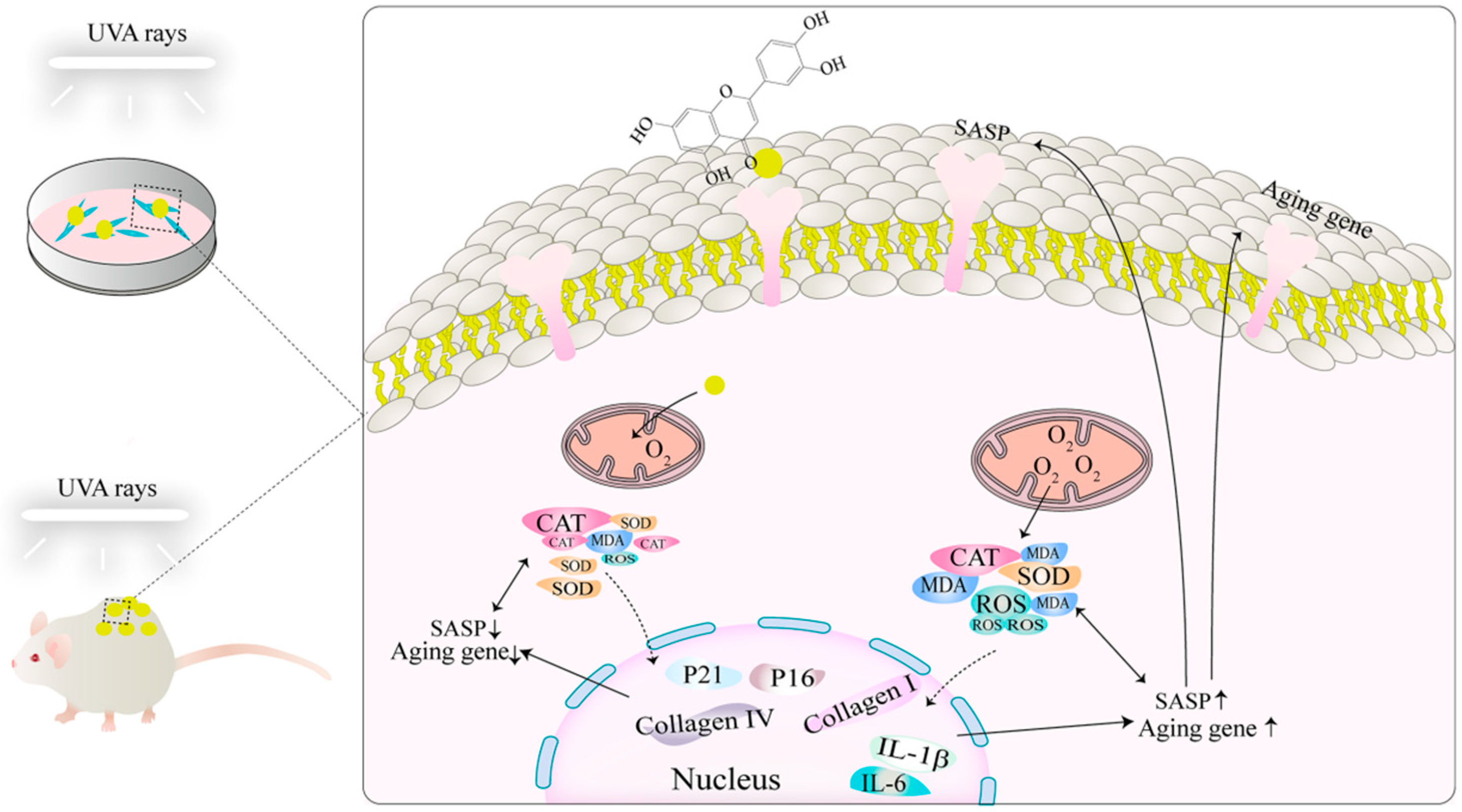
1. Introduction
Skin photoaging is chronic damage to the skin caused by ultraviolet (UV) rays which manifests itself as dryness, wrinkles, roughness, irregular pigmentation, and loss of skin elasticity, etc. In severe cases, it can result in deep plough furrows, milia, open acne, actinic purpura, and the thickening of the epidermis and dermis, and can even lead to the development of cutaneous malignant tumors [
1,
2,
3,
4]. In recent years, skin photoaging has emerged as a key area of interest in the fields of dermatology and medical aesthetic care [
5,
6,
7]. The treatments for skin photoaging include drugs, surgery, and stem cells [
8,
9]. However, these treatments often involve high costs, adverse drug reactions, or postoperative complications. Given the unavoidable exposure to ultraviolet radiation in daily life, the prevention of skin photoaging is crucial.
Similar to naturally aging skin, photoaged skin exhibits a decrease in collagen and elastin, leading to wrinkle formation, loss of elasticity, hyperpigmentation, and dryness of the skin [
10]. However, photoaged skin exposed to ultraviolet radiation (UV) also exhibits a thickening of the epidermal layer and hyperkeratosis [
8,
11]. Aging is a major risk factor for the development of chronic diseases, and skin aging occurs with age or exposure to environmental factors such as UV. This process can propagate the aging phenotype from the skin to other tissues and organs through SASPs, thereby contributing to overall systemic aging [
12]. Mitochondrial dysfunction is considered one of the determinants of aging, leading to the accumulation of reactive oxygen species (ROS) which, in turn, promotes cellular senescence [
13].
Luteolin (3′, 4′, 5′, 7-tetrahydroxyflavones, Lut) is a flavone with various pharmacological properties such as anti-inflammatory, antibacterial, antioxidant, and antitumor activities [
14]. It comes from a wide range of sources and is found in many medicinal plants, fruit, flowers, vegetables, spices, and purgative and detoxifying drugs such as grape, carrot, orange, amaranth, etc. Moreover, Lut is relatively easy to isolate and obtain. At micromolar concentrations, Lut demonstrates a strong anti-inflammatory activity [
15,
16], with its most significant function being its potent antioxidant capacity [
17]. Lut is considered a safer antioxidant substance. Recent studies have shown that Lut exhibits photoprotective effects against ultraviolet B (UVB)-induced photodamage in zebrafish, primarily through DNA protection, antioxidant, and anti-inflammatory effects [
18]. However, ultraviolet B primarily affects the epidermal layer and the superficial dermis, whereas ultraviolet A (UVA), which accounts for 90–95% of the total UV spectrum, is considered to be the main cause of skin photoaging [
4]. Therefore, the photoaging damage caused by UVA to the skin is more significant and lasting, making the prevention of UVA-induced skin photoaging particularly significant. Studies have confirmed that flavonoids play a significant role in preventing UVA-induced skin photoaging [
19]. As one of the flavones, the potential protective effect of Lut against UVA-induced skin photoaging and its mechanism have not been fully elucidated. Therefore, in order to explore the application value of Lut in preventing skin photoaging, this study aims to systematically explore the protective effect of Lut on UVA-induced fibroblast senescence and its mechanism in a mammal model.
