


p21 Promoter Methylation Is Vital for the Anticancer Activity of Withaferin A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
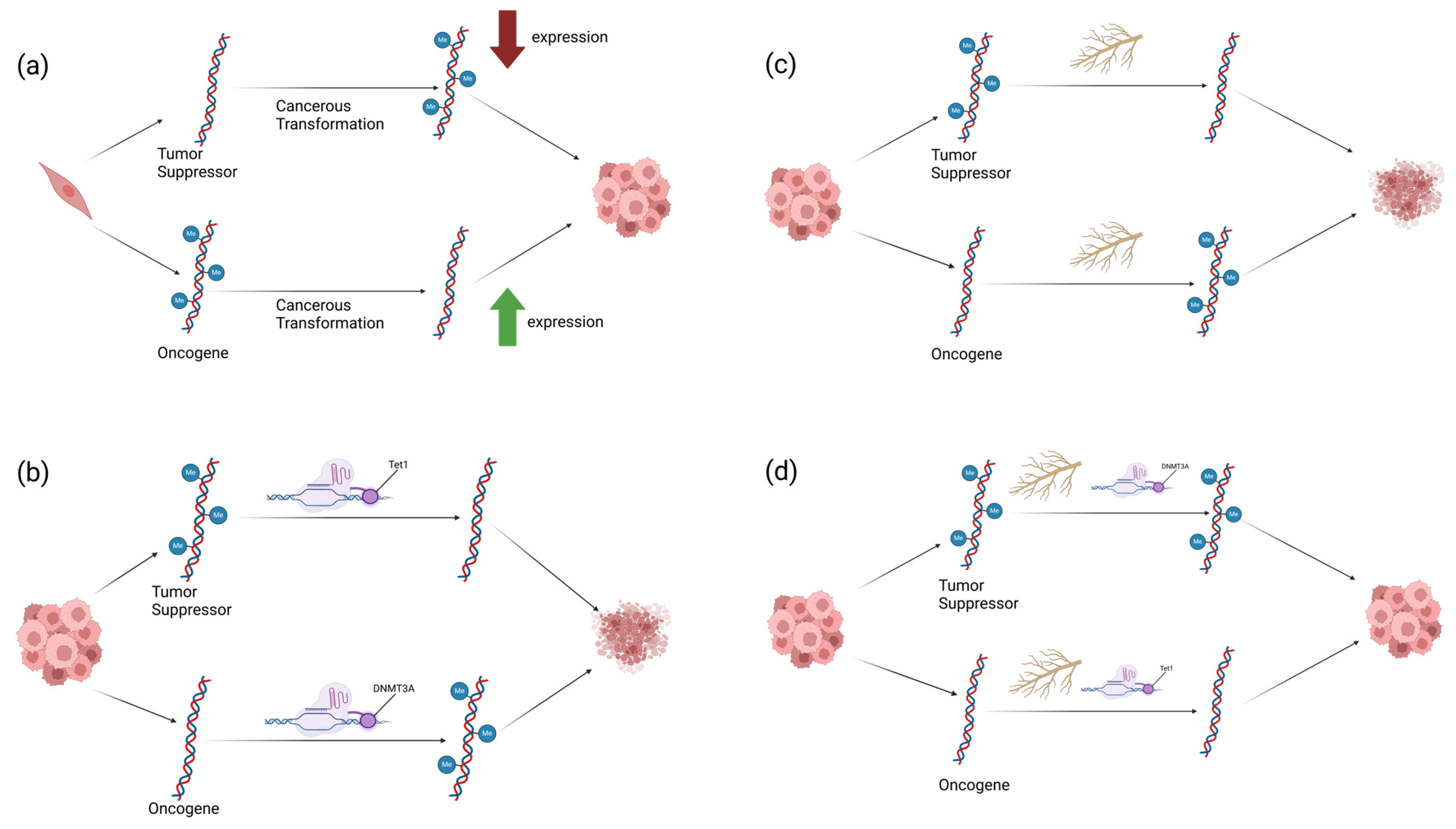
1. Introduction
2. Results
2.1. p21, p53, and CCND1 Are Differentially Methylated in Breast Cancer Patients Versus Normal Breast Tissue
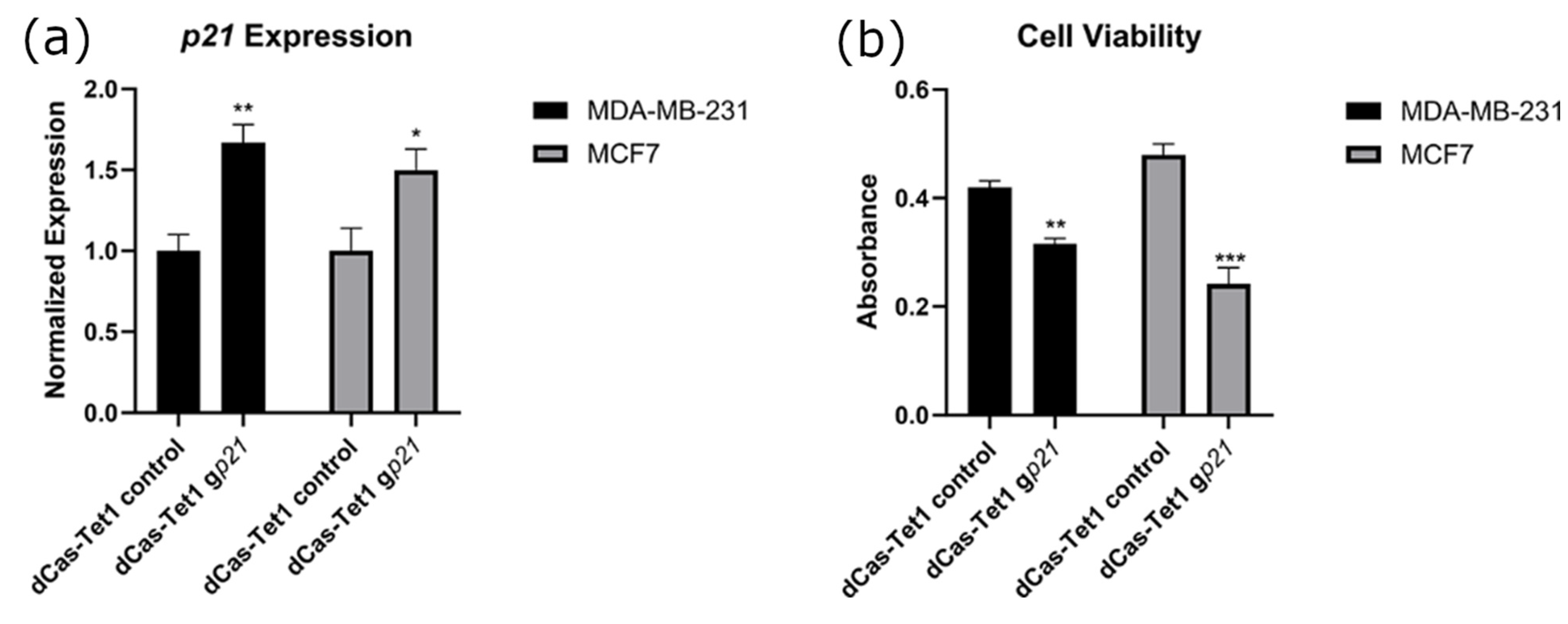
