
Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention



Abstract
1. Introduction
2. Emerging Mechanisms of Immune Evasion
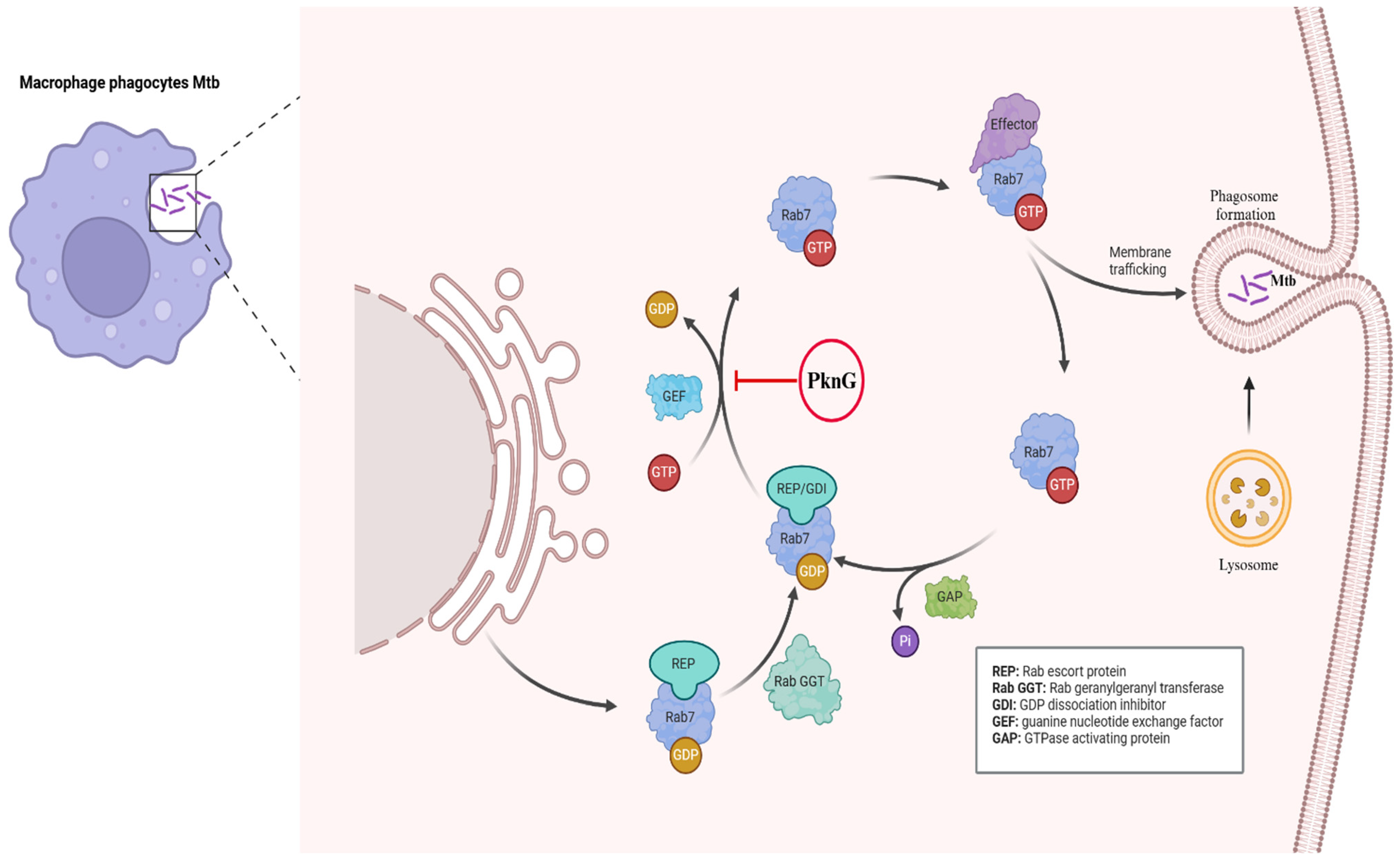
2.1. Phagosome Maturation and Survival
2.2. Alternative Mechanisms of Phagosomal Escape
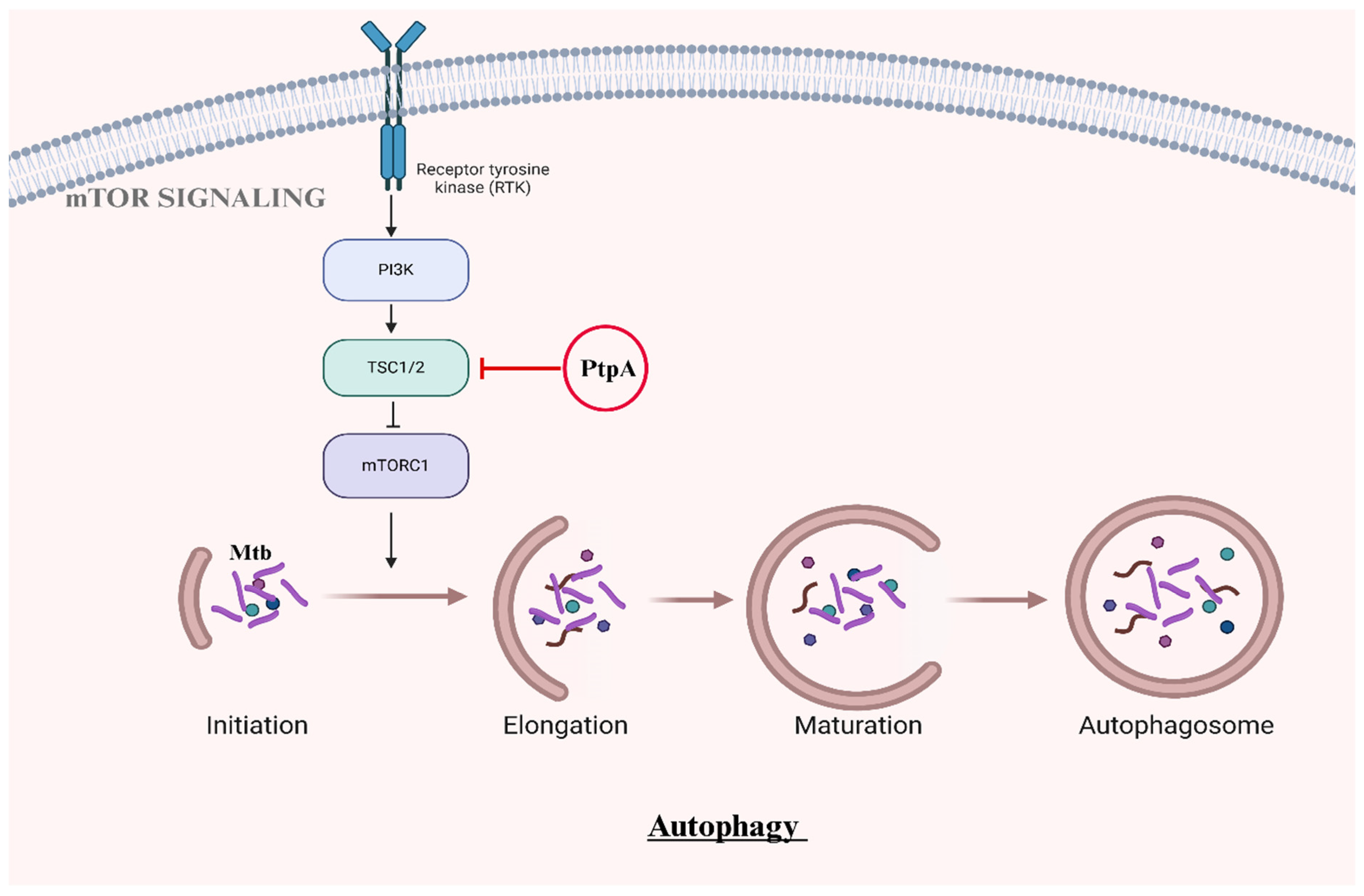
2.3. Autophagosome Maturation and Survival
2.4. Manipulation of Host Metabolism
3. Granuloma Dynamics: New Perspectives
4. Innovative Therapies
4.1. Host-Directed Therapies (HDTs)
4.2. Cytokine-Based Immunotherapies
4.3. Epigenetic Modulation
4.4. Targeting Granuloma Dynamics
5. Future Directions and Challenges
5.1. Addressing Latent TB and Drug Resistance
5.2. Integration of Advanced Technologies
5.3. Vaccine Development
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization, WHO. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Ge, P.; Lei, Z.; Yu, Y.; Lu, Z.; Qiang, L.; Chai, Q.; Zhang, Y.; Zhao, D.; Li, B.; Pang, Y.; et al. M. tuberculosis PknG manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy 2022, 18, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Shrivastva, R.; Mukhopadhyay, S. Mycobacterial PknG Targets the Rab7l1 Signaling Pathway To Inhibit Phagosome–Lysosome Fusion. J. Immunol. 2018, 201, 1421–1433. [Google Scholar] [CrossRef]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis Virulence Is Mediated by PtpA Dephosphorylation of Human Vacuolar Protein Sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Poirier, V.; Bach, H.; Av-Gay, Y. Mycobacterium tuberculosis Promotes Anti-apoptotic Activity of the Macrophage by PtpA Protein-dependent Dephosphorylation of Host GSK3α. J. Biol. Chem. 2014, 289, 29376–29385. [Google Scholar] [CrossRef]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis protein tyrosine phosphatase A disrupts phagosome acidification by exclusion of host vacuolar H+-ATPase. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.V.; Reddy, P.V.; Tyagi, A.K. Secreted Acid Phosphatase (SapM) of Mycobacterium tuberculosis Is Indispensable for Arresting Phagosomal Maturation and Growth of the Pathogen in Guinea Pig Tissues. PLoS ONE 2013, 8, e70514. [Google Scholar] [CrossRef]
- Vergne, I.; Chua, J.; Lee, H.-H.; Lucas, M.; Belisle, J.; Deretic, V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2005, 102, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.T.; Belisle, J.T. Secretion of an Acid Phosphatase (SapM) by Mycobacterium tuberculosis That Is Similar to Eukaryotic Acid Phosphatases. J. Bacteriol. 2000, 182, 6850–6853. [Google Scholar] [CrossRef]
- Nagdev, P.K.; Agnivesh, P.K.; Roy, A.; Sau, S.; Kalia, N.P. Exploring and exploiting the host cell autophagy during Mycobacterium tuberculosis infection. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 1297–1315. [Google Scholar] [CrossRef]
- Foulon, M.; Listian, S.A.; Soldati, T.; Barisch, C. Conserved Mechanisms Drive Host-Lipid Access, Import, and Utilization in Mycobacterium Tuberculosis and M. marinum. In Biology of Mycobacterial Lipids; Elsevier: Amsterdam, The Netherlands, 2022; pp. 133–161. [Google Scholar]
- Simeone, R.; Bobard, A.; Lippmann, J.; Bitter, W.; Majlessi, L.; Brosch, R.; Enninga, J. Phagosomal Rupture by Mycobacterium tuberculosis Results in Toxicity and Host Cell Death. PLOS Pathog. 2012, 8, e1002507. [Google Scholar] [CrossRef]
- Augenstreich, J.; Haanappel, E.; Ferré, G.; Czaplicki, G.; Jolibois, F.; Destainville, N.; Guilhot, C.; Milon, A.; Astarie-Dequeker, C.; Chavent, M. The conical shape of DIM lipids promotes Mycobacterium tuberculosis infection of macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 25649–25658. [Google Scholar] [CrossRef] [PubMed]
- van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef]
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766. [Google Scholar] [CrossRef]
- Liu, S.; Guan, L.; Peng, C.; Cheng, Y.; Cheng, H.; Wang, F.; Ma, M.; Zheng, R.; Ji, Z.; Cui, P.; et al. Mycobacterium tuberculosis suppresses host DNA repair to boost its intracellular survival. Cell Host Microbe 2023, 31, 1820–1836.e10. [Google Scholar] [CrossRef]
- Shariq, M.; Quadir, N.; Alam, A.; Zarin, S.; Sheikh, J.A.; Sharma, N.; Samal, J.; Ahmad, U.; Kumari, I.; Hasnain, S.E.; et al. The exploitation of host autophagy and ubiquitin machinery by Mycobacterium tuberculosis in shaping immune responses and host defense during infection. Autophagy 2022, 19, 3–23. [Google Scholar] [CrossRef]