3. Discussion
UVA radiation is the main pathogenic factor of skin photoaging and can accelerate the aging process of the skin and even cause cancer by inducing mechanisms such as DNA damage, oxidative stress, and cell senescence [
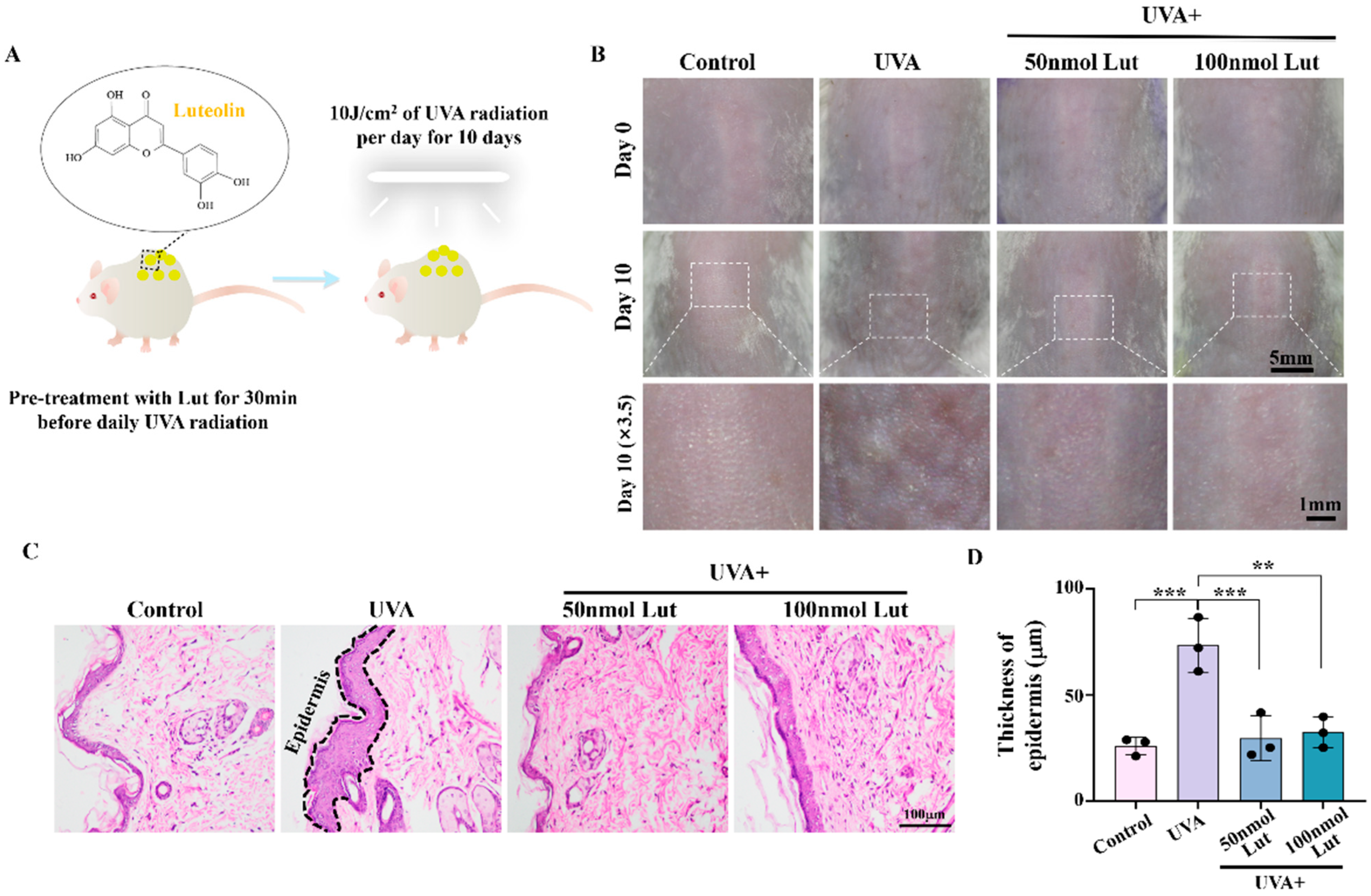
24]. Lut is a natural compound that has been found to have multiple pharmacological activities, but its role and potential mechanism in preventing UVA-induced skin photoaging have not been fully clarified. In this study, we first demonstrated that Lut can effectively alleviate UVA-induced skin photoaging in vivo. By subjecting BALB/c mice to 10 J/cm
2 of UVA radiation for 10 consecutive days, we observed increased wrinkles and epidermal thickening in the dorsal skin of the mice, successfully establishing a photoaging model. These changes are consistent with known signs of photoaging, including structural changes in the dermis and epidermis of the skin. They are primarily due to a compensatory thickening of the stratum corneum following UVA exposure which helps to block part of the ultraviolet rays. Moreover, ultraviolet rays activate tyrosinase to protect cells, resulting in a decrease in the dermal collagen content and reduced skin elasticity. After the Lut pretreatment, these UVA-induced changes were significantly reduced, indicating that Lut has a protective effect against UVA-induced photoaging damage (
Figure 1). Previous studies have shown that flavonoid natural compounds can protect against UV-induced skin damage to a certain extent [
25,
26,
27]. However, most of these studies have primarily focused on UVB. In this study, we demonstrate the therapeutic efficacy of Lut against UVA-induced skin photoaging, highlighting its potential as a more comprehensive photoprotective agent.
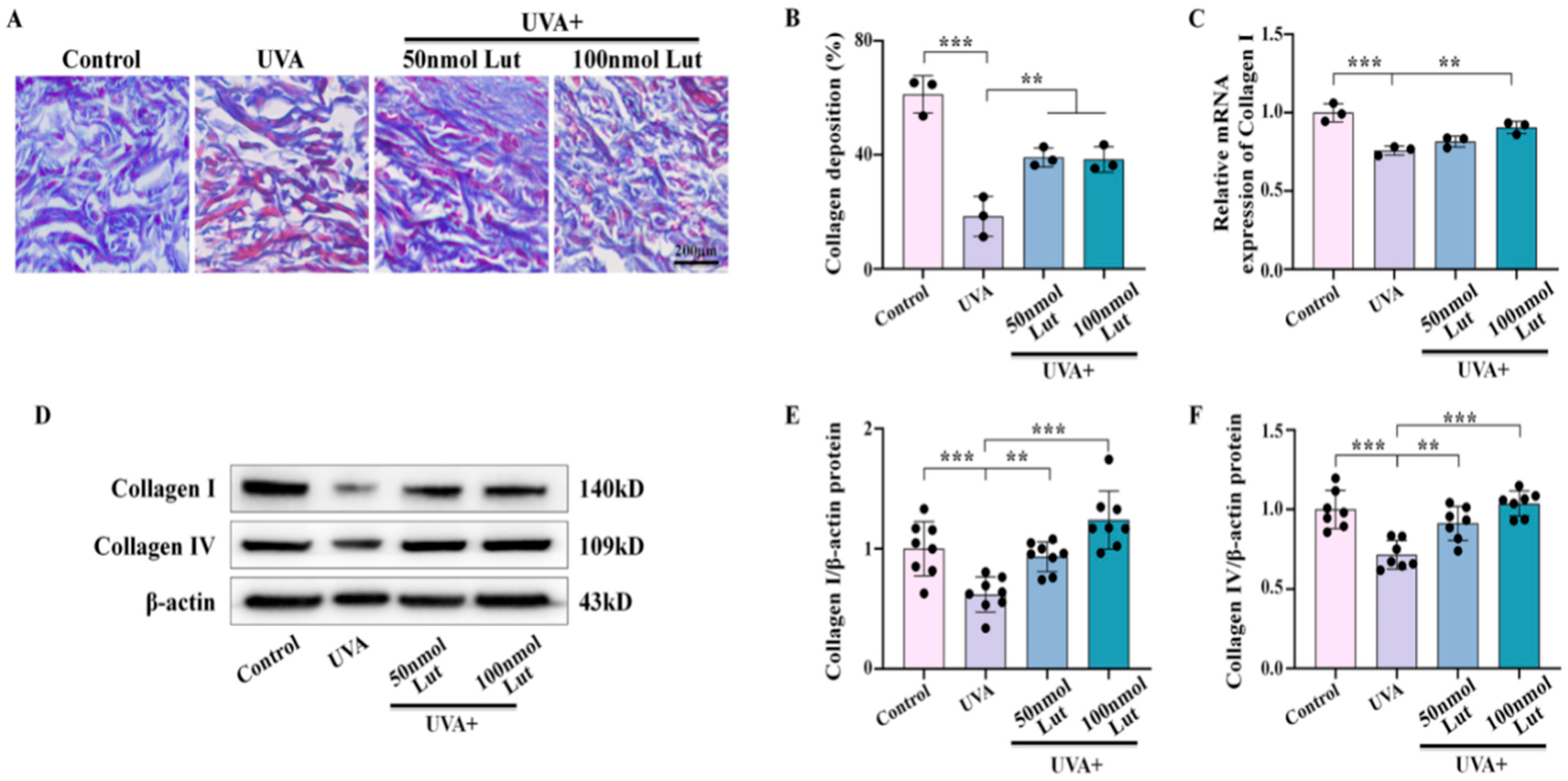
Collagen is a key component of the skin structure and function, and its reduction in content is an important marker of photoaging. Our results show that the Lut pretreatment significantly inhibited UVA-induced collagen degradation. Previous studies have confirmed that, in photoaging, overexpression of MMPs leads to aggravated collagen degradation and damaged skin structure [