2.2. Targeted Demethylation of the p21 Promoter Increased p21 Expression and Decreased Cancer Cell Viability
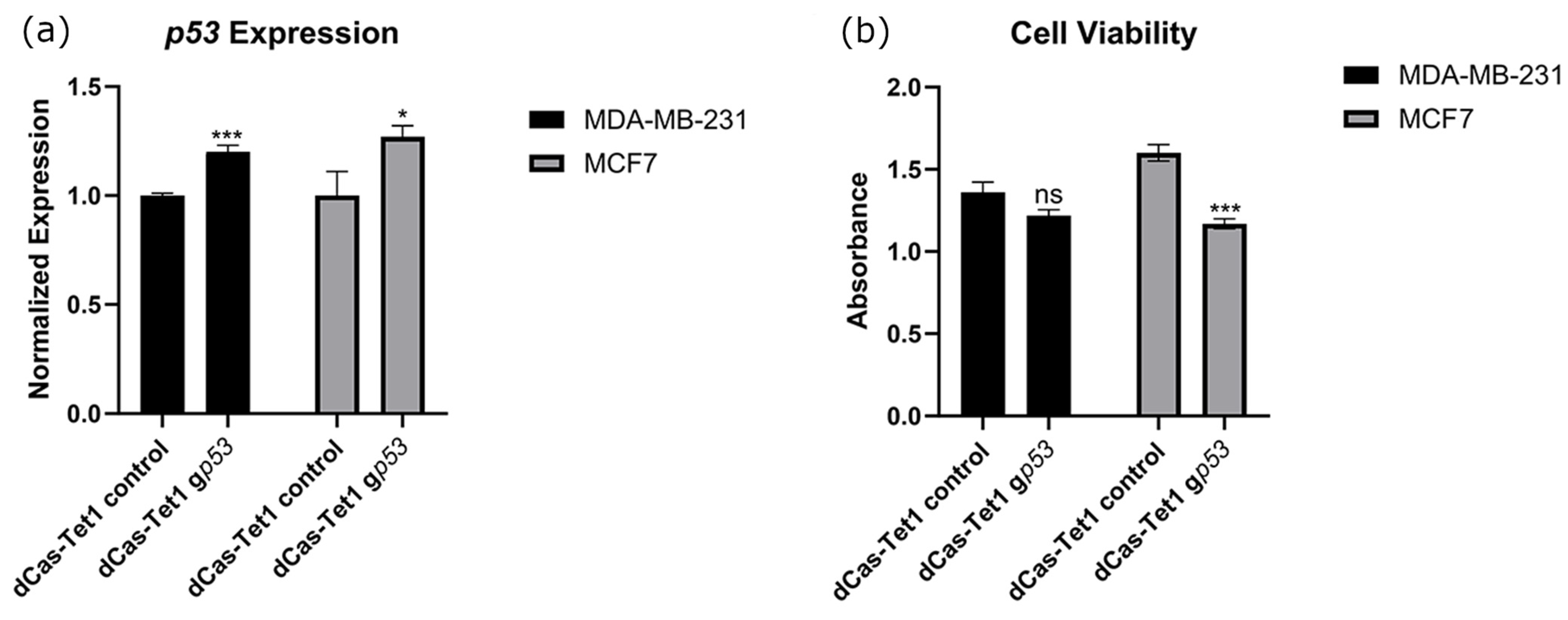
2.3. Targeted Demethylation of the p53 Promoter Increased p53 Expression and Decreased Cancer Cell Viability
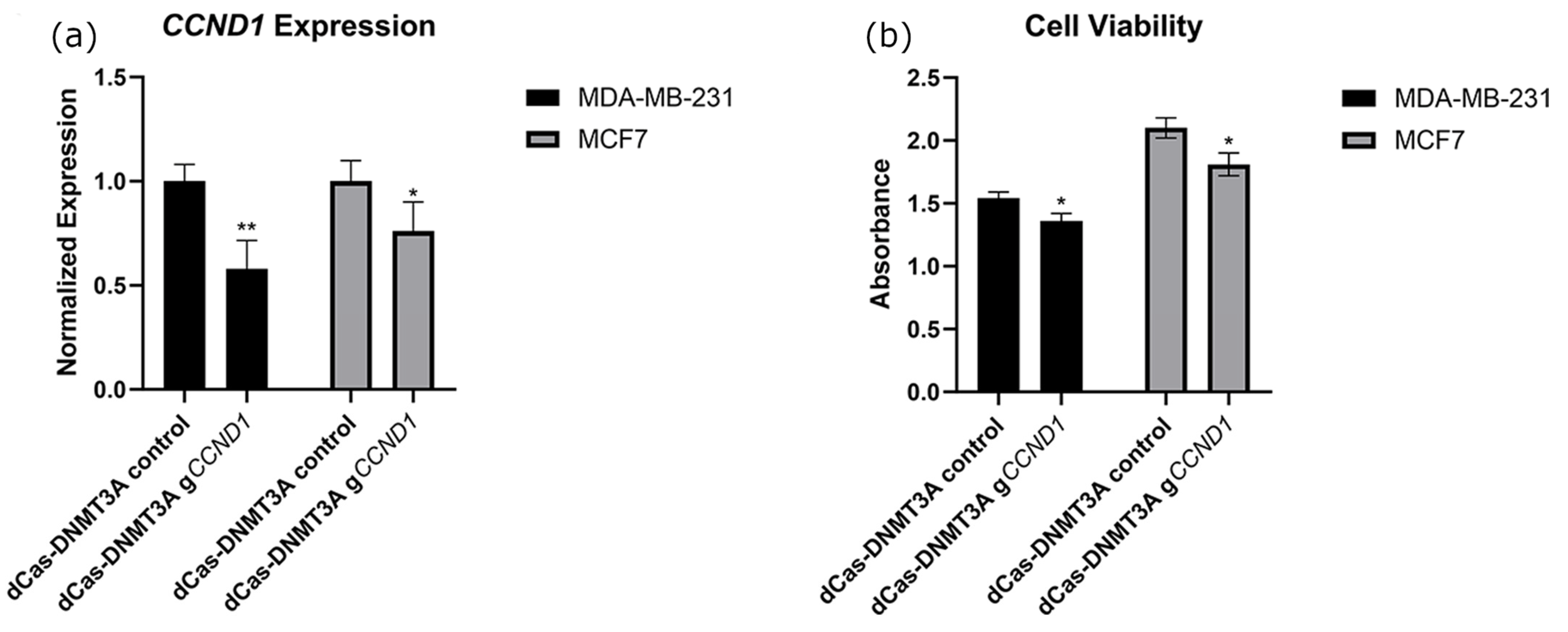
2.4. Targeted Methylation of the CCND1 Promoter Decreased CCND1 Expression and Decreased Cancer Cell Viability
