
Comparison of Lysis and Amplification Methodologies for Optimal 16S rRNA Gene Profiling for Human and Mouse Microbiome Studies

 and
and
Abstract
1. Introduction
2. Results
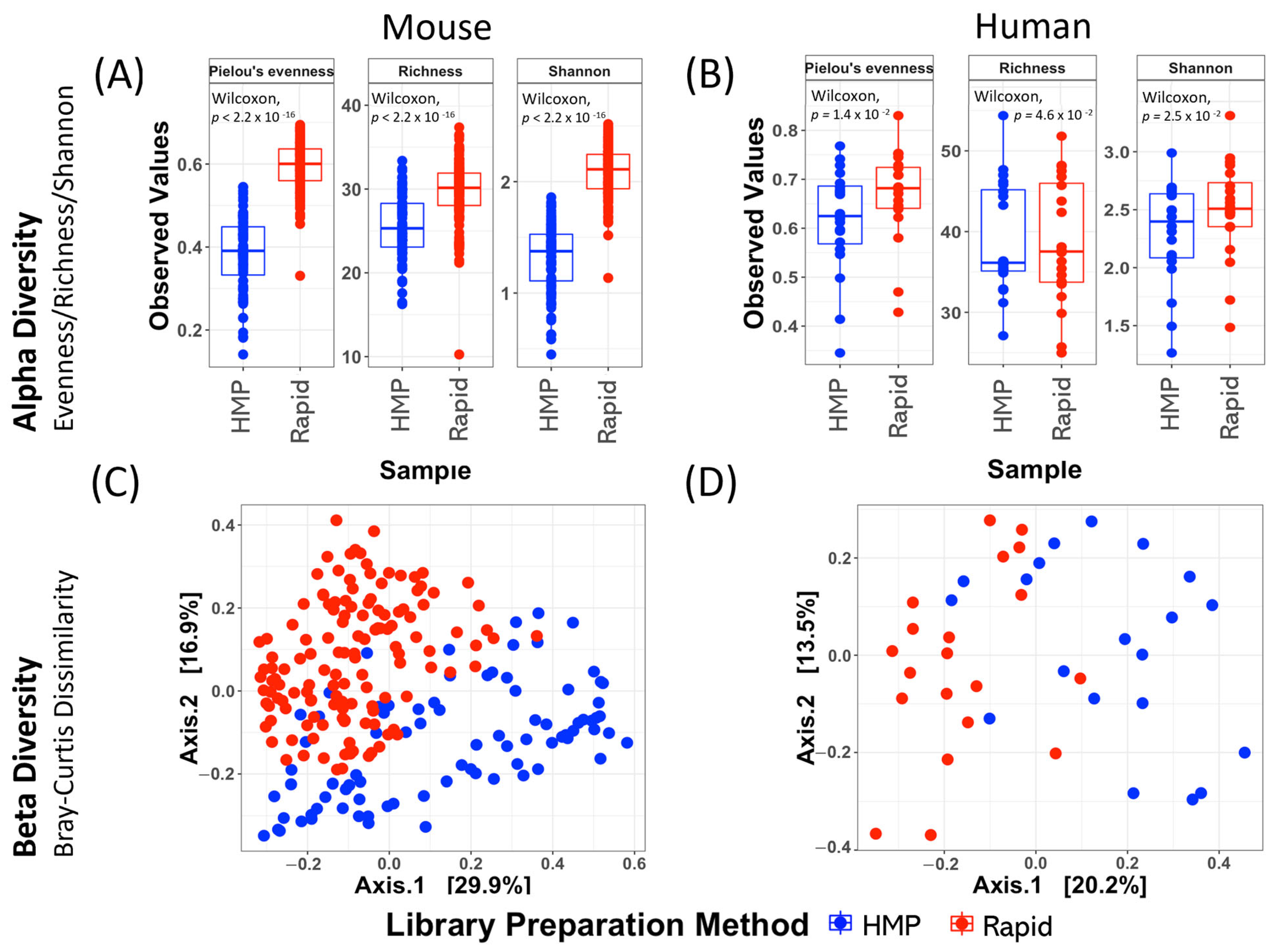
2.1. The Intus Rapid Technique Detects Greater Diversity in Fecal Samples than Established HMP Methods
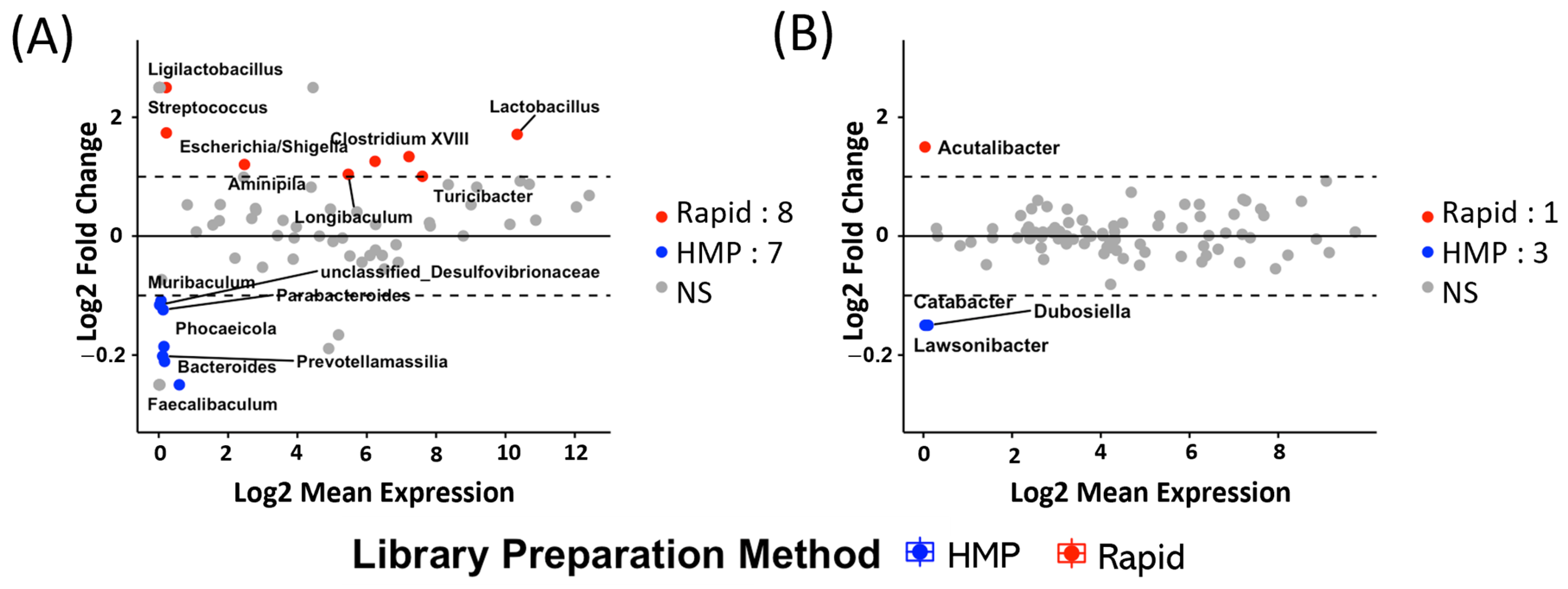
2.2. Different 16S rDNA Preparation Methods Are Biased Towards Different Organisms in Mice and Humans
2.3. A Combined Significance Ranking Score Identifies the Most Consistent Genera That Discriminate Between Rapid and HMP Protocols
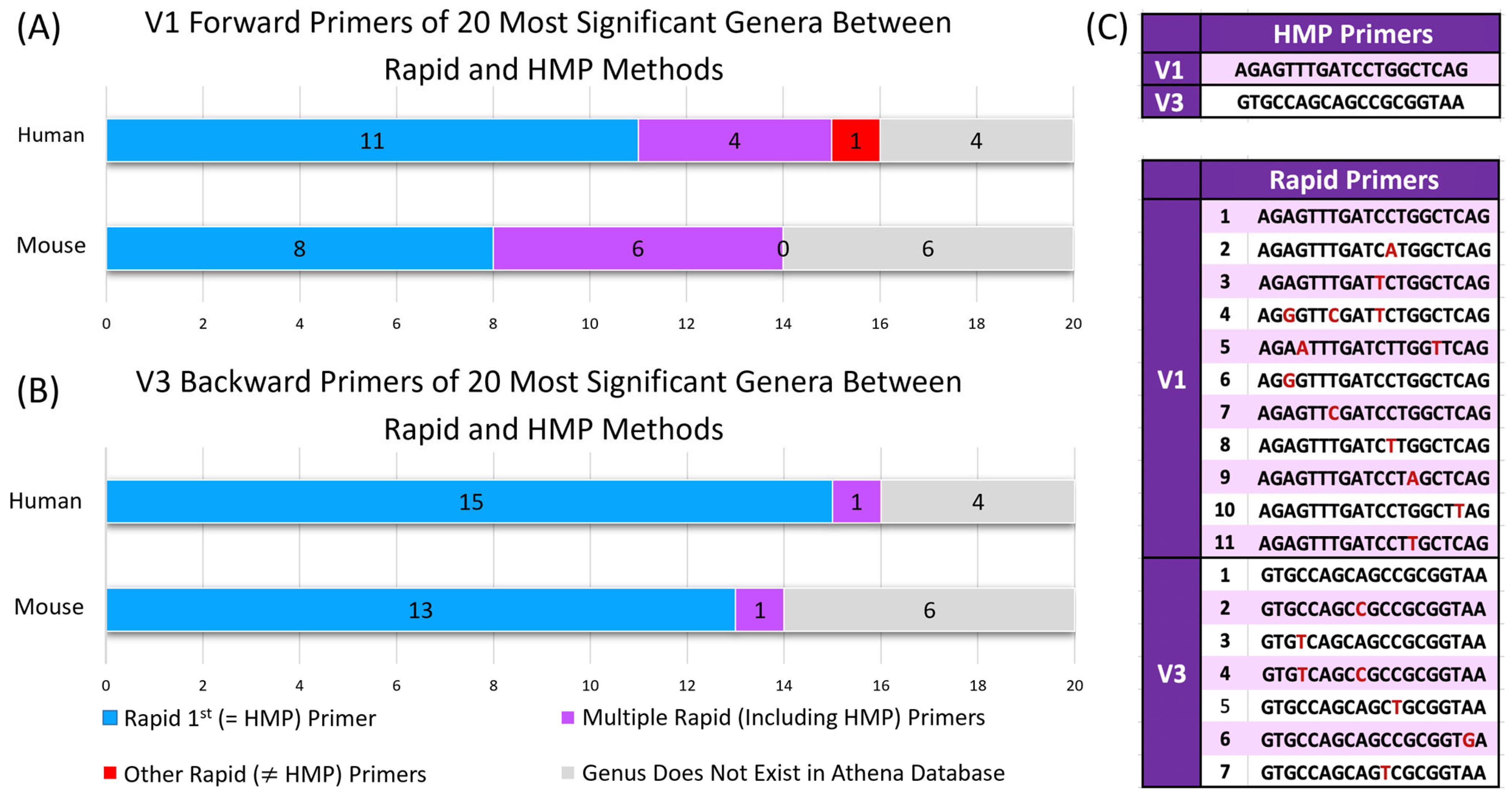
2.4. Rapid V1–V3 Primers Contain Fewer Mismatches to Binding Sites in 16S rRNA Genes of Reference Taxa
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.1.1. Mouse Fecal Microbiome Samples
4.1.2. Human Oral Microbiome Samples
4.2. DNA Extraction, 16S rRNA Gene Amplification, and Sequencing
4.2.1. Qiagen PowerSoil Kit Protocol (HMP)
4.2.2. Intus Biosciences V1–V3-Illumina Kit Protocol (Rapid) [24]
4.3. 16S rRNA Gene Data Processing
4.4. Statistical Analysis and Graphical Display
4.5. Quality Assurance
4.6. Statistical Comparison Between the Rapid and HMP Methods
4.7. Understanding Primer Bias and Optimization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Figueroa-Bossi, N.; Balbontín, R.; Bossi, L. Preparing bacterial genomic DNA. Cold Spring Harb. Protoc. 2022, 2022, pdb-prot107853. [Google Scholar] [CrossRef] [PubMed]