28]. Lut extracted from plants can inhibit the expression or activity of MMPs, which is one of the important anti-aging strategies [
29]. Collagen I and collagen IV are the main components of the dermal fibrous structure and the reticular fibrous structure of the basement membrane, respectively. In this study, we confirmed that Lut significantly inhibits UVA-induced degradation of collagen I and further demonstrated its protective effect on collagen IV (
Figure 2). This finding expands our understanding of the potential mechanism of Lut in the prevention and treatment of photoaging, indicating that Lut can comprehensively enhance the skin’s ability to resist UVA by maintaining the content of multiple key collagens in the dermis.
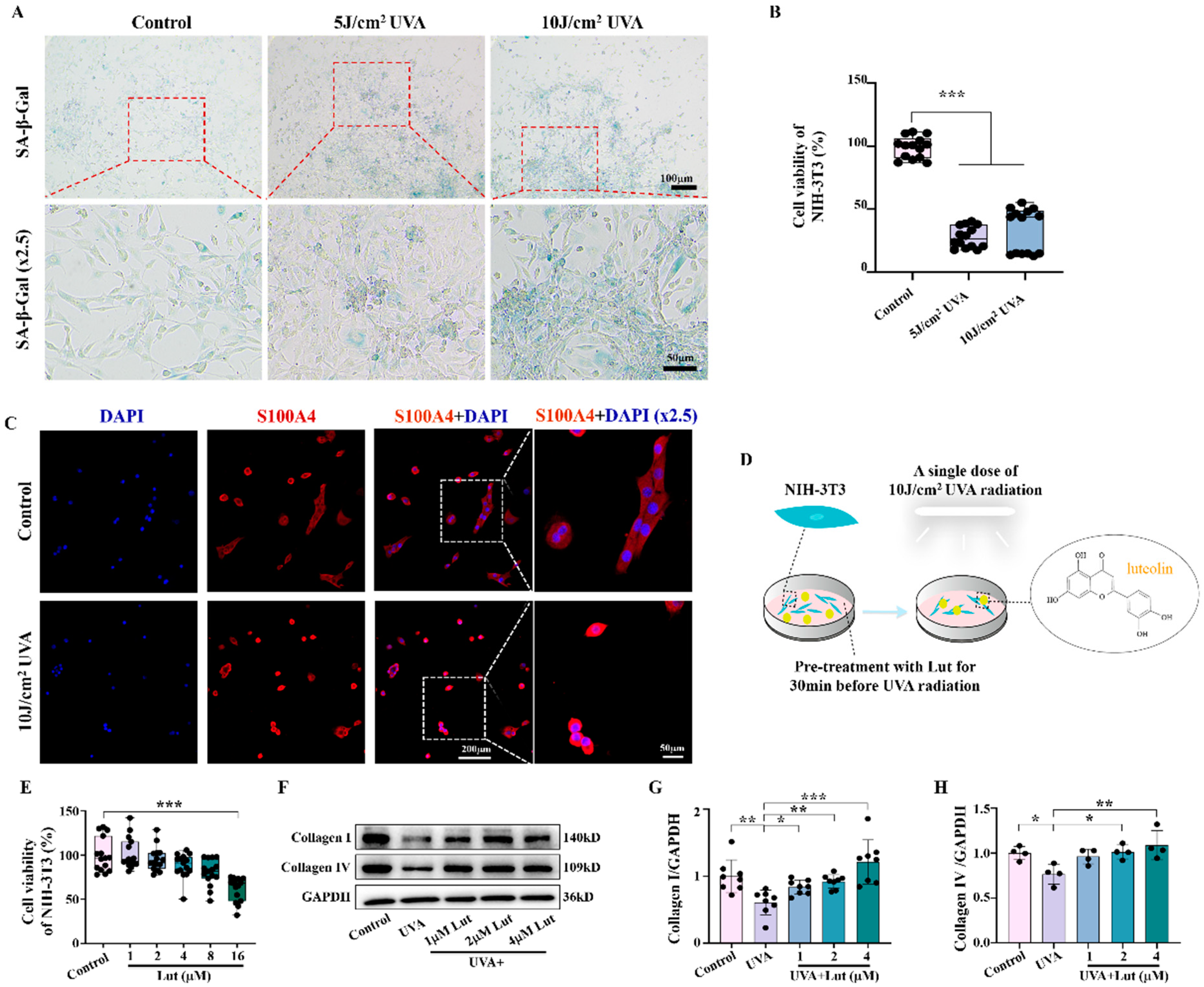
Fibroblasts are the primary cells that maintain the dermal matrix. When their function is impaired, it will lead to reduced collagen synthesis, which, in turn, accelerates skin photoaging. Through our study on NIH-3T3 fibroblasts, we found that Lut can significantly reduce UVA-induced fibroblast senescence. We further confirmed the anti-photoaging activity of Lut using Res, a compound that has been shown to have significant anti-photoaging effects, as a positive control (
Supplementary Figure S1). Furthermore, we confirmed that Lut could restore the collagen synthesis ability of fibroblasts, including the promotion of collagen I and collagen IV. These findings suggest that fibroblasts are the main target cells for Lut to exert its anti-photoaging effect. Therefore, Lut may protect the skin from photoaging damage by improving fibroblast function (
Figure 3). This is consistent with an emerging hypothesis on skin aging proposed by Meinhard Wlaschek et al. which suggests that senescent fibroblasts have an intrinsic ability to escape apoptosis and elimination by the adaptive immune system, thus playing a major role in the irreversible skin aging process [
30].
