 0.3467 | 0.7710 |
| PubChem | IBk | 561 | 98.2759 | 0.0229 | 0.1003 | 0.9660 | 84.8276 | 0.1527 | 0.3738 | 0.6980 | 87.9310 | 0.1603 | 0.3695 | 0.7710 |
| PubChem | LR | 561 | 98.2759 | 0.0201 | 0.1003 | 0.9660 | 71.7241 | 0.2849 | 0.5273 | 0.4380 | 84.4828 | 0.1568 | 0.3811 | 0.7020 |
| PubChem | AdaBoost | 561 | 85.8621 | 0.1838 | 0.3109 | 0.7180 | 86.2069 | 0.1932 | 0.3308 | 0.7240 | 86.2069 | 0.1863 | 0.3436 | 0.7420 |
| PubChem | OneR | 561 | 85.8621 | 0.1414 | 0.3760 | 0.7180 | 85.8621 | 0.1414 | 0.3760 | 0.7180 | 84.4828 | 0.1552 | 0.3939 | 0.7130 |
| Substructure | RF | 101 | 94.1379 | 0.1102 | 0.2039 | 0.8850 | 86.8966 | 0.1971 | 0.3188 | 0.7380 | 82.7586 | 0.2200 | 0.3383 | 0.6570 |
| Substructure | IBk | 101 | 94.1379 | 0.0744 | 0.1901 | 0.8850 | 86.2069 | 0.1590 | 0.3434 | 0.7260 | 81.0345 | 0.2063 | 0.4109 | 0.6320 |
| Substructure | LR | 101 | 94.1379 | 0.0735 | 0.1916 | 0.8840 | 78.2759 | 0.2127 | 0.4242 | 0.5660 | 81.0345 | 0.1938 | 0.4169 | 0.6210 |
| Substructure | AdaBoost | 101 | 84.1379 | 0.2782 | 0.3489 | 0.6940 | 79.3103 | 0.2974 | 0.3793 | 0.5970 | 67.2414 | 0.3210 | 0.4155 | 0.4130 |
| Substructure | OneR | 101 | 70.6897 | 0.2931 | 0.5414 | 0.4990 | 68.2759 | 0.3172 | 0.5632 | 0.4140 | 72.4138 | 0.2759 | 0.5252 | 0.5030 |
| Substructure count | RF | 105 | 98.2759 | 0.0699 | 0.1355 | 0.9660 | 89.3103 | 0.1791 | 0.3072 | 0.7870 | 87.9310 | 0.1848 | 0.3112 | 0.7710 |
| Substructure count | IBk | 105 | 98.2759 | 0.0219 | 0.0974 | 0.9660 | 85.1724 | 0.1592 | 0.3885 | 0.7030 | 82.7586 | 0.1721 | 0.4001 | 0.6630 |
| Substructure count | LR | 105 | 97.5862 | 0.0307 | 0.1237 | 0.9520 | 78.2759 | 0.2250 | 0.4655 | 0.5670 | 82.7586 | 0.1799 | 0.4177 | 0.6550 |
| Substructure count | AdaBoost | 105 | 83.4483 | 0.2148 | 0.3275 | 0.6710 | 81.7241 | 0.2224 | 0.3559 | 0.6350 | 82.7586 | 0.2270 | 0.3629 | 0.6720 |
| Substructure count | OneR | 105 | 83.4483 | 0.1655 | 0.4068 | 0.6690 | 81.0345 | 0.1897 | 0.4355 | 0.6220 | 81.0345 | 0.1897 | 0.4355 | 0.6420 |
| Klekota–Roth | RF | 1094 | 98.6207 | 0.0763 | 0.1341 | 0.9730 | 87.2414 | 0.1966 | 0.3180 | 0.7450 | 86.2069 | 0.1782 | 0.3026 | 0.7420 |
| Klekota–Roth | IBk | 1094 | 98.6207 | 0.0186 | 0.0881 | 0.9730 | 86.8966 | 0.1402 | 0.3600 | 0.7380 | 84.4828 | 0.1486 | 0.3551 | 0.7020 |
| Klekota–Roth | LR | 1094 | 98.6207 | 0.0155 | 0.0881 | 0.9720 | 71.7241 | 0.2803 | 0.5205 | 0.4370 | 79.3103 | 0.2098 | 0.4438 | 0.6130 |
| Klekota–Roth | AdaBoost | 1094 | 86.2069 | 0.1946 | 0.3082 | 0.7330 | 86.5517 | 0.2017 | 0.3186 | 0.7310 | 87.9310 | 0.1894 | 0.3274 | 0.7710 |
| Klekota–Roth | OneR | 1094 | 78.2759 | 0.2172 | 0.4661 | 0.5730 | 74.8276 | 0.2517 | 0.5017 | 0.5030 | 77.5862 | 0.2241 | 0.4734 | 0.5510 |
| Klekota–Roth count | RF | 1097 | 98.6207 | 0.0730 | 0.1330 | 0.9730 | 87.2414 | 0.1874 | 0.3168 | 0.7450 | 84.4828 | 0.1868 | 0.3144 | 0.7020 |