- Koshy, L.; Anju, A.L.; Harikrishnan, S.; Kutty, V.R.; Jissa, V.T.; Kurikesu, I.; Jayachandran, P.; Jayakumaran Nair, A.; Gangaprasad, A.; Nair, G.M.; et al. Evaluating genomic DNA extraction methods from human whole blood using endpoint and real-time PCR assays. Mol. Biol. Rep. 2017, 44, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Corcoll, N.; Österlund, T.; Sinclair, L.; Eiler, A.; Kristiansson, E.; Backhaus, T.; Eriksson, K.M. Comparison of four DNA extraction methods for comprehensive assessment of 16S rRNA bacterial diversity in marine biofilms using high-throughput sequencing. FEMS Microbiol. Lett. 2017, 364, fnx139. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef]
- Birnboim, H.C.; Doly, J. A Rapidalkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef]
- Gibbons, S.M.; Gilbert, J.A. Microbial diversity—Exploration of natural ecosystems and microbiomes. Curr. Opin. Genet. Dev. 2015, 35, 66–72. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA PCR primers for amplification of complex microbial communities. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Fujiyoshi, S.; Muto-Fujita, A.; Maruyama, F. Evaluation of PCR conditions for characterizing bacterial communities with full-length 16S rRNA genes using a portable nanopore sequencer. Sci. Rep. 2020, 10, 12580. [Google Scholar] [CrossRef]
- Graf, J.; Ledala, N.; Caimano, M.J.; Jackson, E.; Gratalo, D.; Fasulo, D.; Driscoll, M.D.; Coleman, S.; Matson, A.P. High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel rRNA Amplicon. mBio 2021, 12, e03656-20. [Google Scholar] [CrossRef]