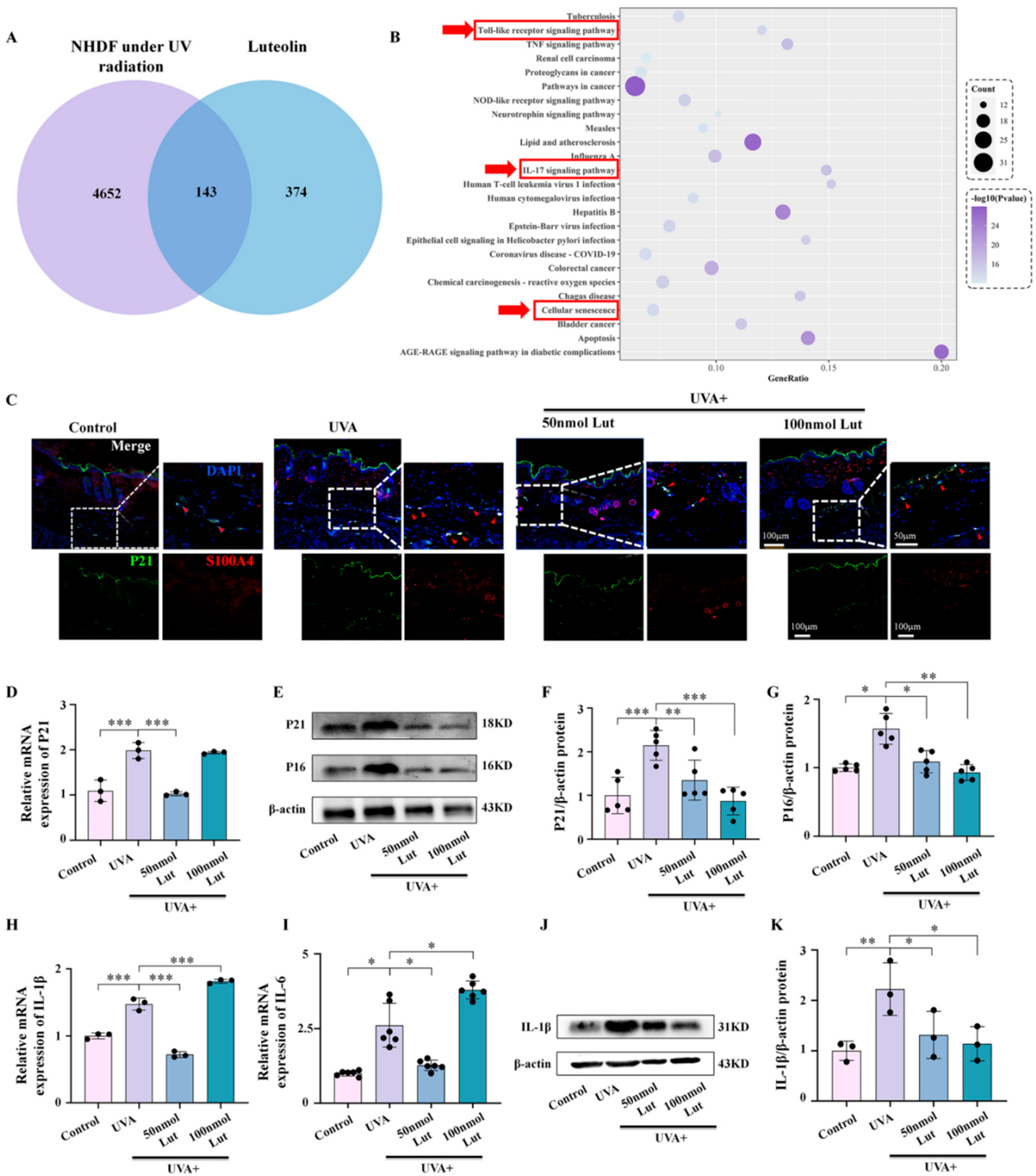
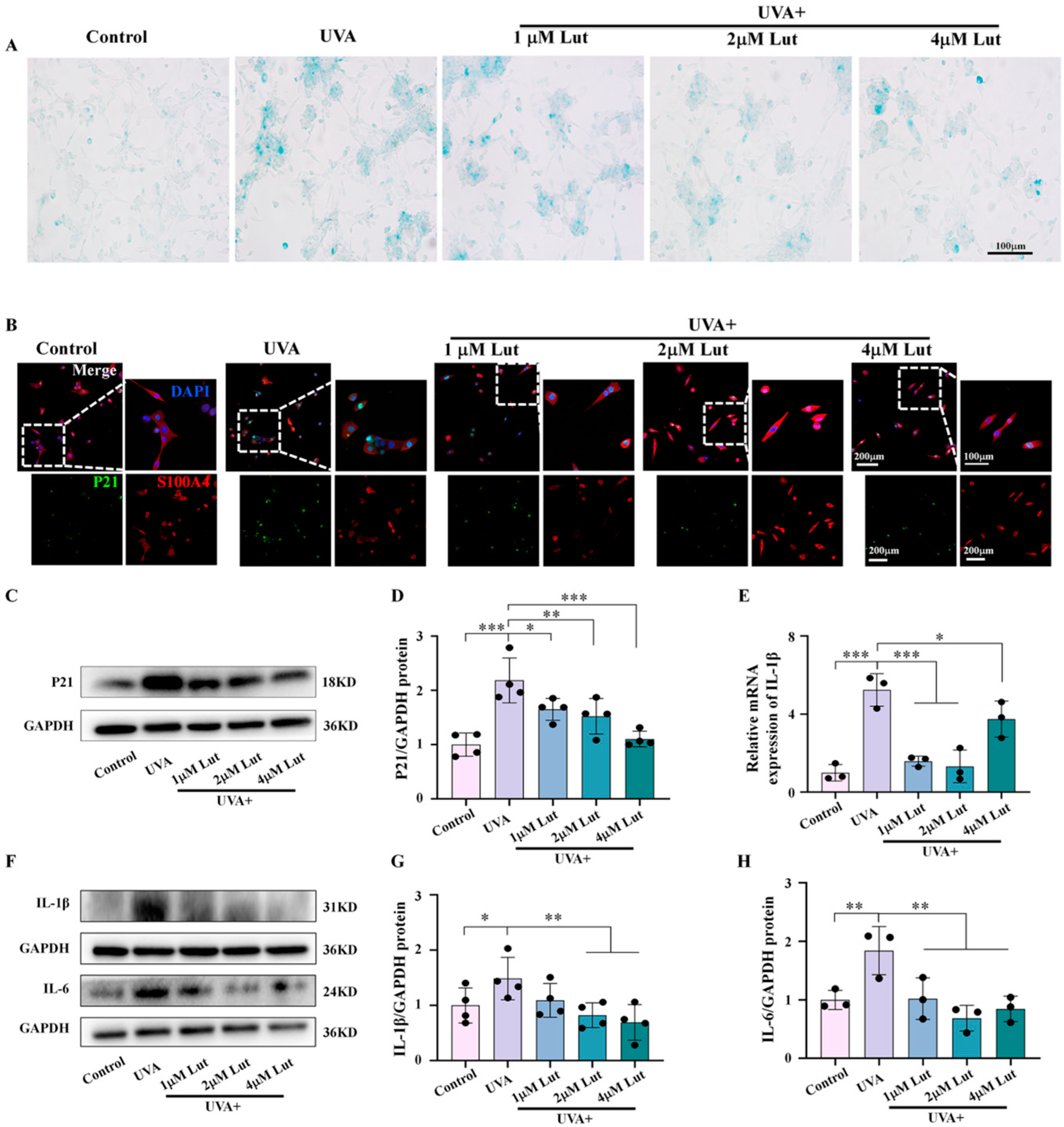
The relationship between cellular senescence and inflammatory responses is close and complex. They interact in multiple biological and pathophysiological processes, forming a self-reinforcing feedback loop. Cellular senescence is accompanied by SASP. One of its characteristics is that senescent cells secrete a large number of inflammatory factors, cytokines, chemokines, and proteases. SASP triggers and maintains inflammatory responses at local and systemic levels through these secreted substances, and this inflammatory environment induces more cells to enter a state of senescence. We explored the potential mechanism of Lut in anti-photoaging through bioinformatics methods. A total of 607 differentially expressed genes in human dermal fibroblasts after ultraviolet radiation exposure were screened out through the GEO database. By integrating these data with the Lut target gene database analysis, we identified 143 common target genes between Lut and photoaged human dermal fibroblasts. These genes are involved in multiple key signaling pathways closely related to inflammation and cellular senescence, such as the toll-like receptor signaling pathway, the IL-17 signaling pathway, and the cellular senescence pathway. At the same time, we further confirmed, through in vivo and in vitro experiments, that the Lut pretreatment can significantly reduce the expression levels of the UVA-induced fibroblast senescence markers P21 and P16, as well as the expression of the inflammatory factors IL-1β and IL-6 (
Figure 4 and
Figure 5).
