
Preimplantation Genetic Testing (PGT) to Reduce the Risk for GBA-Related Parkinson’s Disease: Expanding the Applications for Embryo Selection


 ,
,  ,
,  and
and {kind=link}
Abstract
1. Introduction
1.1. Gaucher Disease
1.2. Prenatal Genetic Counseling for GD Patients and Carriers for the Risk of PD in the Fetus
1.3. “Manifesting Heterozygotes”: Risks Associated with Carrying One Mutation of Autosomal Recessive Disorders
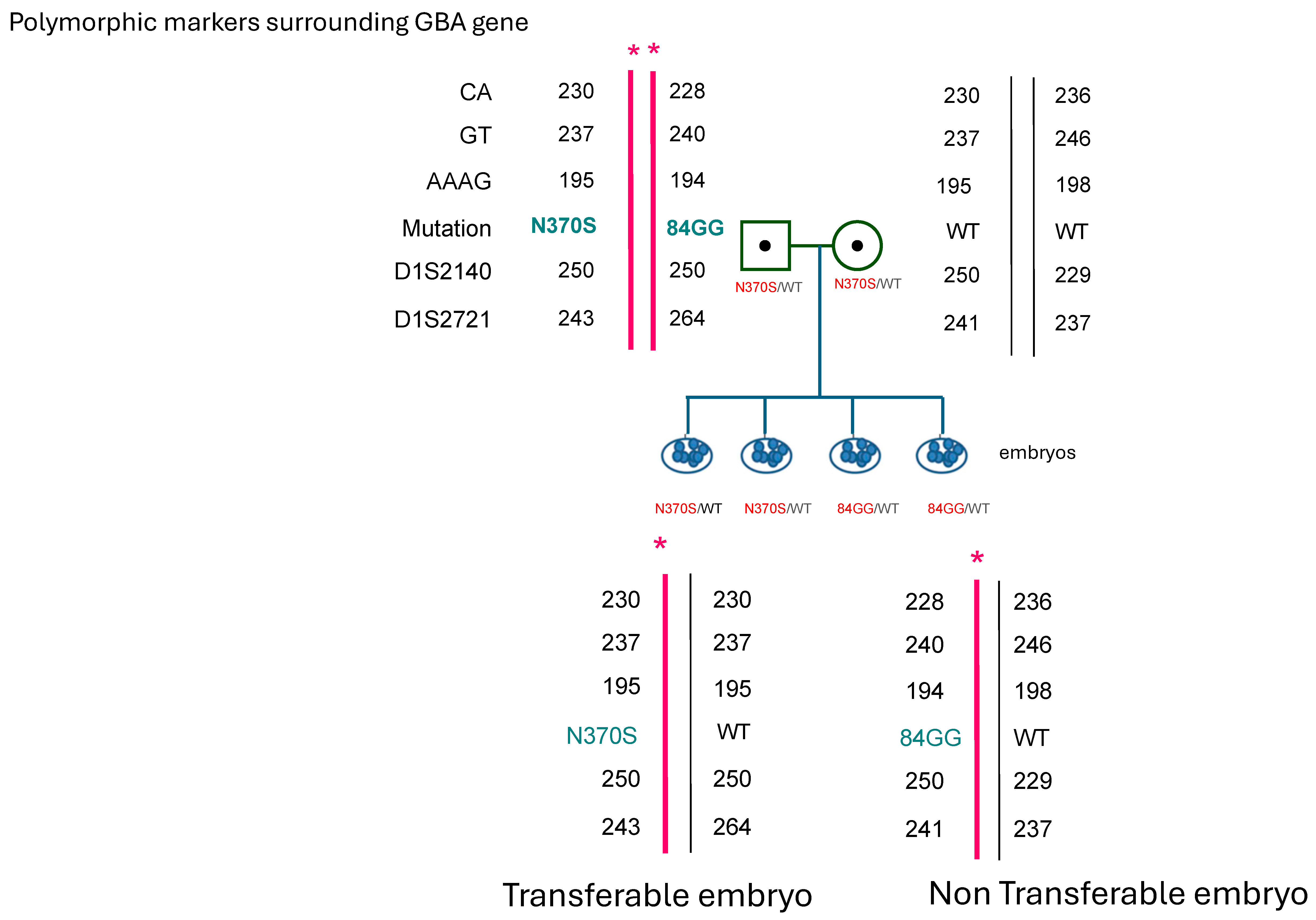
2. Case Report
2.1. Ovarian Stimulation and Egg Retrieval
2.2. Blastomere Biopsy, ICSI, and Embryo Cultures
2.3. Molecular Analysis
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Poorthuis, B.J.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.; de Jong, J.G.; van Weely, S.; Niezen-Koning, K.E.; van Diggelen, O.P. The frequency of lysosomal storage diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Gelbart, T.; Kuhl, W.; Zimran, A.; West, C. Mutations in Jewish patients with Gaucher disease. Blood 1992, 79, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Nguyen, N.J.; Henneberger, M.W.; Smolec, J.M.; McPherson, R.A.; West, C.; Gelbart, T. Gaucher disease: Gene frequencies in the Ashkenazi Jewish population. Am. J. Hum. Genet. 1993, 52, 85–88. [Google Scholar]
- Azuri, J.; Elstein, D.; Lahad, A.; Abrahamov, A.; Hadas-Halpern, I.; Zimran, A. Asymptomatic Gaucher disease implications for large-scale screening. Genet. Test. 1998, 2, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Scott, C.R.; Zeigler, M.; Abrahamov, A.; Zimran, A. Phenotypic heterogeneity in patients with Gaucher disease and the N370S/V394L genotype. Genet. Test. 2005, 9, 26–29. [Google Scholar] [CrossRef]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.J.; Giraldo, P.; Capablo, J.L.; Alfonso, P.; Irun, P.; Garcia-Rodriguez, B.; Pocovi, M.; Pastores, G.M. Greater risk of parkinsonism associated with non-N370S GBA1 mutations. J. Inherit. Metab. Dis. 2013, 36, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Mulhern, M.; Bier, L.; Alcalay, R.N.; Balwani, M. Patients’ Opinions on Genetic Counseling on the Increased Risk of Parkinson Disease among Gaucher Disease Carriers. J. Genet. Couns. 2018, 27, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Thein, S.L. The carrier state for sickle cell disease is not completely harmless. Haematologica 2019, 104, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef]
