


Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy

 , ,
, ,  , , , , , , and
, , , , , , and
Abstract

1. Introduction
2. Results
2.1. Production of Stable Lentiviral Transduced Human Cell Lines
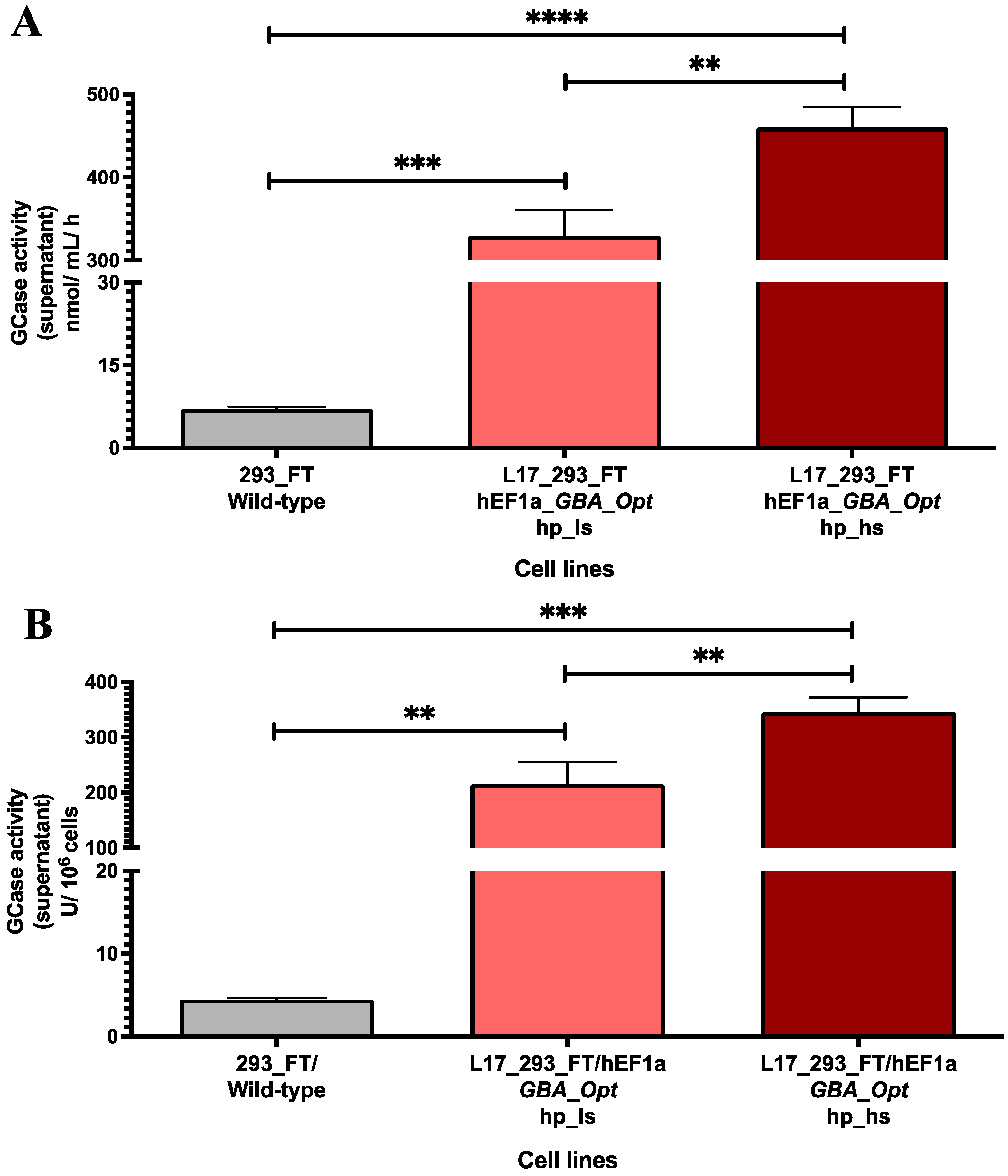
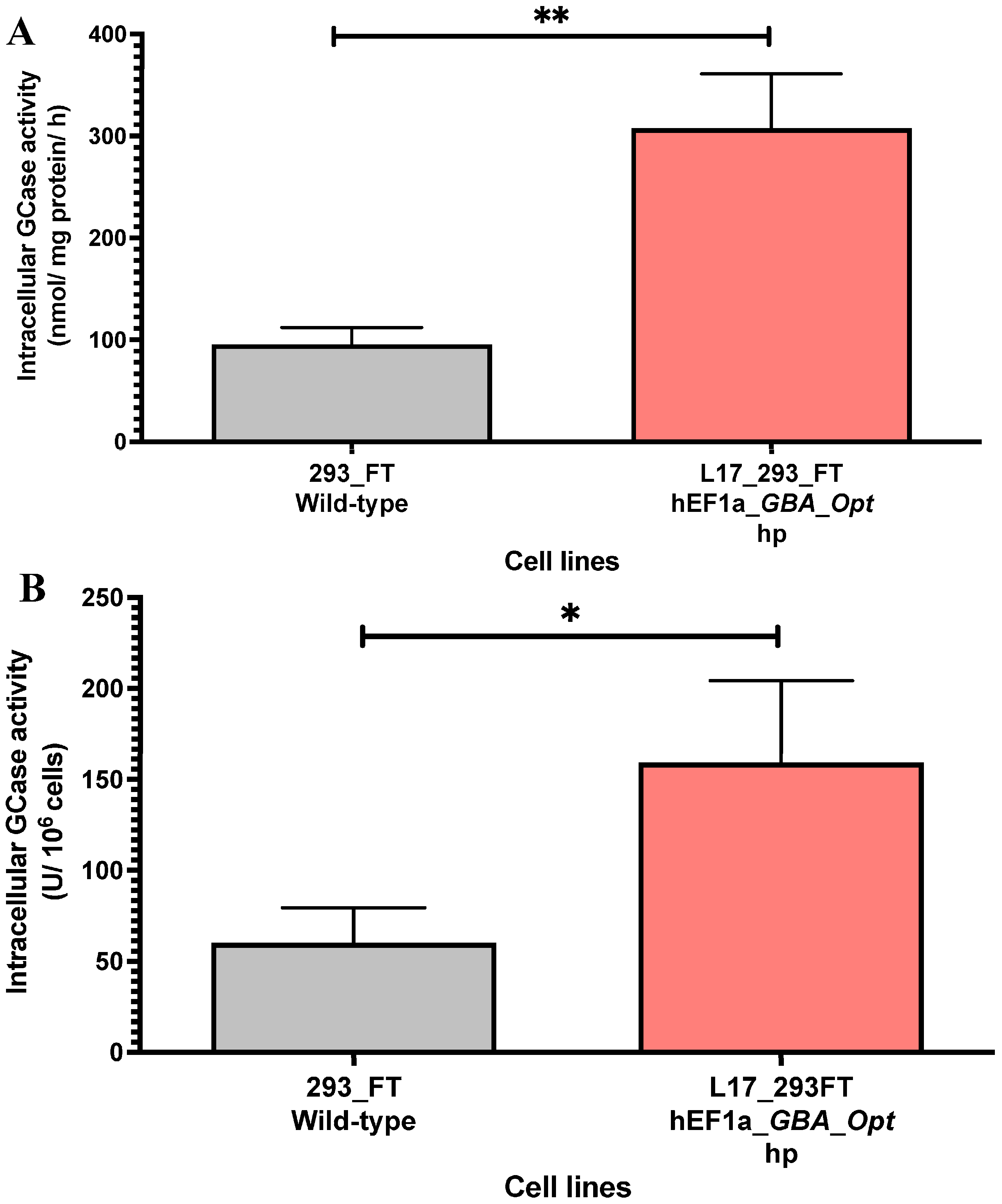
2.2. GCase Activity in Lentiviral Transduced and Puromycin-Selected L17_293FT_GBA_OPT_HP Cells
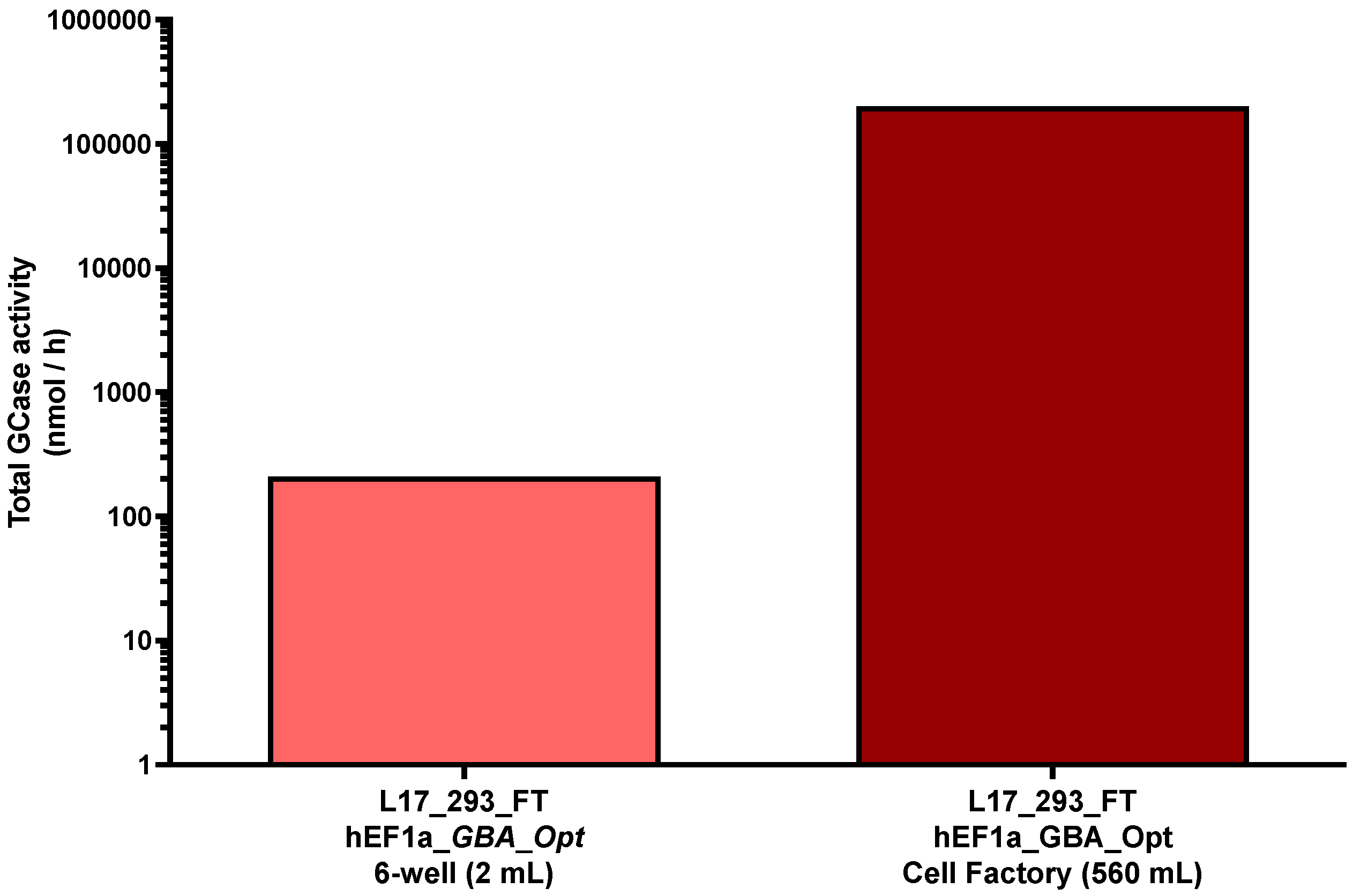
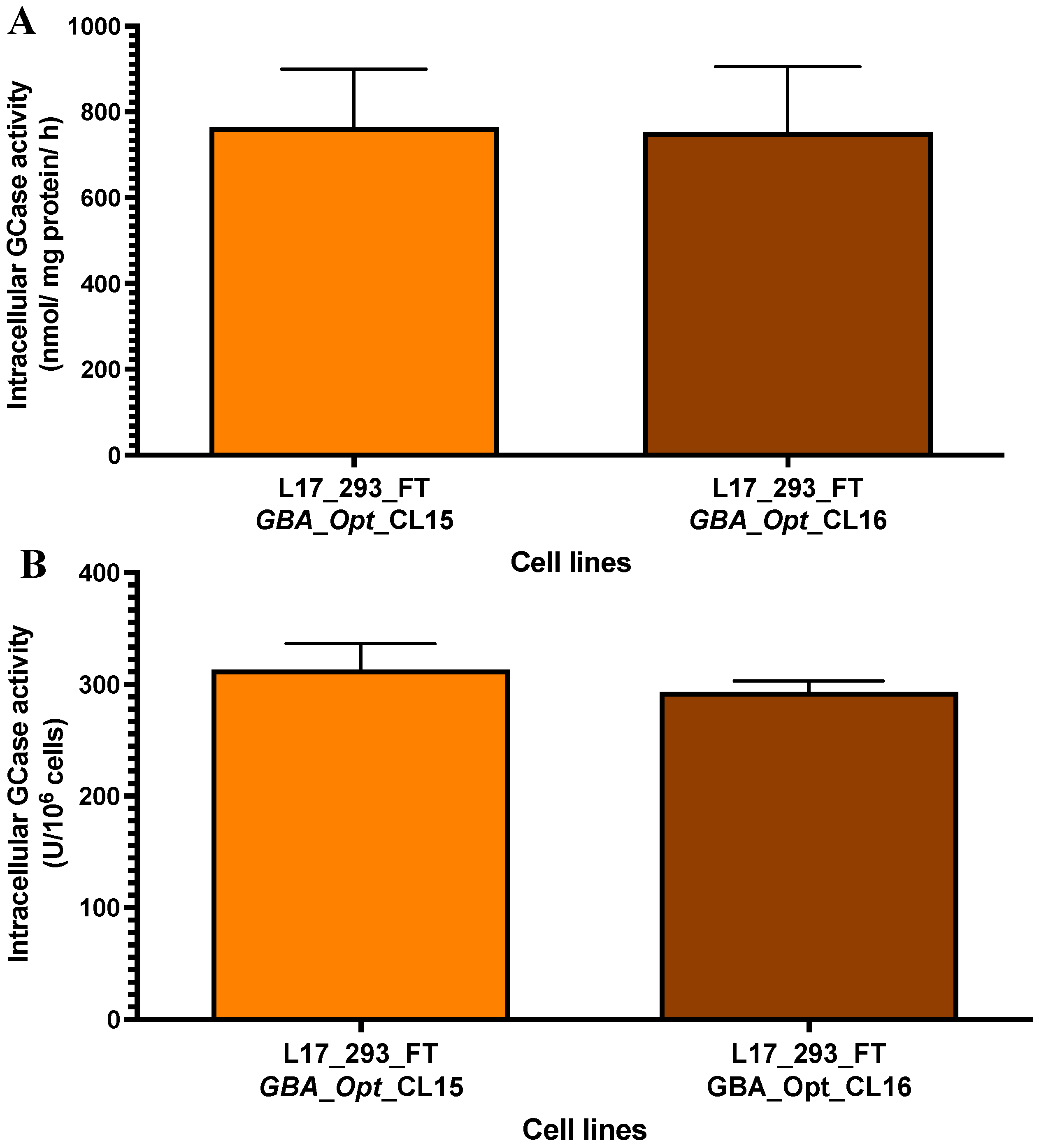
2.3. High-Producer Clone Selection from Puromycin-Selected Population
3. Discussion
Limitations and Future Directions
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cell Culture
4.3. Production of Lentiviral Particles
4.4. Lentiviral Transduction and Establishment of Stable Transduced Cell Lines
4.5. Puromycin Treatment of L17_293FT_GBA_OPT_HP Heterogeneous Population
4.6. GCase Activity Analysis: Secreted and Intracellular (GCase-Specific Activity)
4.7. Scaling of GCase Production in L17_293FT_GBA_OPT_HP Cell Supernatants
4.8. Clone Cell Selection (Isolation)
4.9. Biological Activity by Fluorimetric Assay
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| CHO | Chinese hamster ovary cell |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ERT | Enzyme replacement therapy |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | Fetal bovine serum |
| FDA | Food and Drug Administration |
| GBA1 | Glucocerebrosidase, Glucosylceramidase beta 1 |
| GCase | Glucocerebrosidase, Glucosylceramidase beta 1 |
| GD | Gaucher disease |
| GlcCer | Glucosylceramide |
| 293_FT | Human embryonic kidney 293 cells |
| HP | Heterogeneous population |
| HS | High stringency |
| ICGG | International Collaborative Gaucher Group |
| L17 | Lineage 17 |
| LS | Low stringency |
| LV | Lentiviral vector |
| LTR | Long terminal repeat |
| MOI | Multiplicity of infection |
| MTX | Methotrexate |
| 4MU | 4-methylumbiliferone |
| 4MUG | 4-methylumbiliferon-β-D-glucopyranoside |
| qPCR | Real-time quantitative PCR |
| SUS | Unified Health System (Sistema Único de Saúde) |
| TDC | Sodium taurodeoxycholate hydrate |
References
- Hughes, D.A.; Pastores, G.M. Gaucher Disease. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Belmatoug, N.; Bembi, B.; Deegan, P.; Fernandez-Sasso, D.; Giraldo, P.; Goker-Alpan, O.; Hughes, D.; Lau, H.; Lukina, E.; et al. Twelve Years of the Gaucher Outcomes Survey (GOS): Insights, Achievements, and Lessons Learned from a Global Patient Registry. J. Clin. Med. 2024, 13, 3588. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, G.; Ruan, J.; Fattahi, N.; Mehta, S.; Boddupalli, C.S.; Mistry, P.K.; Nair, S. Modeling bone marrow microenvironment and hematopoietic dysregulation in Gaucher disease through VavCre mediated Gba deletion. Hum. Mol. Genet. 2025, 34, 952–966. [Google Scholar] [CrossRef] [PubMed]