- Golovkine, G.R.; Roberts, A.W.; Morrison, H.M.; Rivera-Lugo, R.; McCall, R.M.; Nilsson, H.; Garelis, N.E.; Repasy, T.; Cronce, M.; Budzik, J.; et al. Autophagy restricts Mycobacterium tuberculosis during acute infection in mice. Nat. Microbiol. 2023, 8, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-A.; Ma, S.; Li, P.; Xie, J. The dynamics of Mycobacterium tuberculosis phagosome and the fate of infection. Cell. Signal. 2023, 108, 110715. [Google Scholar] [CrossRef]
- Pellegrini, J.M.; Tateosian, N.L.; Morelli, M.P.; García, V.E. Shedding Light on Autophagy During Human Tuberculosis. A Long Way to Go. Front. Cell. Infect. Microbiol. 2022, 11, 820095. [Google Scholar] [CrossRef]
- Trong, E.J.; Ng, T.W.; Porcelli, S.A.; Lee, S. Mycobacterium tuberculosis PE_PGRS20 and PE_PGRS47 Proteins Inhibit Autophagy by Interaction with Rab1A. mSphere 2021, 6, e0054921. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Zhou, T.; Pan, J.; Ma, X.; Shi, G.; Jiang, B.; Xiang, Y.-G. Identifying autophagy-related genes as potential targets for immunotherapy in tuberculosis. Int. Immunopharmacol. 2023, 118, 109956. [Google Scholar] [CrossRef]
- Deretic, V.; Wang, F. Autophagy is part of the answer to tuberculosis. Nat. Microbiol. 2023, 8, 762–763. [Google Scholar] [CrossRef] [PubMed]
- Typas, D. Autophagy counteracts Mycobacterium tuberculosis infection at early stages. Nat. Struct. Mol. Biol. 2023, 30, 720. [Google Scholar] [CrossRef]
- Lam, A.; Prabhu, R.; Gross, C.M.; Riesenverg, L.A.; Singh, V.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L218–L229. [Google Scholar] [CrossRef]
- Mittal, E.; Prasad, G.V.R.K.; Upadhyay, S.; Sadadiwala, J.; Olive, A.J.; Yang, G.; Sassetti, C.M.; Philips, J.A. Mycobacterium tuberculosis virulence lipid PDIM inhibits autophagy in mice. Nat. Microbiol. 2024, 9, 2970–2984. [Google Scholar] [CrossRef] [PubMed]
- Strong, E.J.; Jurcic Smith, K.L.; Saini, N.K.; Ng, T.W.; Porcelli, S.A.; Lee, S. Identification of autophagy-inhibiting factors of Mycobacterium tuberculosis by high-throughput loss-of-function screening. Infect. Immun. 2020, 88, 10–128. [Google Scholar] [CrossRef]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. ESAT-6 a Major Virulence Factor of Mycobacterium tuberculosis. Biomolecules 2023, 13, 968. [Google Scholar] [CrossRef] [PubMed]
- Passos, B.B.; Araújo-Pereira, M.; Vinhaes, C.L.; Amaral, E.P.; Andrade, B.B. The role of ESAT-6 in tuberculosis immunopathology. Front. Immunol. 2024, 15, 1383098. [Google Scholar] [CrossRef] [PubMed]
- Koiri, D.; Nandi, M.; Hameem, P.M.A.; Bhausaheb, A.J.; Meher, G.; Behura, A.; Kumar, A.; Choudhary, V.; Choubey, S.; Saleem, M. Real-time visualization reveals Mycobacterium tuberculosis ESAT-6 disrupts phagosome via fibril-mediated vesiculation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Venkatesan, A.; Palaniyandi, K.; Sharma, D.; Bisht, D.; Narayanan, S. Functional characterization of PknI-Rv2159c interaction in redox homeostasis of Mycobacterium tuberculosis. Front. Microbiol. 2016, 7, 1654. [Google Scholar] [CrossRef]
- Šlachtová, V.; Šebela, M.; Torfs, E.; Oorts, L.; Cappoen, D.; Berka, K.; Bazgier, V.; Brulíková, L. Novel thiazolidinedione-hydroxamates as inhibitors of Mycobacterium tuberculosis virulence factor Zmp1. Eur. J. Med. Chem. 2020, 185, 111812. [Google Scholar] [CrossRef] [PubMed]
- Dak, M.; Šlachtová, V.; Šebela, M.; Bazgier, V.; Berka, K.; Smiejkowska, N.; Oorts, L.; Cappoen, D.; Brulíková, L. Novel heterocyclic hydroxamates as inhibitors of the mycobacterial zinc metalloprotease Zmp1 to probe its mechanism of function. Eur. J. Med. Chem. 2022, 244, 114831. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Happonen, L.; Khakzad, H.; Malmström, L.; Malmström, J. Structural proteomics, electron cryo-microscopy and structural modeling approaches in bacteria–human protein interactions. Med. Microbiol. Immunol. 2020, 209, 265–275. [Google Scholar] [CrossRef]
- Aylan, B.; Bernard, E.M.; Pellegrino, E.; Botella, L.; Fearns, A.; Athanasiadi, N.; Bussi, C.; Santucci, P.; Gutierrez, M.G. ATG7 and ATG14 restrict cytosolic and phagosomal Mycobacterium tuberculosis replication in human macrophages. Nat. Microbiol. 2023, 8, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef]