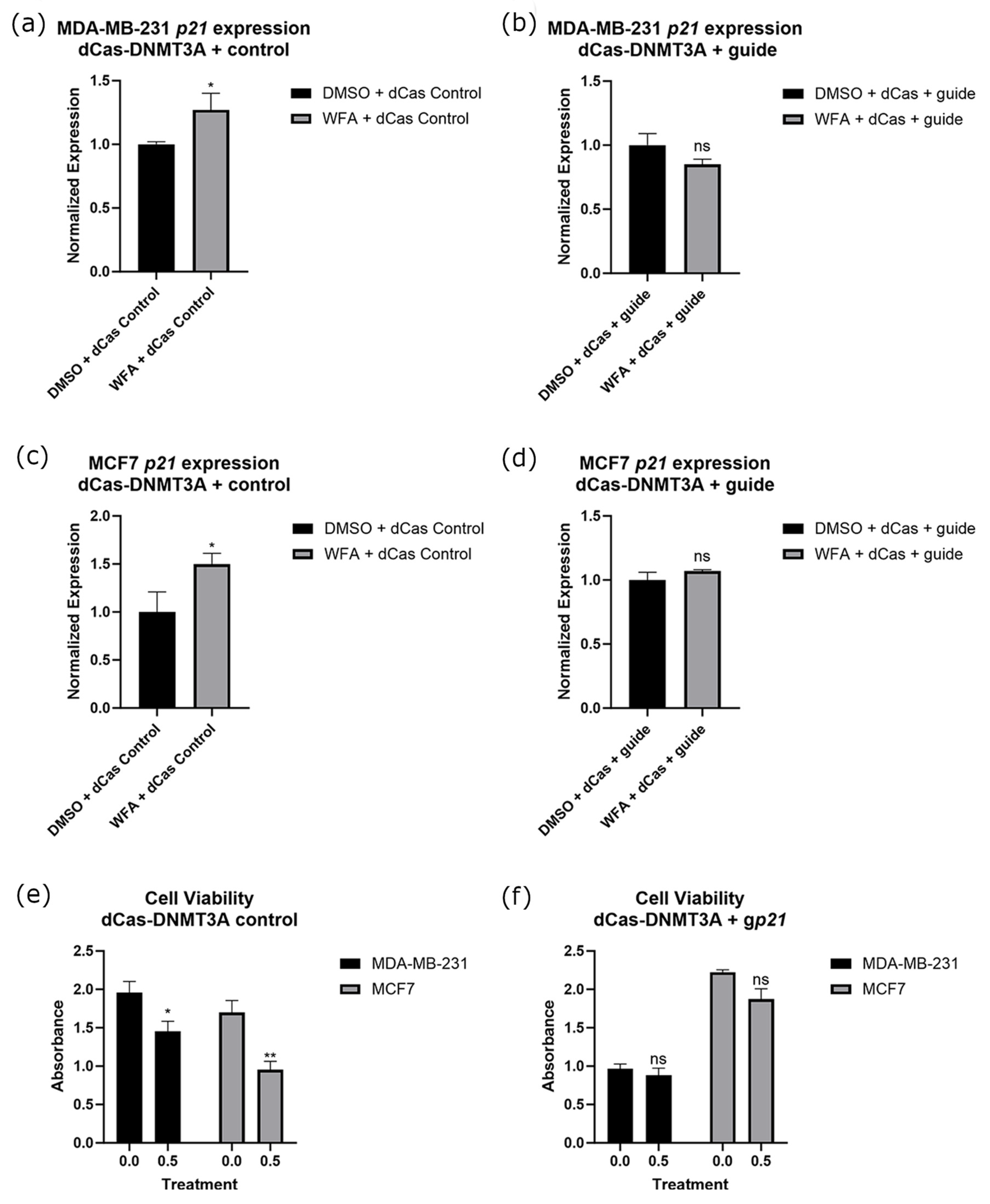
2.5. Targeted Methylation of the p21 Promoter in Combination with WFA Ablates p21 Expression Changes and Resulted in a Loss of WFA Anticancer Function
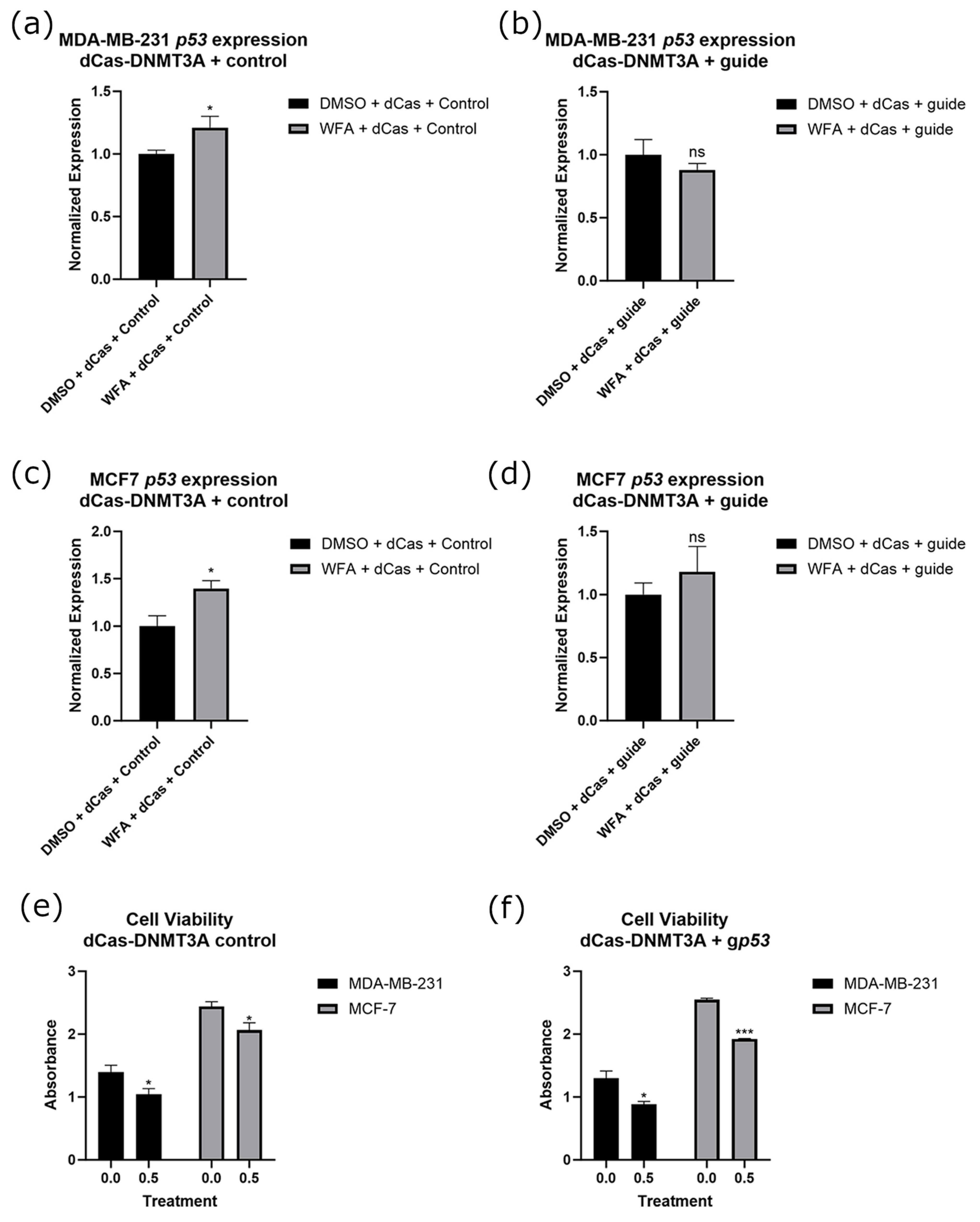
2.6. Targeted Methylation of the p53 Promoter in Combination with WFA Ablates p53 Expression Changes with No Significant Loss of WFA Anticancer Function