- Ducatez, F.; Berger, M.G.; Pilon, C.; Plichet, T.; Lesueur, C.; Berger, J.; Belmatoug, N.; Marret, S.; Bekri, S.; Tebani, A. Deciphering metabolic shifts in Gaucher disease type 1: A multi-omics study. J. Mol. Med. 2025, 103, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Levy, Y.; Bar-Ziv, Y.; Revel-Vilk, S.; Zimran, A.; Lebel, E. Simultaneous Bilateral Femoral Osteonecrosis in Gaucher Disease. Life 2023, 13, 1135. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Antommaria, A.H.M.; Kolodny, E.H.; Mistry, P.K. Gaucher disease: Basic and translational science needs for more complete therapy and management. Mol. Genet. Metab. 2021, 132, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Kishnani, P.S.; Alcalay, R.N.; Prakalapakorn, S.G.; Rosenbloom, B.E.; Tuason, D.A.; Weinreb, N.J. Challenges in Gaucher disease: Perspectives from an expert panel. Mol. Genet. Metab. 2025, 145, 109074. [Google Scholar] [CrossRef] [PubMed]
- Furderer, M.L.; Hertz, E.; Lopez, G.J.; Sidransky, E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int. J. Mol. Sci. 2022, 23, 5842. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.; Chen, Y.; Sidransky, E. Gaucher disease provides a unique window into Parkinson disease pathogenesis. Nat. Rev. Neurol. 2024, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, G.; Ledda, C.; Romagnolo, A.; Covolo, A.; Lopiano, L.; Artusi, C.A. Neurological symptoms in adults with Gaucher disease: A systematic review. J. Neurol. 2024, 271, 3897–3907. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Motta, I.; Delbini, P.; Scaramellini, N.; Ghiandai, V.; Duca, L.; Nava, I.; Nascimbeni, F.; Lugari, S.; Consonni, D.; Trombetta, E.; et al. Enzyme replacement therapy improves erythropoiesis and iron dysregulation in Gaucher disease. Ann. Hematol. 2024, 103, 5113–5121. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.; Lau, H.; Elstein, D.; Fernandez-Sasso, D.; Giraldo, P.; Hughes, D.; Zimran, A.; Istaiti, M.; Gadir, N.; Botha, J.; et al. Long-Term Treatment of Gaucher Disease with Velaglucerase Alfa in ERT-Naive Patients from the Gaucher Outcome Survey (GOS) Registry. J. Clin. Med. 2024, 13, 2782. [Google Scholar] [CrossRef] [PubMed]
- Revel-Vilk, S.; Mansfield, R.; Feder-Krengel, N.; Machtiger-Azoulay, N.; Kuter, D.; Szer, J.; Rosenbaum, H.; Ferreira, D.C.; Ruhrman-Shahar, N.; Wajnrajch, M.; et al. Real-World Experiences with Taliglucerase Alfa Home Infusions for Patients with Gaucher Disease: A Global Cohort Study. J. Clin. Med. 2023, 12, 5913. [Google Scholar] [CrossRef] [PubMed]
- Ain, N.U.; Saith, A.; Ruan, A.; Yang, R.; Burton, A.; Mistry, P.K. Eliglustat substrate reduction therapy in children with Gaucher disease type 1. Front. Pediatr. 2025, 13, 1543136. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Balwani, M.; Charrow, J.; Lorber, J.; Niederau, C.; Carwile, J.L.; Oliveira-Dos-Santos, A.; Perichon, M.G.; Uslu Cil, S.; Kishnani, P.S. Long-term effectiveness of eliglustat treatment: A real-world analysis from the International Collaborative Gaucher Group Gaucher Registry. Am. J. Hematol. 2024, 99, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Camelo, J.S., Jr.; Charrow, J.; McClain, M.R.; Mistry, P.; Belmatoug, N. International Collaborative Gaucher Group Gaucher Registry i: Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol. Genet. Metab. 2021, 132, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Belmatoug, N.; vom Dahl, S.; Giugliani, R. Understanding the natural history of Gaucher disease. Am. J. Hematol. 2015, 90, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Kiec-Wilk, B.; Lampe, C.; Tangeraas, T.; Cefalo, G.; Belmatoug, N.; Francisco, R.; Del Toro, M.; Wagner, L.; Lauridsen, A.G.; et al. Challenges in Transition From Childhood to Adulthood Care in Rare Metabolic Diseases: Results From the First Multi-Center European Survey. Front. Med. 2021, 8, 652358. [Google Scholar] [CrossRef] [PubMed]