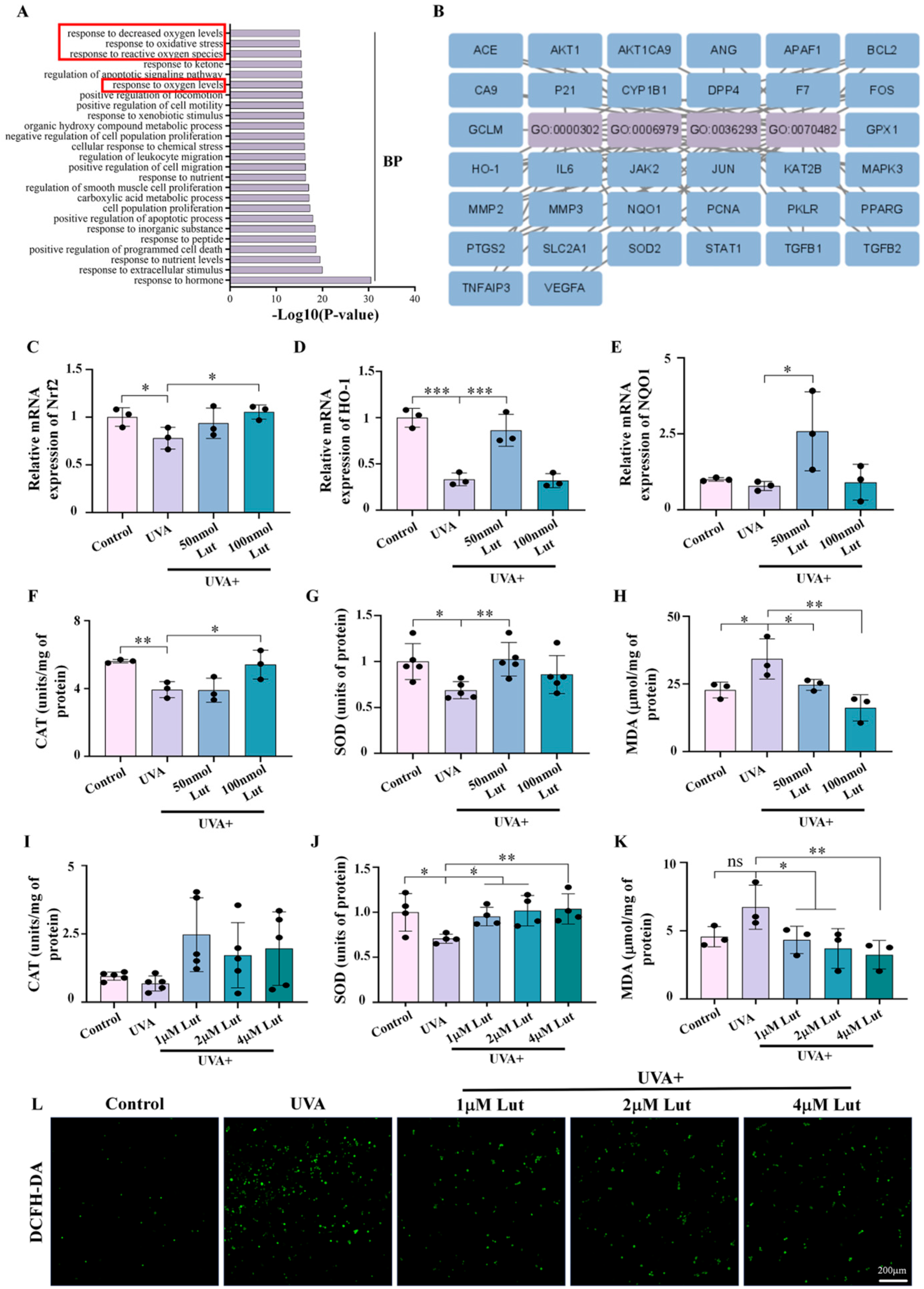
Further investigation into the potential mechanism of Lut’s anti-photoaging effects revealed that Lut significantly mitigates UVA-induced oxidative stress by regulating the expression of key antioxidant genes, including Nrf2, HO-1, and NQO1, and by increasing the levels of antioxidant enzymes such as CAT and SOD (
Figure 6). This is consistent with the antioxidant capacity of Lut reported in previous studies. Lut is involved in protecting skin cells from UVB radiation-induced cellular senescence through the SIRT3/ROS/MAPK axis [
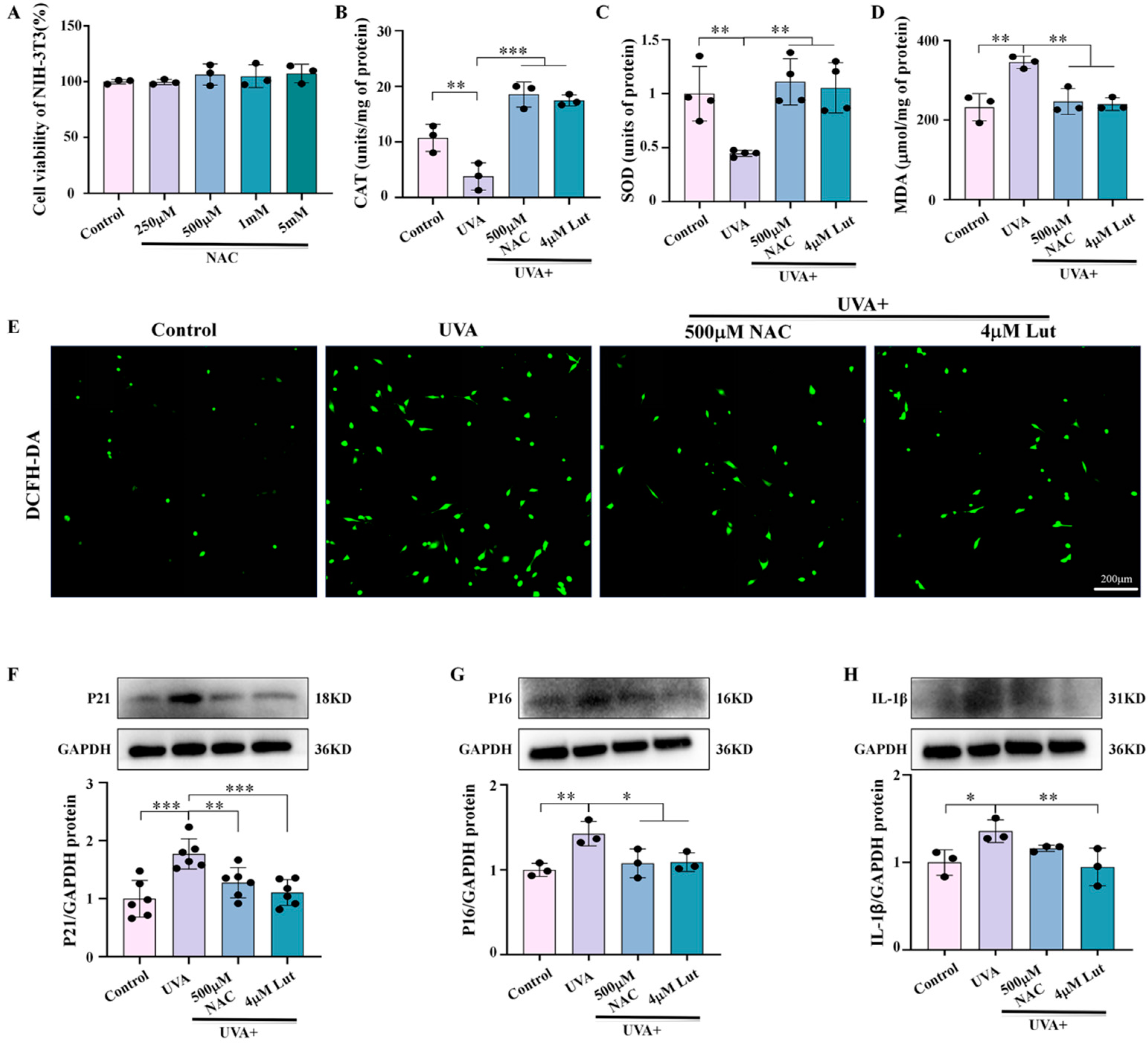