- Mastenbroek, S.; Twisk, M.; van Echten-Arends, J.; Sikkema-Raddatz, B.; Korevaar, J.C.; Verhoeve, H.R.; Vogel, N.E.; Arts, E.G.; de Vries, J.W.; Bossuyt, P.M.; et al. In vitro fertilization with preimplantation genetic screening. N. Engl. J. Med. 2007, 357, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Verlinsky, Y.; Kuliev, A. Micromanipulation and biopsy of polar bodies and blastomeres. In Atlas of Preimplantation Genetic Diagnosis; Taylor & Francis: London, UK, 2005; pp. 15–22. [Google Scholar]
- Thornhill, A.R.; McGrath, J.A.; Eady, R.A.; Braude, P.R.; Handyside, A.H. A comparison of different lysis buffers to assess allele dropout from single cells for preimplantation genetic diagnosis. Prenat. Diagn. 2001, 21, 490–497. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Altarescu, G.; Renbaum, P.; Brooks, P.B.; Margalioth, E.J.; Ben Chetrit, A.; Munter, G.; Levy-Lahad, E.; Eldar-Geva, T. Successful polar body-based preimplantation genetic diagnosis for achondroplasia. Reprod. Biomed. Online 2008, 16, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, A.R.; deDie-Smulders, C.E.; Geraedts, J.P.; Harper, J.C.; Harton, G.L.; Lavery, S.A.; Moutou, C.; Robinson, M.D.; Schmutzler, A.G.; Scriven, P.N.; et al. ESHRE PGD Consortium ‘Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)’. Hum. Reprod. 2005, 20, 35–48. [Google Scholar] [CrossRef]
- Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Varshower, I.; Brooks, B.; Beeri, R.; Margalioth, E.J.; Levy-Lahad, E.; Elstein, D.; Zimran, A. Preimplantation genetic diagnosis (PGD) for a treatable disorder: Gaucher disease type 1 as a model. Blood Cells Mol. Dis. 2011, 46, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Protection GFMoJaC. German Embryo Protection Act. Embryonenschutzgesetz vom 13 Dezember 1990 (BGBl I S 2746). Federal Law Gazette. 13 December 1990. [Google Scholar]
- Authority UHFaE: Code of Practice: Embryo Testing and Sex Selection. 2020. Available online: https://www.hfea.gov.uk/about-us/news-and-press-releases/2018/latest-statement-on-sex-selection/ (accessed on 10 January 2025).