- Serene, L.G.; Webber, K.; Champion, P.A.; Schorey, J.S. Mycobacterium tuberculosis SecA2-dependent activation of host Rig-I/MAVs signaling is not conserved in Mycobacterium marinum. PLoS ONE 2024, 19, e0281564. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Luing, L.A.; Palacios, Y.; Ruiz, A.; Téllez-Navarrete, N.A.; Chavez-Galan, L. Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens 2023, 12, 839. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Koster, S.; Sakowski, E.; Ramkhelawon, B.; van Solingen, C.; Oldebeken, S.; Karunakaran, D.; Portal-Celhay, C.; Sheedy, F.J.; Ray, T.D.; et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 2016, 17, 677–686. [Google Scholar] [CrossRef]
- Ahsan, F.; Maertzdorf, J.; Guhlich-Bornhof, U.; Kaufmann, S.H.E.; Moura-Alves, P. IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 1520. [Google Scholar] [CrossRef]
- Stutz, M.D.; Clark, M.P.; Doerflinger, M.; Pellegrini, M. Mycobacterium tuberculosis: Rewiring host cell signaling to promote infection. J. Leukoc. Biol. 2018, 103, 259–268. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Yuk, J.-M.; Jo, E.-K. AMP-Activated Protein Kinase and Host Defense against Infection. Int. J. Mol. Sci. 2018, 19, 3495. [Google Scholar] [CrossRef] [PubMed]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.-F.; Marchou, B.; Cardona, P.-J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Agarwal, P.; Gordon, S.; Martinez, F.O. Foam Cell Macrophages in Tuberculosis. Front. Immunol. 2021, 12, 775326. [Google Scholar] [CrossRef] [PubMed]
- Laval, T.; Chaumont, L.; Demangel, C. Not too fat to fight: The emerging role of macrophage fatty acid metabolism in immunity to Mycobacterium tuberculosis. Immunol. Rev. 2021, 301, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.-J.; Kim, M.-J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef]
- Chen, Z.; Kong, X.; Ma, Q.; Chen, J.; Zeng, Y.; Liu, H.; Wang, X.; Lu, S. The impact of Mycobacterium tuberculosis on the macrophage cholesterol metabolism pathway. Front. Immunol. 2024, 15, 1402024. [Google Scholar] [CrossRef]
- Liu, Y.; Larrouy-Maumus, G. Lipids and Glycolipids as Biomarkers of Mycobacterial Infections. In Biology of Mycobacterial Lipids; Elsevier: Amsterdam, The Netherlands, 2022; pp. 83–104. [Google Scholar]
- Gong, Y.; Wang, J.; Li, F.; Zhu, B. Polysaccharides and glycolipids of Mycobacterium tuberculosis and their induced immune responses. Scand. J. Immunol. 2023, 97, e13261. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.; Heyckendorf, J.; Marwitz, F.; Zehethofer, N.; Linnemann, L.; Gisch, N.; Karaköse, H.; Reimann, M.; Kranzer, K.; Kalsdorf, B.; et al. Tuberculostearic Acid-Containing Phosphatidylinositols as Markers of Bacterial Burden in Tuberculosis. ACS Infect. Dis. 2022, 8, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Fujiwara, N.; Kim, J.-Y.; Kang, M.; Yang, J.S.; Yim, J.-J.; Whang, J.; Kwak, N. The Impact of Trehalose Dimycolate on the Clinical Course of Mycobacterium avium Complex Pulmonary Disease. Ann. Am. Thorac. Soc. 2024, 21, 1015–1021. [Google Scholar] [CrossRef]
- Wang, H.; Liu, D.; Zhou, X. Effect of Mycolic Acids on Host Immunity and Lipid Metabolism. Int. J. Mol. Sci. 2023, 25, 396. [Google Scholar] [CrossRef]
- Lyu, J.; Narum, D.E.; Baldwin, S.L.; Larsen, S.E.; Bai, X.; Griffith, D.E.; Dartois, V.; Naidoo, T.; Steyn, A.J.C.; Coler, R.N.; et al. Understanding the development of tuberculous granulomas: Insights into host protection and pathogenesis, a review in humans and animals. Front. Immunol. 2024, 15, 1427559. [Google Scholar] [CrossRef] [PubMed]
- Weeratunga, P.; Moller, D.R.; Ho, L.-P. Immune mechanisms of granuloma formation in sarcoidosis and tuberculosis. J. Clin. Investig. 2024, 134. [Google Scholar] [CrossRef] [PubMed]
- Madden, K.; Hamra, R.E.; Berton, S.; Felker, J.; Alvarez, G.G.; Blais, A.; Sun, J. Mycobacterium tuberculosis infection triggers epigenetic changes that are enriched in a type I IFN signature. Microlife 2023, 4, uqad006. [Google Scholar] [CrossRef] [PubMed]