31]. In addition, NAC is a widely used antioxidant, and its antioxidant properties have been confirmed in multiple studies. Through comparative experiments with the classic antioxidant NAC, we found that Lut exhibits better antioxidant and anti-photoaging effects. Our study shows that Lut is significantly more effective than classic antioxidants in dealing with skin photoaging induced by UVA (
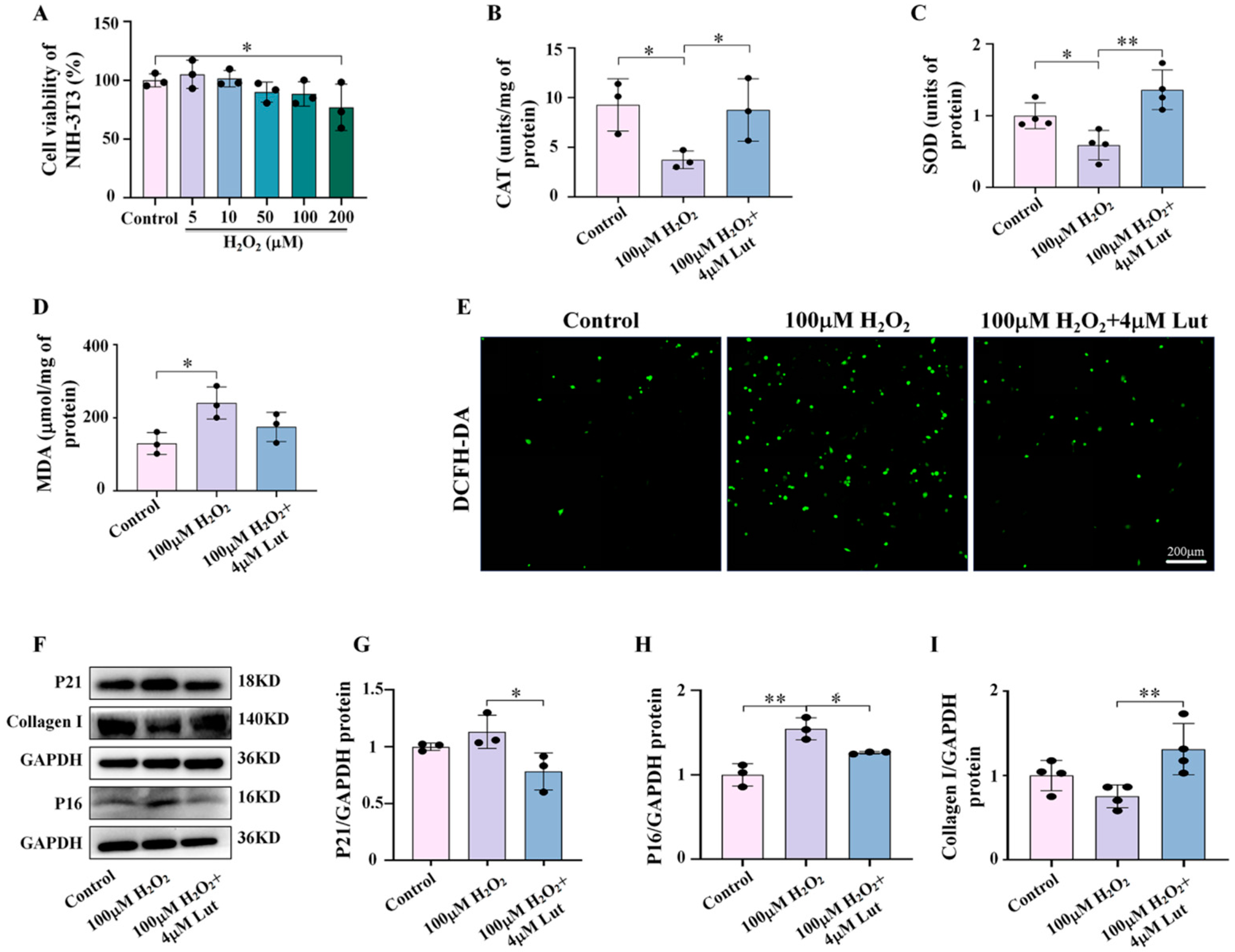
Figure 7). Similarly, our study confirmed the central role of oxidative stress in Lut’s anti-photoaging function through H
2O
2 activation experiments (
Figure 8). This finding has important clinical implications as it provides strong evidence for Lut as a more effective natural anti-photoaging agent.