| Klekota–Roth count | IBk | 1097 | 98.6207 | 0.0186 | 0.0881 | 0.9730 | 81.3793 | 0.1907 | 0.4268 | 0.6300 | 84.4828 | 0.1635 | 0.3947 | 0.7130 |
| Klekota–Roth count | LR | 1097 | 98.6207 | 0.0155 | 0.0881 | 0.9720 | 78.2759 | 0.2213 | 0.4632 | 0.5680 | 81.0345 | 0.1891 | 0.4124 | 0.6420 |
| Klekota–Roth count | AdaBoost | 1097 | 86.8966 | 0.1872 | 0.3072 | 0.7390 | 84.1379 | 0.2188 | 0.3557 | 0.6840 | 84.4828 | 0.2100 | 0.3430 | 0.7020 |
| Klekota–Roth count | OneR | 1097 | 83.1034 | 0.1690 | 0.4111 | 0.6670 | 76.5517 | 0.2345 | 0.4842 | 0.5310 | 72.4138 | 0.2759 | 0.5252 | 0.4480 |
| 2D atom pairs | RF | 298 | 95.8621 | 0.1053 | 0.1920 | 0.9170 | 87.9310 | 0.2053 | 0.3174 | 0.7590 | 86.2069 | 0.2278 | 0.3290 | 0.7240 |
| 2D atom pairs | IBk | 298 | 95.8621 | 0.0603 | 0.1707 | 0.9180 | 87.5862 | 0.1533 | 0.3405 | 0.7520 | 87.9310 | 0.1821 | 0.3584 | 0.7590 |
| 2D atom pairs | LR | 298 | 94.8276 | 0.0670 | 0.1837 | 0.8970 | 76.2069 | 0.2579 | 0.4781 | 0.5260 | 79.3103 | 0.2533 | 0.4565 | 0.5860 |
| 2D atom pairs | AdaBoost | 298 | 84.1379 | 0.2434 | 0.3322 | 0.6960 | 82.7586 | 0.2636 | 0.3609 | 0.6660 | 82.7586 | 0.3157 | 0.3990 | 0.6630 |
| 2D atom pairs | OneR | 298 | 74.1379 | 0.2586 | 0.5085 | 0.5050 | 74.1379 | 0.2586 | 0.5085 | 0.5050 | 77.5862 | 0.2241 | 0.4734 | 0.5530 |
| 2D atom pairs count | RF | 301 | 98.6207 | 0.0709 | 0.1318 | 0.9730 | 87.2414 | 0.1720 | 0.3025 | 0.7450 | 87.9310 | 0.1842 | 0.3265 | 0.7710 |
| 2D atom pairs count | IBk | 301 | 98.6207 | 0.0186 | 0.0881 | 0.9730 | 84.8276 | 0.1548 | 0.3836 | 0.6970 | 84.4828 | 0.1636 | 0.3947 | 0.6900 |
| 2D atom pairs count | LR | 301 | 98.6207 | 0.0155 | 0.0881 | 0.9730 | 73.4483 | 0.2634 | 0.5072 | 0.4700 | 84.4828 | 0.1567 | 0.3861 | 0.6890 |
| 2D atom pairs count | AdaBoost | 301 | 86.5517 | 0.1729 | 0.2977 | 0.7320 | 84.8276 | 0.1978 | 0.3420 | 0.6980 | 81.0345 | 0.2026 | 0.3752 | 0.6320 |
| 2D atom pairs count | OneR | 301 | 86.5517 | 0.1345 | 0.3667 | 0.7330 | 81.0345 | 0.1897 | 0.4355 | 0.6210 | 82.7586 | 0.1724 | 0.4152 | 0.6720 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atasever, S. Enhancing HCV NS3 Inhibitor Classification with Optimized Molecular Fingerprints Using Random Forest. Int. J. Mol. Sci. 2025, 26, 2680. https://doi.org/10.3390/ijms26062680
Atasever S. Enhancing HCV NS3 Inhibitor Classification with Optimized Molecular Fingerprints Using Random Forest. International Journal of Molecular Sciences. 2025; 26(6):2680. https://doi.org/10.3390/ijms26062680
Chicago/Turabian StyleAtasever, Sema. 2025. "Enhancing HCV NS3 Inhibitor Classification with Optimized Molecular Fingerprints Using Random Forest" International Journal of Molecular Sciences 26, no. 6: 2680. https://doi.org/10.3390/ijms26062680
APA StyleAtasever, S. (2025). Enhancing HCV NS3 Inhibitor Classification with Optimized Molecular Fingerprints Using Random Forest. International Journal of Molecular Sciences, 26(6), 2680. https://doi.org/10.3390/ijms26062680