- Borin, M.C.; Alvares-Teodoro, J.; Acurcio, F.A.; Guerra, A.A., Jr. Gaucher disease in Brazil: A comprehensive 16 year retrospective study on survival, cost, and treatment insights. Front. Pharmacol. 2024, 15, 1433970. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Barton, N.W.; Pastores, G.; Dambrosia, J.M.; Banerjee, T.K.; McKee, M.A.; Parker, C.; Schiffmann, R.; Hill, S.C.; Brady, R.O. Enzyme therapy in type 1 Gaucher disease: Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann. Intern. Med. 1995, 122, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Elstein, D.; Levy-Lahad, E.; Zevin, S.; Hadas-Halpern, I.; Bar-Ziv, Y.; Foldes, J.; Schwartz, A.J.; Abrahamov, A. Replacement therapy with imiglucerase for type 1 Gaucher’s disease. Lancet 1995, 345, 1479–1480. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Altarescu, G.; Philips, M.; Attias, D.; Jmoudiak, M.; Deeb, M.; Wang, N.; Bhirangi, K.; Cohn, G.M.; Elstein, D. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010, 115, 4651–4656. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A. Velaglucerase alfa: A new option for Gaucher disease treatment. Drugs Today 2011, 47, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Brill-Almon, E.; Chertkoff, R.; Petakov, M.; Blanco-Favela, F.; Munoz, E.T.; Solorio-Meza, S.E.; Amato, D.; Duran, G.; Giona, F.; et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011, 118, 5767–5773. [Google Scholar] [CrossRef] [PubMed]
- van Dussen, L.; Zimran, A.; Akkerman, E.M.; Aerts, J.M.; Petakov, M.; Elstein, D.; Rosenbaum, H.; Aviezer, D.; Brill-Almon, E.; Chertkoff, R.; et al. Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Brasil; Ministério da Saúde; Secretaria de Atenção à Saúde. Protocolo Clínico e Diretrizes Terapêuticas da Doença de Gaucher; Ministério da Saúde: Brasília, Brazil, 2014; (atualizado em 2017).
- Liu, C.; Bahnson, A.B.; Dunigan, J.T.; Watkins, S.C.; Barranger, J.A. Long-term expression and secretion of human glucocerebrosidase by primary murine and human myoblasts and differentiated myotubes. J. Mol. Med. 1998, 76, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Hong, Y.B.; Lai, Z.; Kim, H.J.; Cho, Y.H.; Brady, R.O.; Jung, S.C. Expression and secretion of human glucocerebrosidase mediated by recombinant lentivirus vectors in vitro and in vivo: Implications for gene therapy of Gaucher disease. Biochem. Biophys. Res. Commun. 2004, 318, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, S.G.; Poletto, E.; Lucot, K.L.; Colella, P.; Sheikali, A.; Montine, T.J.; Porteus, M.H.; Gomez-Ospina, N. Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun. 2020, 11, 3327. [Google Scholar] [CrossRef] [PubMed]
- Novo, J.B.; Morganti, L.; Moro, A.M.; Paes Leme, A.F.; Serrano, S.M.; Raw, I.; Ho, P.L. Generation of a Chinese hamster ovary cell line producing recombinant human glucocerebrosidase. J. Biomed. Biotechnol. 2012, 2012, 875383. [Google Scholar] [CrossRef] [PubMed]