4. Materials and Methods
4.1. Animal Experiment
BALB/c mice (5–6 weeks old, weighing 18–22 g) were purchased from Guangdong Pharmachem Biotechnology Co., Ltd. (Foshan, China) (License No: SYXK (GD) 2018-0182). All mice were housed in SPF-grade conditions at a temperature of 25 ± 2 °C, with a 12/12 h light–dark cycle and free access to water. Subsequently, the mice were randomly divided into four groups (n = 6): control group, UVA group (the mice were exposed to 10 J/cm2 UVA for 10 days), 50 nmol Lut + UVA group, and 100 nmol Lut + UVA group. Specifically, we first dissolved Lut powder with DMSO (D12345, ThermoFisher, Waltham, MA, USA) and then further diluted it to 50 nmol or 100 nmol with ultrapure water, ensuring that the dilution factor was greater than 1000 times to avoid the potential toxicity of DMSO. Before exposing the mice’s back skin to 10 J/cm2 UVA every day, we pretreated the skin with 200 μL of 50 nmol or 100 nmol Lut for 30 min. To ensure that Lut was evenly distributed on the skin, we thoroughly vortexed Lut and cleaned the mouse back skin to remove surface dirt and oil before each treatment. Then, Lut was evenly applied to the designated area of the skin to ensure a full distribution and penetration. This procedure was repeated once daily and, after 10 consecutive days of treatment, the skin samples were photographed and collected. A UVA lamp (TL-K40W10R, Philips, Amsterdam, The Netherlands) was used to irradiate the mouse skin, and the radiation energy was calculated using a UV energy meter (UV-150, Shenzhen Speedre Technology, Shenzhen, China). All animal experiment procedures were approved by the Animal Experiment Center of Guangzhou University of Chinese Medicine (Animal Ethics No: 20230522009, Approval Date: 22 May 2023).
4.2. Tissue Preparation and Histological Staining
After the mice skin tissue was collected and fixed in 4% paraformaldehyde (BL539A, Biosharp, Hefei, China) overnight, the tissue was dehydrated in 50%, 70%, 80%, 90%, 95%, and 100% ethanol in sequence and then transparentized with xylene. The tissue block was immersed in paraffin at 60 °C and placed in an embedding box for paraffin embedding. The embedded tissue was cut into 4–6 μm with a paraffin slicer (RM2235, Leica, Wetzlan, Germany) for subsequent staining.
Paraffin sections were stained with H&E [
32] and Masson [
33] staining following previously established protocols. Specifically, after dewaxing and hydration, skin tissue sections were stained with hematoxylin (FD7443, FUDE Biology, Hangzhou, China) and eosin (FD7443, FUDE Biology, China) for H&E staining. Masson staining was performed with the Masson staining kit (C0189S, Beyotime, Shanghai, China). All sections were then mounted with neutral gum (G6590G, Biotopped, Beijing, China) and imaged using a Leica DM57 light microscope (DM57, Leica, Wetzlan, Germany).
4.3. Cell Culture and Treatment
NIH-3T3 cells (FH0983, Fuheng Biology, Shanghai, China) were cultured using Dulbecco’s modified eagle’s medium (DMEM, C11995500BT, Gibco, Grand Island, NY, USA), containing 10% fetal bovine serum (16410071, Gibco, Grand Island, NY, USA) and 1% streptomycin and penicillin (15140122, Gibco, USA), in an incubator at 37 °C with 5% CO2. The medium was changed every 2 days. When the cells reached the appropriate density, they were treated with or without different concentrations of Lut, resveratrol (Res), N-acetylcysteine (NAC), and/or H2O2, according to experimental needs.
4.4. Immunofluorescent Staining
Tissue sections were deparaffinized in xylene for 1 h and then hydrated by immersion in anhydrous ethanol, 95% ethanol, 80% ethanol, 70% ethanol, and ultrapure water for 10 min each. The membrane was repaired using sodium citrate antigen repair solution (AR0024, Boster, Wuhan, China) at 60 °C overnight. Subsequently, the membrane was washed with PBS, then permeabilized with 0.5% Triton X-100 for 30 min, and blocked with 10% goat serum for 30 min. It was then titrated with the primary antibodies: P21 (1:200, 28248-1-AP, Proteintech, Wuhan, China) and S100A4 (1:200, 66489-1-Ig, Proteintech, Wuhan, China) for overnight incubation at 4 °C. After washing, PBST (0.1% Tween-20) was used. Fluorescent secondary antibodies, either Alexa Fluor 488 (1:500, AB150077, Abcam, Cambridge, MA, USA) or Alexa Fluor 647 (1:500, AB150115, Abcam, USA), were applied and incubated at room temperature in the dark for 1 h. DAPI (P0131-25ml, Beyotime, Shanghai, China) was added for 5 min. The samples were observed and imaged using a confocal microscope (ZEISS, Oberkochen, Germany).
NIH-3T3 cells were planted on cell crawls (5 × 103 cells/cm2) and pretreated with or without different drugs 30 min before 10 J/cm2 UVA irradiation. The cells were divided into a control group, a UVA group, a 10 μM Res + UVA group, a 1 μM Lut + UVA group, a 2 μM Lut + UVA group, a 4 μM Lut + UVA group, a 500 mM NAC + UVA group, a 100 mM H2O2 group, and a 100 mM H2O2 + 4 μM Lut group. The cells were fixed with 4% paraformaldehyde for 30 min and then permeabilized for 30 min. The following steps were consistent with the tissue immunofluorescence as described above.
4.5. SA-β-Gal Experiments
Staining experiments were performed according to the instructions of the kit (C0602, Beyotime, Shanghai, China). Cells were cultured in six-well dishes (8 × 104 cells/cm2) and pretreated with or without different concentration of Lut 30 min before UVA irradiation. The cells were divided into a control group, a 5 J/cm2 UVA group (cells received UVA radiation at 5 J/cm2 per day for 3 days), a 10 J/cm2 UVA group (cells received a single dose of UVA radiation at 10 J/cm2), a 1 μM Lut + UVA group (10 J/cm2 UVA irradiation), a 2 μM Lut + UVA group (10 J/cm2 UVA irradiation), and a 4 μM Lut + UVA group (10 J/cm2 UVA irradiation). The cells were fixed for 30 min and then incubated with a staining solution at 37 °C in an oven overnight. Subsequently, sections were sealed with anti-fluorescence quencher (P0126-5ml, Beyotime, Shanghai, China) for 12 h and then imaged using a Leica DM57 optical microscope.
4.6. CCK-8 Experiment
NIH-3T3 cells were inoculated in 96-well plates (1.5 × 103 cells/cm2), and were divided into the following groups: a control group, a 5 J/cm2 UVA group, a 10 J/cm2 UVA group, different concentrations of Lut (1 μM, 2 μM, 4 μM, 8 μM, 16 μM) groups, different concentrations of oxidative stress inhibitor N-acetylcysteine (NAC) (S1623, Selleck, Houston, TX, USA) (250 μM, 500 μM, 1 mM, 5 mM) groups, and different concentrations of oxidative stress agonist hydrogen peroxide (H2O2) (AAPR555-CA100, Pythonbio, Guangzhou, China) (5 μM, 10 μM, 50 μM, 100 μM, 200 μM) groups. The cells were cultured for 24 h and then experiments were carried out using the CCK-8 kit (C0038, Beyotime, Shanghai, China). Following a 30 min incubation at 37 °C with 5% CO2, the optical density (OD) values at 450 nm were measured using a Bio-Rad 450 microplate reader (Bio-Rad, Hercules, CA, USA).