- Sounderrajan, V.; Rajadas, S.E.; Thangam, T.; Rao, S.S.; Parthasarathy, K.; Tamilanban, R.; Harshavardhan, S. Host-Directed Immunotherapy for Tuberculosis. In Translational Research in Biomedical Sciences: Recent Progress and Future Prospects; Springer: New York, NY, USA, 2024; pp. 323–335. [Google Scholar]
- Kalra, R.; Tiwari, D.; Dkhar, H.K.; Bhagyaraj, E.; Kumar, R.; Bhardwaj, A.; Gupta, P. Host factors subverted by Mycobacterium tuberculosis: Potential targets for host directed therapy. Int. Rev. Immunol. 2021, 42, 43–70. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Xu, J.; Pi, J.; Zheng, B. Roles of HIF-1α signaling in Mycobacterium tuberculosis infection: New targets for anti-TB therapeutics? Biochem. Biophys. Res. Commun. 2024, 711, 149920. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wang, Y.; Dong, Y.; Liu, Y.; Ge, X.; Chen, Y.; Long, M.; Zhou, X. Impact of Hypoxia-Inducible Factor-1α on Host Immune Metabolism and Tissue Damage During Mycobacterium bovis Infection. J. Infect. Dis. 2024, jiae305. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, S.; Kabashima, K. Metabolic reprogramming and macrophage polarization in granuloma formation. Int. Immunol. 2024, 36, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qian, Y.; Wang, N.; Qiu, D.; Cao, H.; Wang, Y.; Luo, H.; Shen, X.; Cui, H.; Wang, J.; et al. The functions and applications of extracellular vesicles derived from Mycobacterium tuberculosis. Biomed. Pharmacother. 2023, 168, 115767. [Google Scholar] [CrossRef] [PubMed]
- Alipoor, S.D.; Elieh-Ali-Komi, D. Significance of extracellular vesicles in orchestration of immune responses in Mycobacterium tuberculosis infection. Front. Cell. Infect. Microbiol. 2024, 14, 1398077. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Flores, L.; Castañeda-Casimiro, J.; Vallejo-Castillo, L.; Álvarez-Jiménez, V.D.; Peregrino, E.S.; García-Martínez, M.; Barreda, D.; Rosales-García, V.H.; Segovia-García, C.D.; Santos-Mendoza, T.; et al. Extracellular vesicles from Mycobacterium tuberculosis–infected neutrophils induce maturation of monocyte-derived dendritic cells and activation of antigen-specific Th1 cells. J. Leukoc. Biol. 2023, 113, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.M.; Gilhotra, R.; Deopa, D.; Bhat, A.A.; Thapa, R.; Singla, N.; Kulshrestha, R.; Gupta, G. Epigenetics of Pulmonary Tuberculosis, In Targeting Epigenetics in Inflammatory Lung Diseases; Springer: New York, NY, USA, 2023; pp. 127–144. [Google Scholar]
- Thomas, S.S.; Abhinand, K.; Menon, A.M.; Nair, B.G.; Kumar, G.B.; Arun, K.B.; Edison, L.K.; Madhavan, A. Epigenetic Mechanisms Induced by Mycobacterium tuberculosis to Promote Its Survival in the Host. Int. J. Mol. Sci. 2024, 25, 11801. [Google Scholar] [CrossRef] [PubMed]
- Meskini, M.; Zamani, M.S.; Amanzadeh, A.; Bouzari, S.; Karimipoor, M.; Fuso, A.; Fateh, A.; Siadat, S.D. Epigenetic modulation of cytokine expression in Mycobacterium tuberculosis-infected monocyte derived-dendritic cells: Implications for tuberculosis diagnosis. Cytokine 2024, 181, 156693. [Google Scholar] [CrossRef]
- Sutter, A.; Landis, D.; Nugent, K. Metformin has immunomodulatory effects which support its potential use as adjunctive therapy in tuberculosis. Indian J. Tuberc. 2024, 71, 89–95. [Google Scholar] [CrossRef]
- Zhao, L.; Fan, K.; Sun, X.; Li, W.; Qin, F.; Shi, L.; Gao, F.; Zheng, C. Host-directed therapy against mycobacterium tuberculosis infections with diabetes mellitus. Front. Immunol. 2024, 14, 1305325. [Google Scholar] [CrossRef]
- Niu, H.; Bai, C.; Zhu, B.; Zhang, Y. Rapamycin improves the long-term T-cell memory and protective efficacy of tuberculosis subunit vaccine. Microb. Pathog. 2024, 190, 106631. [Google Scholar] [CrossRef] [PubMed]
- Abnousian, A.; Vasquez, J.; Sasaninia, K.; Kelley, M.; Venketaraman, V. Glutathione Modulates Efficacious Changes in the Immune Response against Tuberculosis. Biomedicines 2023, 11, 1340. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Lutfy, K.; Venketaraman, V. Challenges of Multidrug-Resistant Tuberculosis Meningitis: Current Treatments and the Role of Glutathione as an Adjunct Therapy. Vaccines 2024, 12, 1397. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Vaughn, C.; Sasaninia, K.; Yeh, C.; Mehta, D.; Khieran, I.; Venketaraman, V. Understanding the relationship between glutathione, TGF-β, and vitamin D in combating Mycobacterium tuberculosis infections. J. Clin. Med. 2020, 9, 2757. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Sahu, G.; Burns, E.; Ensor, A.; Ensor, J.; Pingali, S.R.; Subbiah, V.; Iyer, S.P. Mycobacterial infections due to PD-1 and PD-L1 checkpoint inhibitors. ESMO Open 2020, 5, e000866. [Google Scholar] [CrossRef]