- Hong, B.-Y.; Driscoll, M.; Gratalo, D.; Jarvie, T.; Weinstock, G.M. Improved DNA Extraction and Amplification Strategy for 16S rRNA Gene Amplicon-Based Microbiome Studies. Int. J. Mol. Sci. 2024, 25, 2966. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Carrigg, C.; Rice, O.; Kavanagh, S.; Collins, G.; O’flaherty, V. DNA extraction method affects microbial community profiles from soils and sediment. Appl. Microbiol. Biotechnol. 2007, 77, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Krsek, M.; Wellington, E.M. Comparison of different methods for the isolation and purification of total community DNA from soil. J. Microbiol. Methods 1999, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Shwani, A.; Zuo, B.; Alrubaye, A.; Zhao, J.; Rhoads, D.D. A simple, inexpensive alkaline method for bacterial DNA extraction from environmental samples for PCR surveillance and microbiome analyses. Appl. Sci. 2024, 14, 141. [Google Scholar] [CrossRef]
- Mysara, M.; Vandamme, P.; Props, R.; Kerckhof, F.M.; Leys, N.; Boon, N.; Raes, J.; Monsieurs, P. Reconciliation between operational taxonomic units and species boundaries. FEMS Microbiol. Ecol. 2017, 93, fix029. [Google Scholar] [CrossRef]
- Graspeuntner, S.; Loeper, N.; Künzel, S.; Baines, J.F.; Rupp, J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 2018, 8, 9678. [Google Scholar] [CrossRef]
- Matsuo, Y.; Komiya, S.; Yasumizu, Y.; Yasuoka, Y.; Mizushima, K.; Takagi, T.; Kryukov, K.; Fukuda, A.; Morimoto, Y.; Naito, Y.; et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 2021, 21, 35. [Google Scholar] [CrossRef]