4.7. Catalase (CAT) Activity Assay
Total protein of skin tissues or NIH-3T3 cells was extracted using PBS, and experiments were performed according to the instructions of the kit (S0051, Beyotime, Shanghai, China). Briefly, tissues and cells were grinded using PBS to collect protein fluids. The tissues were divided into a control group, a UVA group, a 50 nmol Lut + UVA group, and a 100 nmol Lut + UVA group. The cells were divided into a control group, a UVA group (the cells were irradiated with 10 J/cm2 UVA), a 1 μM Lut + UVA group, a 2 μM Lut + UVA group, a 4 μM Lut + UVA group, a 500 μM NAC + UVA group (500 μM NAC was used to pretreat the cells for 30 min before the 10 J/cm2 UVA irradiation), a 100 μM H2O2 group (cells were incubated with 100 μM H2O2 at 37 °C for 30 min), and a 100μM H2O2 + 4μM Lut group (cells were incubated with 100 μM H2O2 + 4 μM Lut at 37 °C for 30 min). After that, the CAT working solution was added and incubated at room temperature for 30 min, and then the absorbance value was measured at 520 nm using a Bio-Rad 450 microplate reader.
4.8. Superoxide Dismutase (SOD) Activity Assay
Total protein of skin tissues or NIH-3T3 cells was extracted using PBS, and experiments were performed according to the instructions of the kit (S0109, Beyotime, Shanghai, China). Briefly, tissues and cells were grinded using PBS to collect protein fluids. The tissues were divided into a control group, a UVA group, a 50 nmol Lut + UVA group, and a 100 nmol Lut + UVA group. The cells were divided into a control group, a UVA group, a 1 μM Lut + UVA group, a 2 μM Lut + UVA group, a 4 μM Lut + UVA group, a 500 μM NAC + UVA group, a 100 μM H2O2 group, and a 100 μM H2O2 + 4 μM Lut group. Subsequently, the SOD assay working solution was added and incubated at 37 °C for 30 min, and then the absorbance value was measured at 560 nm using a Bio-Rad 450 microplate reader.
4.9. Malondialdehyde (MDA) Activity Assay
Total protein of skin tissues or NIH-3T3 cells was extracted using PBS, and experiments were performed according to instructions of the kit (S0131S, Beyotime, Shanghai, China). Briefly, the tissues and NIH-3T3 cells were grinded using PBS to collect protein fluids. The tissues were divided into a control group, a UVA group, a 50 nmol Lut + UVA group, and a 100 nmol Lut + UVA group. The cells were divided into a control group, a UVA group, a 1 μM Lut + UVA group, a 2 μM Lut + UVA group, a 4 μM Lut + UVA group, a 500 μM NAC + UVA group, a 100 μM H2O2 group, and a 100 μM H2O2 + 4 μM Lut group. After that, the MDA assay working solution was added and heated for 15 min. The samples were then allowed to return to room temperature and centrifuged at 1000× g for 10 min, and then the absorbance value was measured at 532 nm using a Bio-Rad 450 microplate reader.
4.10. Dichloro-Dihydro-Fluorescein Diacetate (DCFH-DA) Assay
NIH-3T3 cells were planted in 24-well plates (5 × 103 cells/cm2) and the experiments were performed according to the instructions of the kit (S0033S, Beyotime, Shanghai, China). The cells were divided into a control group, a UVA group, a 4 μM Lut + UVA group, a 500 μM NAC + UVA group, a 100 μM H2O2 group, and a 100 μM H2O2 + 4 μM Lut group. Briefly, the cells were treated with a DCFH-DA working solution and incubated for 20 min at 37 °C in a cell culture incubator, protected from light, and then imaged using an Olympus IX51 inverted fluorescent microscope (IX51, Olympus Corporation, Tokyo, Japan).
4.11. Quantitative Real-Time-PCR (RT-qPCR)
Mouse skin tissues were divided into a control group, a UVA group, a 50 nmol Lut + UVA group, and a 100 nmol Lut + UVA group. The cells were divided into a control group, a UVA group, a 1 μM Lut + UVA group, a 2 μM Lut + UVA group, a 4 μM Lut + UVA group, a 500 μM NAC + UVA group, a 100 μM H
2O
2 group, and a 100 μM H
2O
2 + 4 μM Lut group. In brief, total RNA from skin tissues and cells was extracted using the TRIZOL kit (15596018, Invitrogen, Shanghai, China), cDNA was synthesized using a reverse transcription kit (A230-10, GenStar, Beijing, China), and q-PCR was performed using q-PCR Mix (A301-10, GenStar, China). The sequences are listed in
Supplementary Table S1.
4.12. Western Blotting Analysis
Skin tissues or cellular proteins were lysed using RIPA lysate (FD009, FUDE Biology, Hangzhou, China). Total protein concentration was determined using the BCA method (FD2001, FUDE Biology, Hangzhou, China). All proteins were then subjected to SDS-PAGE and then transferred to preactivated polyvinylidene difluoride membranes (PVDF, Bio-Rad, Hercules, CA, USA). After blocking with 5% skimmed milk (9999S, Cell Signaling Technology, Danvers, MA, USA), the primary antibodies i.e., collagen I (1:1000, 66761-1-Ig, Proteintech, Wuhan, China), collagen IV (1:1000, SD83-03, HUABIO, Hangzhou, China), P21 (1:1000, 10355-1-AP, Proteintech, Wuhan, China), P16 (1:1000, sc-1661, Santa Cruz Biotechnology, USA), IL-1β (1:1000, 12703S, Cell Signaling Technology, Danvers, MA, USA), and IL-6 (1:1000, 12912S, Cell Signaling Technology, Danvers, MA, USA), were incubated overnight at 4 °C in a refrigerator. The secondary antibodies, rabbit (1:1000, 7074S, Cell Signaling Technology, Danvers, MA, USA) or mouse (1:2000, 7076S, Cell Signaling Technology, Danvers, MA, USA), were used every other day to bind to the primary antibodies. Images were developed using a luminescent solution (WBKLS0500, Millipore, Shanghai, China) and captured with a fully automated chemiluminescent fluorescence image analysis system (5200Multi, Tanon, Shanghai, China).
4.13. Gene Enrichment Analysis Based on Bioinformatics and Network Pharmacology
The GEO database (
https://www.ncbi.nlm.nih.gov/geo/, accessed on 20 August 2023) was searched by entering the keyword “photoaging” to obtain 4795 genes from the dataset GSE11900918 (transcriptomic expression analysis of the effects of ultraviolet radiation on normal human dermal fibroblasts). A total of 517 Lut targets were obtained from TCMSP, TCD, PharmMapper, and Swisstarget Prediction19. Gene IDs were converted to gene names using the uniprot website (
https://www.uniprot.org/, accessed on 20 August 2023). Gene intersections were obtained using the VENNY2.1 website (
https://bioinfogp.cnb.csic.es/tools/venny/, accessed on 20 August 2023). The Metascape website (
https://metascape.org/gp/index.html#/main/step1, accessed on 20 August 2023) was used to obtain KEGG-enriched pathways, and KEGG pathway bubble plots were generated using R version 4.2.0. Intersecting genes were analyzed using the GO analysis results from the Metascape website (
https://metascape.org/gp/index.html#/main/step1), and GO bioprocess maps were drawn using GraphPad Prism 8.3.
4.14. Data Analysis
The SPSS 25.0 software and GraphPad Prism 8.3 were used for data analysis and statistical graphing. The experiments were repeated at least three times. One-way analyses of variance or Kruskal–Wallis tests were used to compare multiple groups of data, and the results are expressed as the mean ± standard deviation or median. * p < 0.05 was considered statistically significant, ** p < 0.01 and *** p < 0.001 were considered highly statistically significant.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}