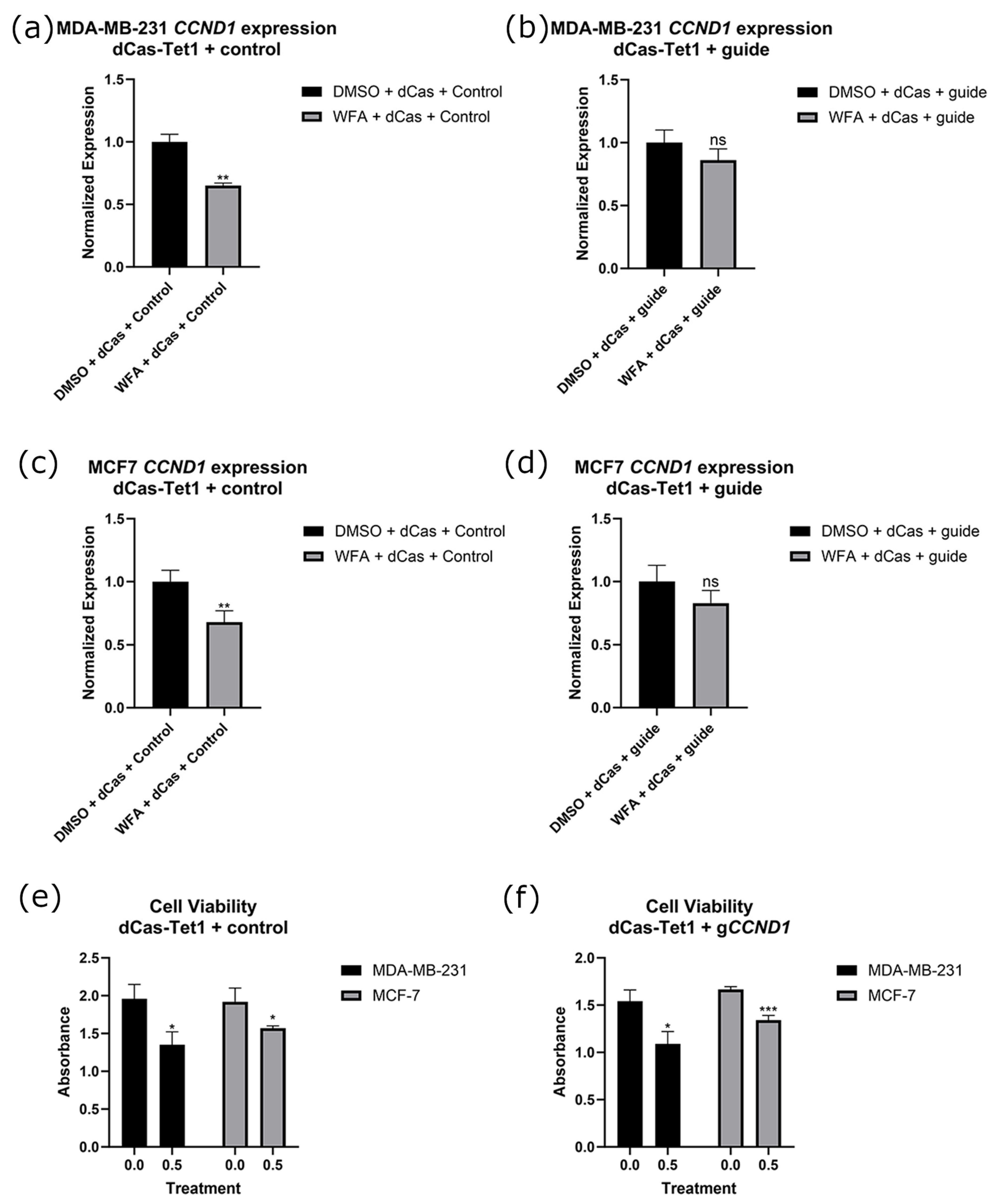
2.7. Targeted Demethylation of the CCND1 Promoter in Combination with WFA Ablates CCND1 Expression Changes with No Significant Loss of WFA Anticancer Function
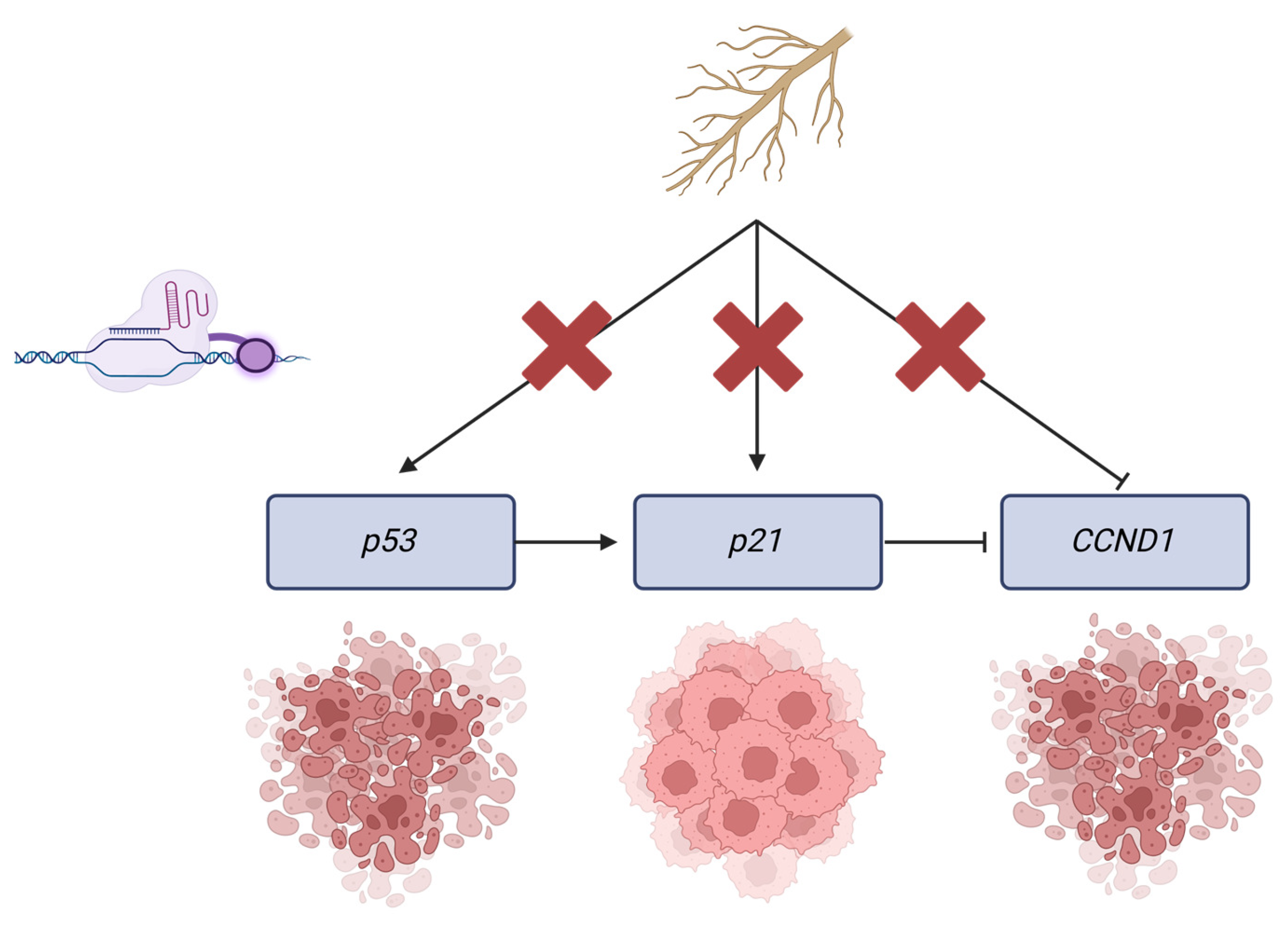
3. Discussion
3.1. p21, p53, and CCND1 as Molecular Targets for Epigenetic Therapies
3.2. Genetic Targets and Their Relationship with WFA’s Function
3.3. Key Findings
3.4. Limitations and Future Directions
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Withaferin A and Cell Treatment
4.3. Isolation and Growth of CRISPR Constructs
4.4. Guide Selection and Cloning
4.5. Nucleic Acid Extraction
4.6. RT-qPCR
4.7. Bisulfite Conversion and Sequencing
4.8. MTT Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Susan, G.; Komen®. Molecular Subtypes of Breast Cancer. 2023. Available online: https://www.komen.org/breast-cancer/diagnosis/molecular-subtypes/ (accessed on 21 November 2024).
- Cancer Stat Facts: Female Breast Cancer Subtypes. SEER. 2023. Available online: https://seer.cancer.gov/statfacts/html/breast-subtypes.html (accessed on 21 November 2024).