- Kim, S.-W.; Suda, W.; Oshima, K.; Fukuda, S.; Ohno, H.; Morita, H.; Hattori, M. Masahira Hattori, Robustness of Gut Microbiota of Healthy Adults in Response to Probiotic Intervention Revealed by High-Throughput Pyrosequencing. DNA Res. 2013, 20, 241–253. [Google Scholar] [CrossRef]
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinION nanopore sequencer. FEBS Open Bio. 2019, 9, 548–557. [Google Scholar] [CrossRef]
- Nadeau, J.H.; Singer, J.B.; Matin, A.; Lander, E.S. Analysing complex genetic traits with chromosome substitution strains. Nat. Genet. 2000, 24, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.B.; Hill, A.E.; Burrage, L.C.; Olszens, K.R.; Song, J.; Justice, M.; O’Brien, W.E.; Conti, D.V.; Witte, J.S.; Lander, E.S.; et al. Genetic dissection of complex traits with chromosome substitution strains of mice. Science 2004, 304, 445–448. [Google Scholar] [CrossRef]
- MO BIO Laboratories, Inc. MO BIO’s PowerSoil DNA Isolation Kit Handbook; MO BIO Laboratories, Inc.: Carlsbad, CA, USA, 2013. [Google Scholar]
- Intus Biosciences, LLC. Shoreline Complete™ V1-V3 Protocol 1 Lyse, Purify, & Amplify Sets A, B (96 Wells) Fecal and Tissue Samples; Intus Biosciences, LLC: Farmington, CT, USA, 2019. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for Rapidassignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Venables, W.N.; Smith, D.M.; Team, R.C. An Introduction to R—Notes on R: A Programming Environment for Data Analysis and Graphics; Network Theory Ltd.: Surrey, UK, 2018. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 20 January 2025).