- Liu, C.-W.; Wu, L.S.-H.; Lin, C.-J.; Wu, H.-C.; Liu, K.-C.; Lee, S.-W. Association of tuberculosis risk with genetic polymorphisms of the immune checkpoint genes PDCD1, CTLA-4, and TIM3. PLoS ONE 2024, 19, e0303431. [Google Scholar] [CrossRef]
- Langan, E.A.; Graetz, V.; Allerheiligen, J.; Zillikens, D.; Rupp, J.; Terheyden, P. Immune checkpoint inhibitors and tuberculosis: An old disease in a new context. Lancet Oncol. 2020, 21, e55–e65. [Google Scholar] [CrossRef]
- Bhat, M.F.; Srdanović, S.; Sundberg, L.-R.; Einarsdóttir, H.K.; Marjomäki, V.; Dekker, F.J. Impact of HDAC inhibitors on macrophage polarization to enhance innate immunity against infections. Drug Discov. Today 2024, 29, 104193. [Google Scholar] [CrossRef] [PubMed]
- Kalsum, S.; Akber, M.; Loreti, M.G.; Andersson, B.; Danielson, E.; Lerm, M.; Brighenti, S. Sirtuin inhibitors reduce intracellular growth of M. tuberculosis in human macrophages via modulation of host cell immunity. Sci. Rep. 2024, 14, 28150. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, F.; Guarnieri, A.; Brancazio, N.; Falcone, M.; Di Naro, M.; Azeem, M.; Zubair, M.; Nicolosi, D.; Di Marco, R.; Petronio, G.P.; et al. The role of Mycobacterium tuberculosis exosomal miRNAs in host pathogen cross-talk as diagnostic and therapeutic biomarkers. Front. Microbiol. 2024, 15, 1441781. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Bastos, R.; Marinho, A.I.; Vieira, R.; Benício, I.; de Noronha, M.A.; Lírio, S.; Brodskyn, C.; Tavares, N.M. Recent advances in the development and clinical application of miRNAs in infectious diseases. Non-coding RNA Res. 2024, 10, 41–54. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Zhang, Z.-C.; Wu, Y.-Y.; Pi, Y.-N.; Lou, S.-H.; Liu, T.-B.; Lou, G.; Yang, C. Bromodomain and extraterminal (BET) proteins: Biological functions, diseases and targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-Y.; Yu, X.-F.; Ji, W.-L. Repression of BRD4 mitigates NLRP3 inflammasome-mediated pyroptosis in Mycobacterium-infected macrophages by repressing endoplasmic reticulum stress. Tuberculosis 2024, 148, 102542. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liang, Y.; Tan, X.; Liu, L. Host long noncoding RNAs in bacterial infections. Front. Immunol. 2024, 15, 1419782. [Google Scholar] [CrossRef] [PubMed]
- Kotey, S.K.; Tan, X.; Kinser, A.L.; Liu, L.; Cheng, Y. Host Long Noncoding RNAs as Key Players in Mycobacteria–Host Interactions. Microorganisms 2024, 12, 2656. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Han, X.; Liu, F.; Long, J.; Jin, Y.; Chen, S.; Duan, G.; Yang, H. Review on Long Non-Coding RNAs as Biomarkers and Potentially Therapeutic Targets for Bacterial Infections. Curr. Issues Mol. Biol. 2024, 46, 7558–7576. [Google Scholar] [CrossRef] [PubMed]
- Jhilta, A.; Jadhav, K.; Singh, R.; Ray, E.; Kumar, A.; Singh, A.K.; Verma, R.K. Breaking the Cycle: Matrix Metalloproteinase Inhibitors as an Alternative Approach in Managing Tuberculosis Pathogenesis and Progression. ACS Infect. Dis. 2024, 10, 2567–2583. [Google Scholar] [CrossRef]
- Maison, D.P. Tuberculosis pathophysiology and anti-VEGF intervention. J. Clin. Tuberc. Other Mycobact. Dis. 2022, 27, 100300. [Google Scholar] [CrossRef] [PubMed]
- McKell, M.C.; Crowther, R.R.; Schmidt, S.M.; Robillard, M.C.; Cantrell, R.; Lehn, M.A.; Janssen, E.M.; Qualls, J.E. Promotion of Anti-Tuberculosis Macrophage Activity by L-Arginine in the Absence of Nitric Oxide. Front. Immunol. 2021, 12, 653571. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carlos, A.; Jacobo-Delgado, Y.; Santos-Mena, A.O.; García-Hernández, M.H.; De Jesus-Gonzalez, L.A.; E Lara-Ramirez, E.; Rivas-Santiago, B. Histone deacetylase (HDAC) inhibitors- based drugs are effective to control Mycobacterium tuberculosis infection and promote the sensibility for rifampicin in MDR strain. Mem. Do Inst. Oswaldo Cruz 2023, 118, e230143. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.; Heater, S.; Peterson, G.J.; Simmons, J.D.; Skerrett, S.J.; Mayanja-Kizza, H.; Stein, C.M.; Boom, W.H.; Hawn, T.R. HDAC3 inhibitor RGFP966 controls bacterial growth and modulates macrophage signaling during Mycobacterium tuberculosis infection. Tuberculosis 2021, 127, 102062. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, T.; An, P.; Ren, Z.; Xi, J.; Tang, B. Important role of DNA methylation hints at significant potential in tuberculosis. Arch. Microbiol. 2024, 206, 177. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Periyasamy, K.M.; Ranganathan, U.D.; Bethunaickan, R. Monocyte and Macrophage miRNA: Potent Biomarker and Target for Host-Directed Therapy for Tuberculosis. Front. Immunol. 2021, 12, 667206. [Google Scholar] [CrossRef] [PubMed]
- Kathamuthu, G.R.; Kumar, N.P.; Moideen, K.; Nair, D.; Banurekha, V.V.; Sridhar, R.; Baskaran, D.; Babu, S. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Are Potential Biomarkers of Pulmonary and Extra-Pulmonary Tuberculosis. Front. Immunol. 2020, 11, 419. [Google Scholar] [CrossRef]
- Datta, M.; Via, L.E.; Dartois, V.; Weiner, D.M.; Zimmerman, M.; Kaya, F.; Walker, A.M.; Fleegle, J.D.; Raplee, I.D.; McNinch, C.; et al. Normalizing granuloma vasculature and matrix improves drug delivery and reduces bacterial burden in tuberculosis-infected rabbits. Proc. Natl. Acad. Sci. USA 2024, 121, e2321336121. [Google Scholar] [CrossRef] [PubMed]
- Oehlers, S.H. Revisiting hypoxia therapies for tuberculosis. Clin. Sci. 2019, 133, 1271–1280. [Google Scholar] [CrossRef]
- Datta, M.; Via, L.E.; Dartois, V.; Xu, L.; Barry, C.E.; Jain, R.K. Leveraging insights from cancer to improve tuberculosis therapy. Trends Mol. Med. 2024, 31, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Zangiabadian, M.; Arabpour, E.; Amini, S.; Khalili, F.; Centis, R.; D'Ambrosio, L.; Denholm, J.T.; Schaaf, H.S.; Boom, M.v.D.; et al. Delamanid-containing regimens and multidrug-resistant tuberculosis: A systematic review and meta-analysis. Int. J. Infect. Dis. 2022, 124, S90–S103. [Google Scholar] [CrossRef]
- Hatami, H.; Sotgiu, G.; Bostanghadiri, N.; Abadi, S.S.D.; Mesgarpour, B.; Goudarzi, H.; Migliori, G.B.; Nasiri, M.J. Bedaquiline-containing regimens and multidrug-resistant tuberculosis: A systematic review and meta-analysis. J. Bras. de Pneumol. 2022, 48, e20210384. [Google Scholar] [CrossRef]
- Yan, F.; Zhao, X.; Li, R.; Han, X.; Yan, Q.; Feng, L.; Xin, X.; Cui, J.; Ma, X. High-throughput fluorescent screening of thioredoxin reductase inhibitors to inhibit Mycobacterium tuberculosis. Chin. Chem. Lett. 2024, 35, 108504. [Google Scholar] [CrossRef]
- Perveen, S.; Negi, A.; Saini, S.; Gangwar, A.; Sharma, R. Identification of Chemical Scaffolds Targeting Drug-Resistant and Latent Mycobacterium tuberculosis through High-Throughput Whole-Cell Screening. ACS Infect. Dis. 2024, 10, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Wu, L.; Li, M.; Dai, J.; Shi, Y.; Wang, Q.; Xu, F.; Zheng, L.; Xiao, X.; Cai, J.; et al. High-throughput nanopore targeted sequencing for efficient drug resistance assay of Mycobacterium tuberculosis. Front. Microbiol. 2024, 15, 1331656. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Gu, R.; Long, J.; Duan, G.; Yang, H. Application of CRISPR–cas-based technology for the identification of tuberculosis, drug discovery and vaccine development. Mol. Biol. Rep. 2024, 51, 466. [Google Scholar] [CrossRef]
- Nanda, P.; Budak, M.; Michael, C.T.; Krupinsky, K.; Kirschner, D.E. Development and analysis of multiscale models for tuberculosis: From molecules to populations. In Predicting Pandemics in a Globally Connected World, Toward a Multiscale, Multidisciplinary Framework Through Modeling and Simulation; Springer: New York, NY, USA, 2024; Volume 2, pp. 11–43. [Google Scholar]
- Qureshi, H.; Shah, Z.; Raja, M.A.Z.; Alshahrani, M.Y.; Khan, W.A.; Shoaib, M. Machine learning investigation of tuberculosis with medicine immunity impact. Diagn. Microbiol. Infect. Dis. 2024, 110, 116472. [Google Scholar] [CrossRef] [PubMed]