- Naphatsamon, U.; Ohashi, T.; Misaki, R.; Fujiyama, K. The Production of Human beta-Glucocerebrosidase in Nicotiana benthamiana Root Culture. Int. J. Mol. Sci. 2018, 19, 1972. [Google Scholar] [CrossRef] [PubMed]
- Uthailak, N.; Kajiura, H.; Misaki, R.; Fujiyama, K. Production of recombinant beta-glucocerebrosidase in wild-type and glycoengineered transgenic Nicotiana benthamiana root cultures with different N-glycan profiles. J. Biosci. Bioeng. 2022, 133, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon bias confers stability to human mRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zheng, Y.; Qureshi, I.; Zin, H.T.; Beck, T.; Bulka, B.; Freeland, S.J. SGDB: A database of synthetic genes re-designed for optimizing protein over-expression. Nucleic Acids Res. 2007, 35, D76–D79. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- VandenDriessche, T.; Chuah, M.K. Hemophilia Gene Therapy: Ready for Prime Time? Hum. Gene Ther. 2017, 28, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Abraham, A.; Aboobacker, F.; Singh, G.; Geevar, T.; Kulkarni, U.; Selvarajan, S.; Korula, A.; Dave, R.G.; Shankar, M.; et al. Lentiviral Gene Therapy with CD34+ Hematopoietic Cells for Hemophilia A. N. Engl. J. Med. 2025, 392, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Charrier, S.; Corre, G.; Gjata, B.; Vignaud, A.; Zhang, F.; Rothe, M.; Schambach, A.; Gaspar, H.B.; Thrasher, A.J.; et al. Preclinical Development of a Lentiviral Vector for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol. Ther. Methods Clin. Dev. 2018, 9, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Konig, J.F.; Gray, S.J.; Tikkanen, R. Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production. Int. J. Mol. Sci. 2017, 18, 706. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, J.M.; Kia, A.; Tam, L.C.S.; McIntosh, J.; Spiewak, J.; Mills, K.; Heywood, W.; Chisari, E.; Castaldo, N.; Verhoef, D.; et al. Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther. 2023, 30, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, G.; Choy, F.Y. Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expr. Purif. 2002, 26, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.L.S.; Junior, W.L.; Goncalves, V.W.S.; Ramos, E.S.; D’Almeida, V.; Souza, L.E.B.; Orellana, M.D.; Abraham, K.J.; Lichtenstein, F.; Bleicher, L.; et al. Engineering Synthetic and Recombinant Human Lysosomal β-Glucocerebrosidase for Enzyme Replacement Therapy for Gaucher Disease. Discov. Appl. Sci. 2024, 6, 527. [Google Scholar] [CrossRef]
- Pimentel, N.; Rodriguez-Lopez, A.; Diaz, S.; Losada, J.C.; Diaz-Rincon, D.J.; Cardona, C.; Espejo-Mojica, A.J.; Ramirez, A.M.; Ruiz, F.; Landazuri, P.; et al. Production and characterization of a human lysosomal recombinant iduronate-2-sulfatase produced in Pichia pastoris. Biotechnol. Appl. Biochem. 