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—expanded options, evolving needs. Nature reviews. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA A Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Van Le, Q.; Yang, H.; Zhang, D.; Gu, H.; Yang, Y.; Sonne, C.; Lam, S.S.; Zhong, J.; Jianguang, Z.; et al. A review of dietary phytochemicals and their relation to oxidative stress and human diseases. Chemosphere 2021, 271, 129499. [Google Scholar] [CrossRef]
- Shankar, S.; Kumar, D.; Srivastava, R.K. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacol. Ther. 2013, 138, 1–17. [Google Scholar] [CrossRef]
- Sultana, T.; Okla, M.K.; Ahmed, M.; Akhtar, N.; Al-Hashimi, A.; Abdelgawad, H.; Haq, I.U. Withaferin A: From Ancient Remedy to Potential Drug Candidate. Molecules 2021, 26, 7696. [Google Scholar] [CrossRef]
- Singh, N.; Bhalla, M.; de Jager, P.; Gilca, M. An overview on ashwagandha: A Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. AJTCAM 2011, 8 (Suppl. S5), 208–213. [Google Scholar] [CrossRef]
- Dutta, R.; Khalil, R.; Green, R.; Mohapatra, S.S.; Mohapatra, S. Withania somnifera (Ashwagandha) and Withaferin A: Potential in Integrative Oncology. Int. J. Mol. Sci. 2019, 20, 5310. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef]
- Szarc vel Szic, K.; Op de Beeck, K.; Ratman, D.; Wouters, A.; Beck, I.M.; Declerck, K.; Heyninck, K.; Fransen, E.; Bracke, M.; De Bosscher, K.; et al. Pharmacological levels of Withaferin A (Withania somnifera) trigger clinically relevant anticancer effects specific to triple negative breast cancer cells. PLoS ONE 2014, 9, e87850. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.V.; Suman, S.; Das, T.P.; Luevano, J.E.; Damodaran, C. Withaferin A, a steroidal lactone from Withania somnifera, induces mitotic catastrophe and growth arrest in prostate cancer cells. J. Nat. Prod. 2013, 76, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Szarc Vel Szic, K.; Declerck, K.; Crans, R.A.J.; Diddens, J.; Scherf, D.B.; Gerhäuser, C.; Vanden Berghe, W. Epigenetic silencing of triple negative breast cancer hallmarks by Withaferin A. Oncotarget 2017, 8, 40434–40453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, Q.; Ren, L.; Liu, J.; Li, W.; Zheng, X.; Wang, J.; Du, G. Withaferin A triggers G2/M arrest and intrinsic apoptosis in glioblastoma cells via ATF4-ATF3-CHOP axis. Cell Prolif. 2020, 53, e12706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vyas, A.R.; Singh, S.V. Molecular targets and mechanisms of cancer prevention and treatment by withaferin a, a naturally occurring steroidal lactone. AAPS J. 2014, 16, 1–10. [Google Scholar] [CrossRef]
- Shuaib, M.; Prajapati, K.S.; Gupta, S.; Kumar, S. Natural Steroidal Lactone Induces G1/S Phase Cell Cycle Arrest and Intrinsic Apoptotic Pathway by Up-Regulating Tumor Suppressive miRNA in Triple-Negative Breast Cancer Cells. Metabolites 2022, 13, 29. [Google Scholar] [CrossRef]
- Moselhy, J.; Suman, S.; Alghamdi, M.; Chandarasekharan, B.; Das, T.P.; Houda, A.; Ankem, M.; Damodaran, C. Withaferin A Inhibits Prostate Carcinogenesis in a PTEN-deficient Mouse Model of Prostate Cancer. Neoplasia 2017, 19, 451–459. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Straughn, A.R.; Kakar, S.S. Withaferin A ameliorates ovarian cancer-induced cachexia and proinflammatory signaling. J. Ovarian Res. 2019, 12, 115. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, S.H.; Hahm, E.R.; Arlotti, J.A.; Samanta, S.K.; Moura, M.B.; Thorne, S.H.; Shuai, Y.; Anderson, C.J.; White, A.G.; Lokshin, A.; et al. Withaferin A inhibits in vivo growth of breast cancer cells accelerated by Notch2 knockdown. Breast Cancer Res. Treat. 2016, 157, 41–54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat Rev. 2020, 86, 102019. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Chandra, U.; Shukla, P.; Verma, S.P.; Suvirya, S. Reticular Skin Rash as an Adverse Effect of 5-Azacitidine. Cureus 2022, 14, e24228. [Google Scholar] [CrossRef]
- De Cicco, P.; Catani, M.V.; Gasperi, V.; Sibilano, M.; Quaglietta, M.; Savini, I. Nutrition and Breast Cancer: A Literature Review on Prevention, Treatment and Recurrence. Nutrients 2019, 11, 1514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pires, N.; Gota, V.; Gulia, A.; Hingorani, L.; Agarwal, M.; Puri, A. Safety and pharmacokinetics of Withaferin-A in advanced stage high grade osteosarcoma: A phase I trial. J. Ayurveda Integr. Med. 2020, 11, 68–72. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, J.; Zeng, B.; Yang, D.; Sun, J.; Yin, X.; Lu, M.; Qiu, Z.; Peng, W.; Xiang, T.; et al. PSMD2 regulates breast cancer cell proliferation and cell cycle progression by modulating p21 and p27 proteasomal degradation. Cancer Lett. 2018, 430, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Brane, A.; Arora, I.; Tollefsbol, T.O. Peripubertal Nutritional Prevention of Cancer-Associated Gene Expression and Phenotypes. Cancers 2023, 15, 674. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ocker, M.; Bitar, S.A.; Monteiro, A.C.; Gali-Muhtasib, H.; Schneider-Stock, R. Epigenetic Regulation of p21cip1/waf1 in Human Cancer. Cancers 2019, 11, 1343. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Zhang, S.; Carlsen, L.; Hernandez Borrero, L.; Seyhan, A.A.; Tian, X.; El-Deiry, W.S. Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer. Biomolecules 2022, 12, 548. [Google Scholar] [CrossRef]