- Nafisah, S.I.; Muhammad, G. Tuberculosis detection in chest radiograph using convolutional neural network architecture and explainable artificial intelligence. Neural Comput. Appl. 2024, 36, 111–131. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, F.; Li, L.; Pang, Y. Clinical utilization of artificial intelligence in predicting therapeutic efficacy in pulmonary tuberculosis. J. Infect. Public Health 2024, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Dutta, S.; Murti, K.; Charan, J.K.; Pandey, K.; Ravichandiran, V.; Dhingra, S. The integration of omics: A promising approach to personalized tuberculosis treatment. Med. Omics 2024, 12, 100033. [Google Scholar] [CrossRef]
- Otchere, I.D.; Aboagye, S.Y.; Arthur, P.K.; Asante-Poku, A. Viewpoint of multi-omics potential in tuberculosis research: Identifying biomarkers for biomanufacturing of efficient control tools. Front. Trop. Dis. 2024, 5, 1443248. [Google Scholar] [CrossRef]
- Ayaz, O.; Ali, A.; Ayaz, A.; Nazir, A.; Ahmad, S.B.; Singh, N.; Wani, A.B.; Bhat, K.A. Multiomics technologies applied to tuberculosis drug discovery. In Biological Insights of Multi-Omics Technologies in Human Diseases; Elsevier: New York, NY, USA, 2024; pp. 253–286. [Google Scholar]

{kind=link}
{kind=link}
| Mtb Effector/Protein | Mechanism of Action | Impact on Autophagy | References |
|---|---|---|---|
| PDIM | Inhibits LC3-associated phagocytosis (LAP) by blocking the phagosome recruitment of NADPH oxidase. | PDIM protects Mtb from LAP and classical autophagy, helping the bacteria survive in non-alveolar macrophages in an autophagy-dependent manner. | [26] |
| EsxH | Interferes with Endosomal Sorting Complex Required for Transport (ESCRT) machinery by targeting the VPS4 subunit. | Prevents autophagosome–lysosome fusion, halting autophagy and allowing Mtb to evade degradation within macrophages. | [27] |
| EsxA (ESAT-6) | Forms pores in the lysosomal membrane, causing dysfunction. Activates the cGAS-STING pathway. | Disrupts lysosomal acidification, inhibits autophagic degradation, and increases type I interferon production, which suppresses autophagy. | [28,29,30] |
| PtpA | Dephosphorylates TSC2, leading to prolonged activation of mTORC1. | Sustained mTORC1 activation inhibits autophagy initiation, promoting bacterial survival and supporting intracellular growth. | [10] |
| Zmp1 | Interacts with mitochondrial membranes and blocks NLRP3 inflammasome activation. | Reduces mitochondrial ROS production, further hindering autophagy induction. | [31,32,33] |
| LpqN | Interferes with actin polymerization by targeting host actin-regulating proteins like Arp2/3. | Disrupts early stages of autophagy by blocking autophagosome membrane formation. | [34] |
| PknG | Manipulates host ubiquitin signaling. | Prevents Mtb from being recognized and degraded by autophagy. Inhibits LC3-associated phagocytosis (LAP), which captures cytosolic bacteria. | [17,23,35,36] |
| CpsA | Suppresses ROS production by inhibiting NADPH oxidase activity. | Limits the oxidative burst required for effective xenophagy (selective autophagy of cytoplasmic pathogens). | [17,23,35,36] |
| SecA2 | Modulates host response by shifting from autophagy to apoptosis. | Induces apoptosis, which is less effective at clearing intracellular bacteria compared to autophagy. | [37,38] |
| Therapy Category | Therapy/Agent | Mechanism of Action | Targeted Mtb Mechanism | Impact on TB Treatment | References |
|---|---|---|---|---|---|
| Host-Directed Therapies. | Autophagy Modulators | Rapamycin (mTOR pathway modulator) and metformin (AMPK activator) enhance autophagy to degrade Mtb. | Blocks Mtb’s evasion of autophagy. | Promotes bacterial clearance, reduces intracellular survival of Mtb. | [67,68,69] |
| Glutathione Supplementation | Enhances macrophage activity, reduces Mtb-induced inflammation, and supports Th1 immune responses. | Modulates oxidative stress, boosts immune reprogramming for Th1 responses. | Enhances T-cell proliferation, promotes bacterial killing, supports chronic infection control. | [70,71,72] | |
| Checkpoint Inhibitors | Inhibits immune checkpoint molecules (PD-1/PD-L1, CTLA-4) to restore T-cell responses. | Mtb manipulates checkpoint molecules to suppress immunity. | Restores T-cell immunity, enhances bacterial clearance. | [73,74,75] | |