2018, 65, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Belur, L.R.; Karlen, A.D.; Erlanson, O.; Podetz-Pedersen, K.M.; McKenzie, J.; Detellis, J.; Gagnidze, K.; Parsons, G.; Robinson, N.; et al. Phenotypic Correction of Murine Mucopolysaccharidosis Type II by Engraftment of Ex vivo Lentiviral Vector-Transduced Hematopoietic Stem and Progenitor Cells. Hum. Gene Ther. 2022, 33, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Sands, S.A.; Yue, Y.; Zhang, K.; LeVine, S.M.; Duan, D. An Engineered Galactosylceramidase Construct Improves AAV Gene Therapy for Krabbe Disease in Twitcher Mice. Hum. Gene Ther. 2019, 30, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Ungari, S.; Montepeloso, A.; Morena, F.; Cocchiarella, F.; Recchia, A.; Martino, S.; Gentner, B.; Naldini, L.; Biffi, A. Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol. Ther. Methods Clin. Dev. 2015, 2, 15038. [Google Scholar] [CrossRef] [PubMed]
- Doerfler, P.A.; Todd, A.G.; Clement, N.; Falk, D.J.; Nayak, S.; Herzog, R.W.; Byrne, B.J. Copackaged AAV9 Vectors Promote Simultaneous Immune Tolerance and Phenotypic Correction of Pompe Disease. Hum. Gene Ther. 2016, 27, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Stok, M.; de Boer, H.; Huston, M.W.; Jacobs, E.H.; Roovers, O.; Visser, T.P.; Jahr, H.; Duncker, D.J.; van Deel, E.D.; Reuser, A.J.J.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2020, 17, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Catalano, F.; Vlaar, E.C.; Pijnenburg, J.M.; Stok, M.; van Helsdingen, Y.; Vulto, A.G.; van der Ploeg, A.T.; van Til, N.P.; Pijnappel, W. IGF2-tagging of GAA promotes full correction of murine Pompe disease at a clinically relevant dosage of lentiviral gene therapy. Mol. Ther. Methods Clin. Dev. 2022, 27, 109–130. [Google Scholar] [CrossRef] [PubMed]
- Ornaghi, F.; Sala, D.; Tedeschi, F.; Maffia, M.C.; Bazzucchi, M.; Morena, F.; Valsecchi, M.; Aureli, M.; Martino, S.; Gritti, A. Novel bicistronic lentiviral vectors correct beta-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: Implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol. Dis. 2020, 134, 104667. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Xu, Q.; Zhang, F.; Buckland, K.F.; Gao, Y.; Du, W.; Ding, Y.; Zhou, L.; Sun, X.; Ma, L.; et al. Preclinical ex vivo IL2RG gene therapy using autologous hematopoietic stem cells as an effective and safe treatment for X-linked severe combined immunodeficiency disease. Genes Dis. 2025, 12, 101445. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.H.; Rothe, M.; Barber, D.L.; McKillop, W.M.; Fraser, G.; Morel, C.F.; Schambach, A.; Auray-Blais, C.; West, M.L.; Khan, A.; et al. Persistent hematopoietic polyclonality after lentivirus-mediated gene therapy for Fabry disease. Mol. Ther. Methods Clin. Dev. 2023, 28, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Mangiameli, E.; Cecchele, A.; Morena, F.; Sanvito, F.; Matafora, V.; Cattaneo, A.; Della Volpe, L.; Gnani, D.; Paulis, M.; Susani, L.; et al. Human iPSC-based neurodevelopmental models of globoid cell leukodystrophy uncover patient- and cell type-specific disease phenotypes. Stem Cell Rep. 2021, 16, 1478–1495. [Google Scholar] [CrossRef] [PubMed]
- Ellison, S.; Buckland, K.; Learmonth, Y.; Day, V.; Kalra, S.; Howe, L.; Roman-Rodriguez, F.J.; Bonafont, J.; Booth, L.; Holley, R.; et al. Design and validation of a GMP stem cell manufacturing protocol for MPSII hematopoietic stem cell gene therapy. Mol. Ther. Methods Clin. Dev. 2024, 32, 101271. [Google Scholar] [CrossRef] [PubMed]