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. vegan: Community Ecology Package. 2022. Available online: https://CRAN.R-project.org/package=vegan (accessed on 20 January 2025).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 20 January 2025).
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 20 January 2025).
- Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef]
- Wickham, H.; Hester, J.; Chang, W.; Bryan, J. devtools: Tools to Make Developing R Packages Easier. 2021. Available online: https://CRAN.R-project.org/package=devtools (accessed on 20 January 2025).
- Ssekagiri, A. microbiomeSeq: Microbial Community Analysis in an Environmental Context. 2022. Available online: http://www.github.com/umerijaz/microbiomeSeq (accessed on 20 January 2025).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Firke, S. janitor: Simple Tools for Examining and Cleaning Dirty Data. 2021. Available online: https://CRAN.R-project.org/package=janitor (accessed on 20 January 2025).
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Moore, J.C. Diversity, Taxonomic versus Functional. In Encyclopedia of Biodiversity, 2nd ed.; Levin, S.A., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 648–656. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Chao, A.; Lee, S.-M. Estimating the number of classes in a rare data set. Biometrics 1992, 48, 827–837. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual comparisons by ranking methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Student. The probable error of a mean. Biometrika 1908, 6, 1–25. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Ranking | Matched Primers with Each Method Based on Athena Database | Number of Detected OTUs | Prevalence Percentage | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| V1 Forward Primer | V3 Reverse Primer | |||||||||
| Final Rank | Significant Genera | Rapid | HMP | Rapid | HMP | Rapid | HMP | Rapid | HMP | |
| Mouse | 1 | Lactobacillus | 1, 9 | Yes | 1 | Yes | 2 | 1 | 93 | 21 |
| 2 | Ihubacter | Doesn’t exist in Athena database | 3 | 1 | 99 | 7 | ||||
| 3 | unclassified_Eggerthellaceae | 1 | Yes | 1 | Yes | 7 | 0 | 61 | 0 | |
| 4 | unclassified_Lachnospiraceae | 1, 7 | Yes | 1 | Yes | 216 | 172 | 99 | 99 | |
| 5 | Duncaniella | Doesn’t exist in Athena database | 23 | 21 | 91 | 89 | ||||
| 6 | Adlercreutzia | 1 | Yes | 1 | Yes | 7 | 1 | 73 | 2 | |
| 7 | Longibaculum | Doesn’t exist in Athena database | 1 | 1 | 49 | 1 | ||||
| 8 | unclassified_Clostridiales | 1, 6, 7, 9 | Yes | 1 | Yes | 27 | 18 | 100 | 86 | |
| 9 | unclassified_Bacteroidales | 1, 9 | Yes | 1 | Yes | 13 | 9 | 100 | 100 | |
| 10 | Clostridium XVIII | Doesn’t exist in Athena database | 1 | 1 | 92 | 35 | ||||
| 11 | unclassified_Firmicutes | 1, 6, 7, 8, 9 | Yes | 1, 6, 7 | Yes | 16 | 6 | 65 | 1 | |
| 12 | unclassified_Ruminococcaceae | 1, 6 | Yes | 1 | Yes | 35 | 28 | 93 | 23 | |
| 13 | Intestinimonas | 1 | Yes | 1 | Yes | 5 | 5 | 97 | 92 | |
| 14 | unclassified_Erysipelotrichaceae | 1 | Yes | 1 | Yes | 7 | 2 | 70 | 89 | |
| 15 | Schaedlerella | Doesn’t exist in Athena database | 1 | 1 | 57 | 1 | ||||