- Europe Co: Steering Committee on Bioethics. 2004. Available online: https://www.coe.int/t/dg3/healthbioethic/cdbi/ (accessed on 10 January 2025).
- Bioethics TPsCo, Washington DC. Reproduction and Responsibility: The Regulation of New Biotechnologies. 2004. Available online: https://bioethicsarchive.georgetown.edu/pcbe/reports/reproductionandresponsibility/fulldoc.html (accessed on 10 January 2025).
- Pennings, G.; de Wert, G.; Shenfield, F.; Cohen, J.; Tarlatzis, B.; Devroey, P. ESHRE Task Force on Ethics and Law 13: The welfare of the child in medically assisted reproduction. Hum. Reprod. 2007, 22, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.G.; King, M.R.; Whitaker, M.I. Who gets born? How did New Zealand’s Bioethics Council arrive at its recommendations? N. Z. Med. J. 2009, 122, 84–91. [Google Scholar]
- Robertson, J.A. Extending preimplantation genetic diagnosis: The ethical debate. Ethical issues in new uses of preimplantation genetic diagnosis. Hum. Reprod. 2003, 18, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kuliev, A.; Verlinsky, Y. Place of preimplantation diagnosis in genetic practice. Am. J. Med. Genet. A 2005, 134A, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Krahn, T. Preimplantation genetic diagnosis: Does age of onset matter (anymore)? Med. Health Care Philos. 2009, 12, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.; Williamson, R. Is there an ethical difference between preimplantation genetic diagnosis and abortion? J. Med. Ethics 2003, 29, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Savulescu, J. Procreative beneficence: Why we should select the best children. Bioethics 2001, 15, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Heyd, D. Male or female, we will create them: The ethics of sex selection for non-medical reasons. Ethical Perspect. 2003, 10, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Malek, J.; Daar, J. The case for a parental duty to use preimplantation genetic diagnosis for medical benefit. Am. J. Bioeth. 2012, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Stock, G. Germinal choice technology and the human future. Reprod. Biomed. Online 2005, 10 (Suppl. S1), 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.L. Assisted procreation and its relationship to genetics and eugenics. Hum. Reprod. Genet. Ethics 2009, 15, 7–27. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, S.; Zeevi, D.A.; Gooldin, S.; Altarescu, G. Acceptable applications of preimplantation genetic diagnosis (PGD) among Israeli PGD users. Eur. J. Hum. Genet. 2017, 25, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Harper, J.; Sharma, A.; Fraser, L.; Burnell, M.; ElMasry, K.; Rodeck, C.; Jacobs, I. Views of BRCA gene mutation carriers on preimplantation genetic diagnosis as a reproductive option for hereditary breast and ovarian cancer. Hum. Reprod. 2007, 22, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Quinn, G.; Vadaparampil, S.; Wilson, C.; King, L.; Choi, J.; Miree, C.; Friedman, S. Attitudes of high-risk women toward preimplantation genetic diagnosis. Fertil. Steril. 2009, 91, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Ormondroyd, E.; Donnelly, L.; Moynihan, C.; Savona, C.; Bancroft, E.; Evans, D.G.; Eeles, R.; Lavery, S.; Watson, M. Attitudes to reproductive genetic testing in women who had a positive BRCA test before having children: A qualitative analysis. Eur. J. Hum. Genet. 