| Cytokine-Based Immunotherapies | Recombinant Cytokines | IL-2, IL-7, IL-12, IL-15, IL-24, and IFN-γ therapies enhance immune responses to clear Mtb. | Boosts immune responses, enhances macrophage and T-cell activation. | Improves sputum conversion, reduces bacterial load, activates CD8+ T-cells. | [71] |
| TLR Agonists | Imiquimod activates Toll-like receptors to promote autophagy and Th1 differentiation. | Stimulates immune pathways to enhance antigen presentation. | Boosts immune responses, enhances autophagic activity, and improves immune function. | [71] | |
| Anti-IL-4 Antibodies | Shifts immune response from Th2 to Th1, enhancing macrophage activity. | Mtb manipulation of cytokine balance (Th1 vs. Th2). | Increases bacterial clearance by promoting Th1 immunity. | [71] | |
| Corticosteroids | Dexamethasone reduces inflammation, potentially reducing TBM-related mortality. | Modulates immune responses and inflammation. | Potential to reduce inflammation, enhance treatment efficacy in TBM. | [71] | |
| Epigenetic Modulation | HDAC Inhibitors | Vorinostat, panobinostat, entinostat restore immune responses by reactivating immune genes. | Reverses Mtb-induced immune suppression by modulating histone acetylation. | Enhances pathogen recognition, improves bacterial clearance, shortens treatment. | [76] |
| DNA Methylation Modifiers | Decitabine inhibits DNA methyltransferases, reversing immune suppression through DNA demethylation. | Silences key immune-related gene promoters. | Restores gene expression of immune regulators like IFN-γ, improves immune response in latent and active TB. | [77] | |
| MicroRNA-Based Modulation | Anti-miRNA therapies block Mtb-induced miRNAs, restoring cytokine production and enhancing bacterial killing. | Suppression of miRNA expression impairs immune signaling. | Improves immune responses, enhances bacterial killing, restores macrophage function. | [78,79] | |
| Histone Modifications (BET Inhibitors) | JQ1 inhibits BET proteins, modulating chromatin structure and immune responses. | Mtb alters histone acetylation and methylation patterns. | Enhances anti-inflammatory responses, improves immune activation. | [80,81] | |
| Long Non-Coding RNAs (lncRNAs) | Targets lncRNAs like MEG3 to enhance immune activation. | Mtb modulates lncRNA to suppress immune responses. | Promotes enhanced resistance to TB by improving immune activation. | [82,83,84] | |
| Targeting Granuloma Dynamics | Matrix Metalloproteinase (MMP) Inhibitors | Doxycycline reduces MMP activity, preserving granuloma structure and preventing bacterial dissemination. | Mtb exploits MMPs for granuloma remodeling. | Reduces tissue destruction, preserves granuloma structure, enhances bacterial clearance. | [85] |
| Anti-Angiogenic Therapies | Bevacizumab (anti-VEGF) disrupts granuloma vasculature, limiting nutrient and oxygen supply to Mtb. | Mtb thrives in vascularized granulomas, promotes survival in hypoxia. | Reduces bacterial load, enhances immune access to granulomas, counters Mtb persistence. | [86] | |
| Hypoxia Modulation | HIF-1α inhibitors or arginine supplementation restore immune function in hypoxic granulomas. | Hypoxia stabilizes HIF-1α, aiding Mtb persistence. | Enhances macrophage bactericidal activity, reduces granuloma necrosis. | [58,87] | |
| Fibrosis Control | TGF-β inhibitors reduce fibrosis in granulomas, enhancing immune cell penetration. | Excessive fibrosis protects Mtb from immune attack. | Improves granuloma stability, enhances immune cell infiltration and bacterial clearance. | [53,72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri, M.J.; Venketaraman, V. Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. Int. J. Mol. Sci. 2025, 26, 1621. https://doi.org/10.3390/ijms26041621
Nasiri MJ, Venketaraman V. Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. International Journal of Molecular Sciences. 2025; 26(4):1621. https://doi.org/10.3390/ijms26041621
Chicago/Turabian StyleNasiri, Mohammad J., and Vishwanath Venketaraman. 2025. "Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention" International Journal of Molecular Sciences 26, no. 4: 1621. https://doi.org/10.3390/ijms26041621
APA StyleNasiri, M. J., & Venketaraman, V. (2025). Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. International Journal of Molecular Sciences, 26(4), 1621. https://doi.org/10.3390/ijms26041621