| 16 | Lachnospiracea_incertae_sedis | Doesn’t exist in Athena database | 1 | 1 | 49 | 5 | ||||
| 17 | Acutalibacter | 1 | Yes | 1 | Yes | 3 | 2 | 46 | 1 | |
| 18 | unclassified_Muribaculaceae | 1 | Yes | 1 | Yes | 16 | 11 | 34 | 5 | |
| 19 | Ruminococcus | 1 | Yes | 1 | Yes | 2 | 0 | 33 | 0 | |
| 20 | Turicibacter | 1 | Yes | 1 | Yes | 1 | 1 | 35 | 35 | |
| Human | 1 | Bacteroides | 1, 9 | Yes | 1, 6 | Yes | 7 | 12 | 70 | 95 |
| 2 | Faecalibacterium | 1 | Yes | 1 | Yes | 5 | 5 | 95 | 85 | |
| 3 | Phocaeicola | 1 | Yes | 1 | Yes | 6 | 8 | 55 | 75 | |
| 4 | Blautia | 1 | Yes | 1 | Yes | 8 | 4 | 100 | 30 | |
| 5 | Anaerobutyricum | Doesn’t exist in Athena database | 3 | 2 | 90 | 10 | ||||
| 6 | Ruminococcus | 1 | Yes | 1 | Yes | 2 | 2 | 60 | 20 | |
| 7 | unclassified_Ruminococcaceae | 1, 6 | Yes | 1 | Yes | 26 | 21 | 95 | 95 | |
| 8 | Dorea | 1 | Yes | 1 | Yes | 1 | 1 | 85 | 55 | |
| 9 | Coprococcus | 1 | Yes | 1 | Yes | 3 | 1 | 55 | 5 | |
| 10 | Collinsella | 7 | No Match | 1 | Yes | 1 | 2 | 65 | 20 | |
| 11 | Romboutsia | Doesn’t exist in Athena database | 1 | 1 | 65 | 50 | ||||
| 12 | Parabacteroides | 1 | Yes | 1 | Yes | 3 | 5 | 25 | 60 | |
| 13 | Anaerostipes | 1 | Yes | 1 | Yes | 2 | 2 | 70 | 25 | |
| 14 | unclassified_Lachnospiraceae | 1, 7 | Yes | 1 | Yes | 22 | 17 | 80 | 50 | |
| 15 | unclassified_Clostridiales | 1, 6, 7, 9 | Yes | 1 | Yes | 12 | 11 | 55 | 55 | |
| 16 | Prevotella | 1 | Yes | 1 | Yes | 4 | 6 | 40 | 55 | |
| 17 | Mediterraneibacter | Doesn’t exist in Athena database | 1 | 0 | 35 | 0 | ||||
| 18 | Alistipes | 1 | Yes | 1 | Yes | 1 | 4 | 5 | 45 | |
| 19 | Faecalibacillus | Doesn’t exist in Athena database | 2 | 2 | 75 | 65 | ||||
| 20 | Roseburia | 1 | Yes | 1 | Yes | 3 | 2 | 60 | 40 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rastegari, F.; Driscoll, M.; Riordan, J.D.; Nadeau, J.H.; Johnson, J.S.; Weinstock, G.M. Comparison of Lysis and Amplification Methodologies for Optimal 16S rRNA Gene Profiling for Human and Mouse Microbiome Studies. Int. J. Mol. Sci. 2025, 26, 1180. https://doi.org/10.3390/ijms26031180
Rastegari F, Driscoll M, Riordan JD, Nadeau JH, Johnson JS, Weinstock GM. Comparison of Lysis and Amplification Methodologies for Optimal 16S rRNA Gene Profiling for Human and Mouse Microbiome Studies. International Journal of Molecular Sciences. 2025; 26(3):1180. https://doi.org/10.3390/ijms26031180
Chicago/Turabian StyleRastegari, Farzaneh, Mark Driscoll, Jesse D. Riordan, Joseph H. Nadeau, Jethro S. Johnson, and George M. Weinstock. 2025. "Comparison of Lysis and Amplification Methodologies for Optimal 16S rRNA Gene Profiling for Human and Mouse Microbiome Studies" International Journal of Molecular Sciences 26, no. 3: 1180. https://doi.org/10.3390/ijms26031180
APA StyleRastegari, F., Driscoll, M., Riordan, J. D., Nadeau, J. H., Johnson, J. S., & Weinstock, G. M. (2025). Comparison of Lysis and Amplification Methodologies for Optimal 16S rRNA Gene Profiling for Human and Mouse Microbiome Studies. International Journal of Molecular Sciences, 26(3), 1180. https://doi.org/10.3390/ijms26031180