2012, 20, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Gietel-Habets, J.J.G.; de Die-Smulders, C.E.M.; Tjan-Heijnen, V.C.G.; Derks-Smeets, I.A.P.; van Golde, R.; Gomez-Garcia, E.; van Osch, L.A.D.M. Professionals’ knowledge, attitude and referral behaviour of preimplantation genetic diagnosis for hereditary breast and ovarian cancer. Reprod. Biomed. Online 2018, 36, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Forzano, F.; Antonova, O.; Clarke, A.; de Wert, G.; Hentze, S.; Jamshidi, Y.; Moreau, Y.; Perola, M.; Prokopenko, I.; Read, A.; et al. The use of polygenic risk scores in pre-implantation genetic testing: An unproven, unethical practice. Eur. J. Hum. Genet. 2022, 30, 493–495. [Google Scholar] [CrossRef]
- Rotshenker-Olshinka, K.; Srebnik Moshe, N.; Weiss, O.; Shaviv, S.; Freireich, O.; Segel, R.; Zeligson, S.; Eldar-Geva, T.; Altarescu, G. Preimplantation genetic testing (PGT) for copy number variants of uncertain significance (CNV-VUS) in the genomic era: To do or not to do? J. Assist. Reprod. Genet. 2021, 38, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Porto, A.; Gaber Caffrey, R.; Crowley-Matoka, M.; Spencer, S.; Li, M.; Propst, L. Offering preimplantation genetic testing for monogenic disorders (PGT-M) for conditions with reduced penetrance or variants of uncertain significance: Ethical insight from U.S. laboratory genetic counselors. J. Genet. Couns. 2022, 31, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.; Bracewell-Milnes, T.; Saso, S.; Jones, B.P.; Almeida, P.A.; Maclaren, K.; Norman-Taylor, J.; Johnson, M.; Nikolaou, D. A review on the motivations, decision-making factors, attitudes and experiences of couples using pre-implantation genetic testing for inherited conditions. Hum. Reprod. Update 2021, 27, 944–966. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Steeves, M.; Bayrak-Toydemir, P.; Benson, K.A.; Coe, B.P.; Conlin, L.K.; Ganapathi, M.; Garcia, J.; Gollob, M.H.; Jobanputra, V.; et al. Recommendations for risk allele evidence curation, classification, and reporting from the ClinGen Low Penetrance/Risk Allele Working Group. Genet. Med. 2024, 26, 101036. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuckerman, S.; Zimran, A.; Szer, J.; Revel-Vilk, S.; Altarescu, G. Preimplantation Genetic Testing (PGT) to Reduce the Risk for GBA-Related Parkinson’s Disease: Expanding the Applications for Embryo Selection. Int. J. Mol. Sci. 2025, 26, 912. https://doi.org/10.3390/ijms26030912
Zuckerman S, Zimran A, Szer J, Revel-Vilk S, Altarescu G. Preimplantation Genetic Testing (PGT) to Reduce the Risk for GBA-Related Parkinson’s Disease: Expanding the Applications for Embryo Selection. International Journal of Molecular Sciences. 2025; 26(3):912. https://doi.org/10.3390/ijms26030912
Chicago/Turabian StyleZuckerman, Shachar, Ari Zimran, Jeff Szer, Shoshana Revel-Vilk, and Gheona Altarescu. 2025. "Preimplantation Genetic Testing (PGT) to Reduce the Risk for GBA-Related Parkinson’s Disease: Expanding the Applications for Embryo Selection" International Journal of Molecular Sciences 26, no. 3: 912. https://doi.org/10.3390/ijms26030912
APA StyleZuckerman, S., Zimran, A., Szer, J., Revel-Vilk, S., & Altarescu, G. (2025). Preimplantation Genetic Testing (PGT) to Reduce the Risk for GBA-Related Parkinson’s Disease: Expanding the Applications for Embryo Selection. International Journal of Molecular Sciences, 26(3), 912. https://doi.org/10.3390/ijms26030912