- Poosari, A.; Nutravong, T.; Namwat, W.; Wasenang, W.; Sa-Ngiamwibool, P.; Ungareewittaya, P. The relationship between P16INK4A and TP53 promoter methylation and the risk and prognosis in patients with oesophageal cancer in Thailand. Sci. Rep. 2022, 12, 10337. [Google Scholar] [CrossRef]
- Saeed, W.H.; Eissa, A.A.; Al-Doski, A.A. Impact Of TP53 Gene Promoter Methylation On Chronic Lymphocytic Leukemia Pathogenesis And Progression. J. Blood Med. 2019, 10, 399–404. [Google Scholar] [CrossRef]
- Hui, L.; Zheng, Y.; Yan, Y.; Bargonetti, J.; Foster, D.A. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 2006, 25, 7305–7310. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mukerji, R.; Samadi, A.K.; Cohen, M.S. Down-regulation of estrogen receptor-alpha and rearranged during transfection tyrosine kinase is associated with withaferin a-induced apoptosis in MCF-7 breast cancer cells. BMC Complement. Altern. Med. 2011, 11, 84. [Google Scholar] [CrossRef]
- Kumar, S.; Mathew, S.O.; Aharwal, R.P.; Tulli, H.S.; Mohan, C.D.; Sethi, G.; Ahn, K.S.; Webber, K.; Sandhu, S.S.; Bishayee, A. Withaferin A: A Pleiotropic Anticancer Agent from the Indian Medicinal Plant Withania somnifera (L.) Dunal. Pharmaceuticals 2023, 16, 160. [Google Scholar] [CrossRef]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin A induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Cheryan, V.T.; Wu, W.; Cui, C.Q.; Polin, L.A.; Pass, H.I.; Dou, Q.P.; Rishi, A.K.; Wali, A. Withaferin A inhibits the proteasome activity in mesothelioma in vitro and in vivo. PLoS ONE 2012, 7, e41214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, J.; Lin, D.I. Oncogenic c-terminal cyclin D1 (CCND1) mutations are enriched in endometrioid endometrial adenocarcinomas. PLoS ONE 2018, 13, e0199688. [Google Scholar] [CrossRef]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- SEER, National Institute of Health. Annual Report to the Nation 2022 National Cancer Statistics. 2022. Available online: https://seer.cancer.gov/report_to_nation/statistics.html?cid=soc_tw_en_sharedlink_arn2023_statistics1 (accessed on 12 October 2023).
- Vanduchova, A.; Anzenbacher, P.; Anzenbacherova, E. Isothiocyanate from Broccoli, Sulforaphane, and Its Properties. J. Med. Food 2019, 22, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Shimizu, M. Possible Mechanisms of Green Tea and Its Constituents against Cancer. Molecules 2018, 23, 2284. [Google Scholar] [CrossRef]
- Atteeq, M. Evaluating anticancer properties of Withaferin A-a potent phytochemical. Front. Pharmacol. 2022, 13, 975320. [Google Scholar] [CrossRef]
- Kumamoto, T.; Yamazaki, F.; Nakano, Y.; Tamura, C.; Tashiro, S.; Hattori, H.; Nakagawara, A.; Tsunematsu, Y. Medical guidelines for Li-Fraumeni syndrome 2019, version 1.1. Int. J. Clin. Oncol. 2021, 26, 2161–2178, Erratum in Int. J. Clin. Oncol. 2022, 27, 262–263. [Google Scholar] [CrossRef]
- Royston, K.J.; Udayakumar, N.; Lewis, K.; Tollefsbol, T.O. A Novel Combination of Withaferin A and Sulforaphane Inhibits Epigenetic Machinery, Cellular Viability and Induces Apoptosis of Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1092. [Google Scholar] [CrossRef]
- Verma, N.; Pan, H.; Dore, L.C.; Shukla, A.; Li, Q.V.; Pelham-Webb, B.; Teijeiro, V.; Gonzalez, F.; Krivtsov, A.; Chang, C.J.; et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet. 2018, 50, 83–95. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic. Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Pulido-Quetglas, C.; Aparicio-Prat, E.; Arnan, C.; Polidori, T.; Hermoso, T.; Palumbo, E.; Ponomarenko, J.; Guigo, R.; Johnson, R. Scalable Design of Paired CRISPR Guide RNAs for Genomic Deletion. PLoS Comput. Biol. 2017, 13, e1005341. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. UCSC Genome Browser. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, M.; Wu, H.; Li, Y.; Tollefsbol, T.O. Maternal Epigenetic Regulation Contributes to Prevention of Estrogen Receptor-negative Mammary Cancer with Broccoli Sprout Consumption. Cancer Prev. Res. 2020, 13, 449–462. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hickey, G.L.; Grant, S.W.; Dunning, J.; Siepe, M. Statistical primer: Sample size and power calculations-why, when and how? Eur. J. Cardiothorac. Surg. 2018, 54, 4–9. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brane, A.; Sutko, M.; Tollefsbol, T.O. p21 Promoter Methylation Is Vital for the Anticancer Activity of Withaferin A. Int. J. Mol. Sci. 2025, 26, 1210. https://doi.org/10.3390/ijms26031210
Brane A, Sutko M, Tollefsbol TO. p21 Promoter Methylation Is Vital for the Anticancer Activity of Withaferin A. International Journal of Molecular Sciences. 2025; 26(3):1210. https://doi.org/10.3390/ijms26031210
Chicago/Turabian StyleBrane, Andrew, Madeline Sutko, and Trygve O. Tollefsbol. 2025. "p21 Promoter Methylation Is Vital for the Anticancer Activity of Withaferin A" International Journal of Molecular Sciences 26, no. 3: 1210. https://doi.org/10.3390/ijms26031210
APA StyleBrane, A., Sutko, M., & Tollefsbol, T. O. (2025). p21 Promoter Methylation Is Vital for the Anticancer Activity of Withaferin A. International Journal of Molecular Sciences, 26(3), 1210. https://doi.org/10.3390/ijms26031210