- Dogan, Y.; Barese, C.N.; Schindler, J.W.; Yoon, J.K.; Unnisa, Z.; Guda, S.; Jacobs, M.E.; Oborski, C.; Maiwald, T.; Clarke, D.L.; et al. Screening chimeric GAA variants in preclinical study results in hematopoietic stem cell gene therapy candidate vectors for Pompe disease. Mol. Ther. Methods Clin. Dev. 2022, 27, 464–487. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.S.; Gurung, S.; Perocheau, D.; Counsell, J.; Baruteau, J. Gene therapy for inherited metabolic diseases. J. Mother Child 2020, 24, 53–64. [Google Scholar] [PubMed]
- Zielske, S.P.; Gerson, S.L. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol. Ther. 2003, 7, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Fontes, A.M.; Davis, B.M.; Encell, L.P.; Lingas, K.; Covas, D.T.; Zago, M.A.; Loeb, L.A.; Pegg, A.E.; Gerson, S.L. Differential competitive resistance to methylating versus chloroethylating agents among five O6-alkylguanine DNA alkyltransferases in human hematopoietic cells. Mol. Cancer Ther. 2006, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fontes, A.M.; Melo, F.U.; Greene, L.J.; Faca, V.M.; Lin, Y.; Gerson, S.L.; Covas, D.T. Production of human factor VIII-FL in 293T cells using the bicistronic MGMT(P140K)-retroviral vector. Genet. Mol. Res. 2012, 11, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Castilho-Fernandes, A.; Fontes, A.M.; Abraham, K.J.; de Freitas, M.C.; da Rosa, N.G.; Picanco-Castro, V.; de Sousa Russo-Carbolante, E.M.; Covas, D.T. Significant differences in integration sites of Moloney murine leukemia virus/Moloney murine sarcoma virus retroviral vector carrying recombinant coagulation factor IX in two human cell lines. Biotechnol. Lett. 2015, 37, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Correa de Freitas, M.C.; Fontes, A.M.; de Castilho Fernandes, A.; Picanco-Castro, V.; de Sousa Russo, E.M.; Covas, D.T. Murine leukemia virus-derived retroviral vector has differential integration patterns in human cell lines used to produce recombinant factor VIII. Rev. Bras. Hematol. Hemoter. 2014, 36, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Trono, D.; Verma, I.M. Lentiviral vectors, two decades later. Science 2016, 353, 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Scotti, C.; Aiuti, A.; Naldini, L. Challenges and solutions to the sustainability of gene and cell therapies. Nat. Rev. Genet. 2025, 26, 437–438. [Google Scholar] [CrossRef]
- al Yacoub, N.; Romanowska, M.; Haritonova, N.; Foerster, J. Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene Med. 2007, 9, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Hirch, T.; Brander, N.; Schenk, F.; Pollmann, S.J.; Reichenbach, J.; Schubert, R.; Modlich, U. Expression of a large coding sequence. Gene therapy vectors for Ataxia Telangiectasia. Sci. Rep. 2023, 13, 19386. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, C.; Ge, X. Large T antigen mediated target gene replication improves site-specific recombination efficiency. Front. Bioeng. Biotechnol. 2024, 12, 1377167. [Google Scholar] [CrossRef] [PubMed]
- Abaandou, L.; Quan, D.; Shiloach, J. Affecting HEK293 Cell Growth and Production Performance by Modifying the Expression of Specific Genes. Cells 2021, 10, 1667. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; Rayat, A. Lentiviral Vector Bioprocessing. Viruses 2021, 13, 268. [Google Scholar] [CrossRef] [PubMed]
- Jargalsaikhan, B.E.; Muto, M.; Been, Y.; Matsumoto, S.; Okamura, E.; Takahashi, T.; Narimichi, Y.; Kurebayashi, Y.; Takeuchi, H.; Shinohara, T.; et al. The Dual-Pseudotyped Lentiviral Vector with VSV-G and Sendai Virus HN Enhances Infection Efficiency through the Synergistic Effect of the Envelope Proteins. Viruses 2024, 16, 827. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Hao, Y.; Zhang, L.; Cao, X.; An, L.; Wang, H.; Ma, Q.; Jin, X.; Ma, X. Development and validation of optimized lentivirus-like particles for gene editing tool delivery with Gag-Only strategy. Eur. J. Med. Res. 2025, 30, 242. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.; Edelstein, H.I.; Glykofrydis, F.; Love, K.S.; Palacios, S.; Tycko, J.; Zhang, M.; Lensch, S.; Shields, C.E.; Livingston, M.; et al. The sound of silence: Transgene silencing in mammalian cell engineering. Cell Syst. 2022, 13, 950–973. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.J.; Jia, Y.L.; Wang, M.; Yi, D.D.; Zhang, W.L.; Wang, X.Y.; Zhang, J.H. Effect of promoter, promoter mutation and enhancer on transgene expression mediated by episomal vectors in transfected HEK293, Chang liver and primary cells. Bioengineered 2019, 10, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Vlaar, E.C.; Pijnenburg, J.M.; Rijkers, E.; Demmers, J.A.A.; Vulto, A.G.; van der Ploeg, A.T.; van Til, N.P.; Pijnappel, W. Lentiviral gene therapy with IGF2-tagged GAA normalizes the skeletal muscle proteome in murine Pompe disease. J. Proteom. 2024, 291, 105037. [Google Scholar] [CrossRef] [PubMed]
- da Rosa, N.G.; Swiech, K.; Picanco-Castro, V.; Russo-Carbolante, E.M.; Soares Neto, M.A.; de Castilho-Fernandes, A.; Faca, V.M.; Fontes, A.M.; Covas, D.T. SK-HEP cells and lentiviral vector for production of human recombinant factor VIII. Biotechnol. Lett. 2012, 34, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Alves, P.; Serra, M.; Brito, C.; Ricardo, C.P.; Cunha, R.; Sousa, M.F.; Sanchez, B.; Bernad, A.; Carrondo, M.J.; Rodriguez-Borlado, L.; et al. In vitro expansion of human cardiac progenitor cells: Exploring ‘omics tools for characterization of cell-based allogeneic products. Transl. Res. 2016, 171, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Vicente, P.; Inocencio, L.R.; Ullate-Agote, A.; Louro, A.F.; Jacinto, J.; Gamelas, B.; Iglesias-Garcia, O.; Martin-Uriz, P.S.; Aguirre-Ruiz, P.; Rios-Munoz, G.R.; et al. Billion-Scale Expansion of Functional hiPSC-Derived Cardiomyocytes in Bioreactors Through Oxygen Control and Continuous Wnt Activation. Adv. Sci. 2025, 12, e2410510. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.T.; Denning, G.; Gautney, R.E.; Dropulic, B.; Roy, A.J.; Baranyi, L.; Gangadharan, B.; Parker, E.T.; Lollar, P.; Doering, C.B. Lentiviral vector platform for production of bioengineered recombinant coagulation factor VIII. Mol. Ther. 2011, 19, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Kutner, R.H.; Zhang, X.Y.; Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Fantacini, D.M.; Fontes, A.M.; de Abreu Neto, M.S.; Covas, D.T.; Picanco-Castro, V. The F309S mutation increases factor VIII secretion in human cell line. Rev. Bras. Hematol. Hemoter. 2016, 38, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Shibata, M.; Brown, B.; Labelle, A.; Hegadorn, C.; Andrews, C.; Hebbel, R.P.; Galipeau, J.; Hough, C.; Lillicrap, D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells 2007, 25, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.P.; Coyle, P.; Glew, R.H. Differentiation of beta-glucocerebrosidase from beta-glucosidase in human tissues using sodium taurocholate. Arch. Biochem. Biophys. 1976, 175, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.B.; Rodrigues, M.D.; Pereira, V.G.; Martins, A.M.; D’Almeida, V. Reference values for lysosomal enzymes activities using dried blood spots samples—A Brazilian experience. Diagn. Pathol. 2010, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | GCase Activity (nmol/mL/h) | GCase Activity (U/106 Cells) |
|---|---|---|
| L17_293FT_GBA_OPT_CL5 | 265.087 | 252.464 |
| L17_293FT_GBA_OPT_CL7 | 89.911 | 128.444 |
| L17_293FT_GBA_OPT_CL8 | 230.045 | 135.320 |
| L17_293FT_GBA_OPT_CL9 | 301.160 | 200.773 |
| L17_293FT_GBA_OPT_CL10 | 118.907 | 72.065 |
| L17_293FT_GBA_OPT_CL11 | 285.561 | 219.662 |
| L17_293FT_GBA_OPT_CL13 | 440.955 | 275.597 |
| L17_293FT_GBA_OPT_CL15 | 585.464 | 390.310 |
| L17_293FT_GBA_OPT_CL16 | 683.952 | 455.968 |
| L17_293FT_GBA_OPT_CL17 | 207.610 | 143.180 |
| L17_293FT_GBA_OPT_CL18 | 167.931 | 108.342 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, A.C.; Wiezel, C.E.V.; de Campos, A.C.; Figueiredo, L.L.S.; Suardi, G.A.M.; de Paula Bernardes, J.; da Cunha Tirapelli, D.P.; Faça, V.M.; Abraham, K.J.; Carlotti-Júnior, C.G.; et al. Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy. Int. J. Mol. Sci. 2025, 26, 7089. https://doi.org/10.3390/ijms26157089
Coelho AC, Wiezel CEV, de Campos AC, Figueiredo LLS, Suardi GAM, de Paula Bernardes J, da Cunha Tirapelli DP, Faça VM, Abraham KJ, Carlotti-Júnior CG, et al. Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy. International Journal of Molecular Sciences. 2025; 26(15):7089. https://doi.org/10.3390/ijms26157089
Chicago/Turabian StyleCoelho, Ana Carolina, Claudia Emília Vieira Wiezel, Alline Cristina de Campos, Lílian Louise Souza Figueiredo, Gabriela Aparecida Marcondes Suardi, Juliana de Paula Bernardes, Daniela Pretti da Cunha Tirapelli, Vitor Marcel Faça, Kuruvilla Joseph Abraham, Carlos Gilberto Carlotti-Júnior, and et al. 2025. "Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy" International Journal of Molecular Sciences 26, no. 15: 7089. https://doi.org/10.3390/ijms26157089
APA StyleCoelho, A. C., Wiezel, C. E. V., de Campos, A. C., Figueiredo, L. L. S., Suardi, G. A. M., de Paula Bernardes, J., da Cunha Tirapelli, D. P., Faça, V. M., Abraham, K. J., Carlotti-Júnior, C. G., Siciliano, V., Weiss, R., Gerson, S., & Fontes, A. M. (2025). Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy. International Journal of Molecular Sciences, 26(15), 7089. https://doi.org/10.3390/ijms26157089