

Transcriptomics Unveil Canonical and Non-Canonical Heat Shock-Induced Pathways in Human Cell Lines



Abstract
1. Introduction
2. Results
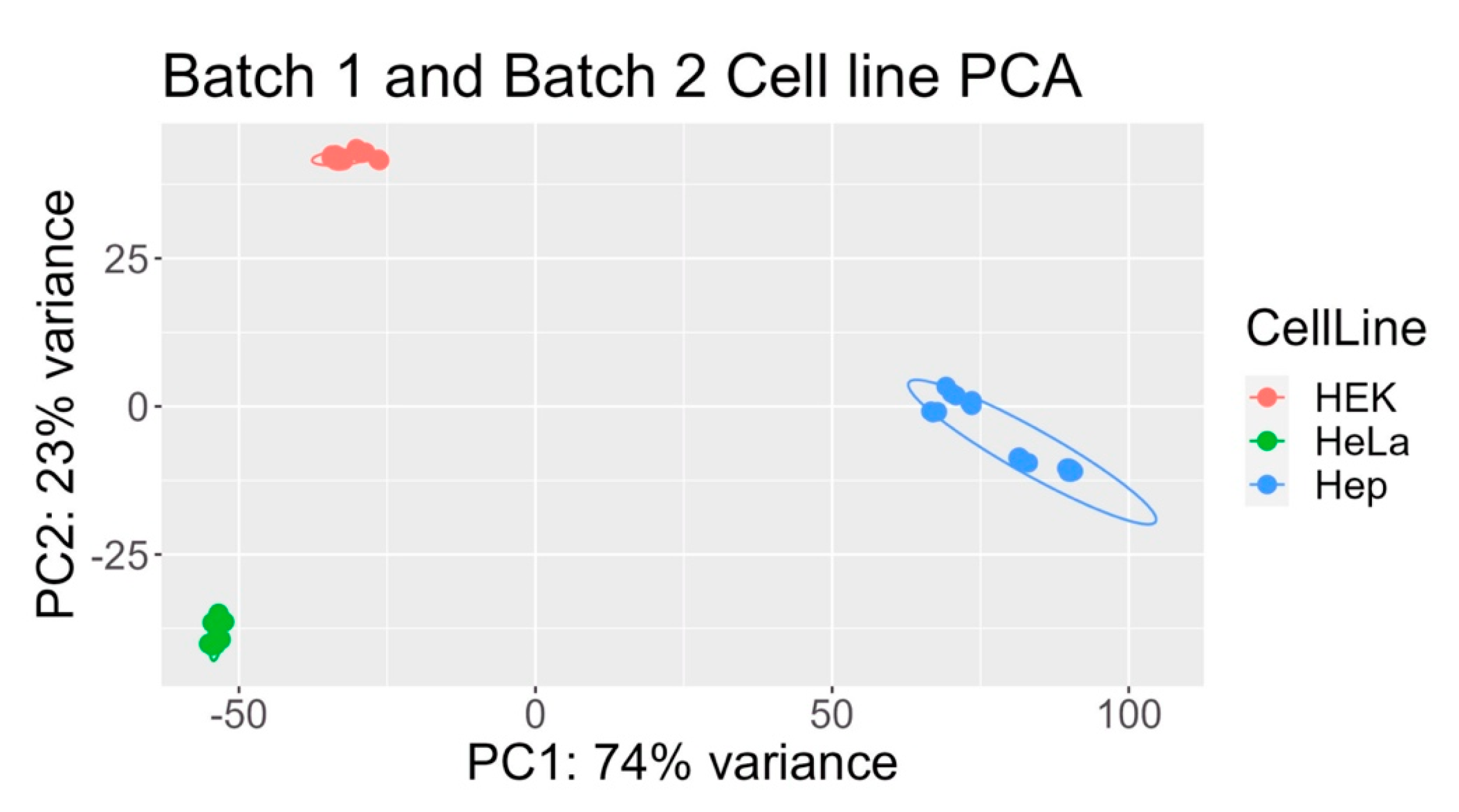
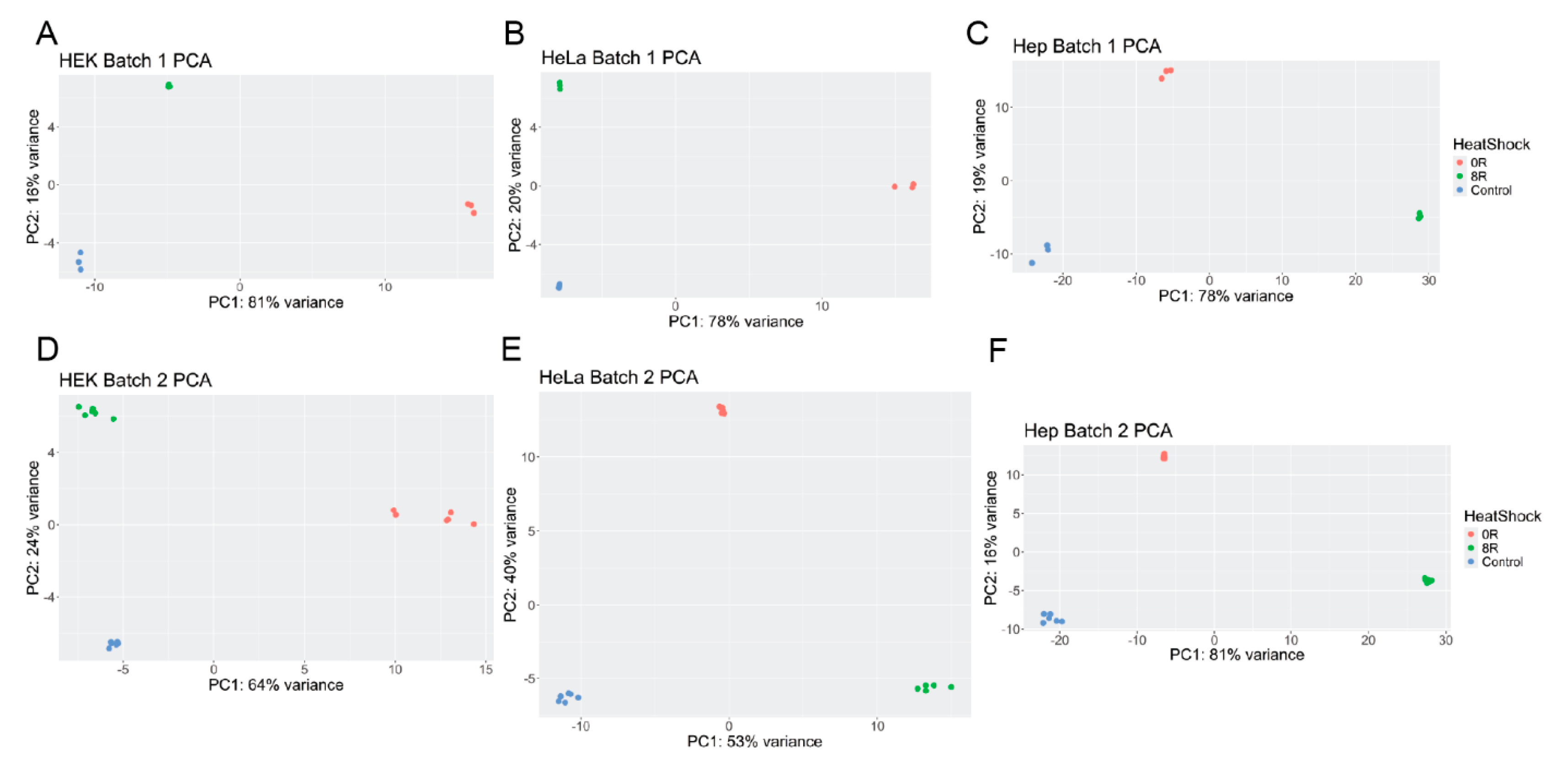
2.1. Principal Component Analysis
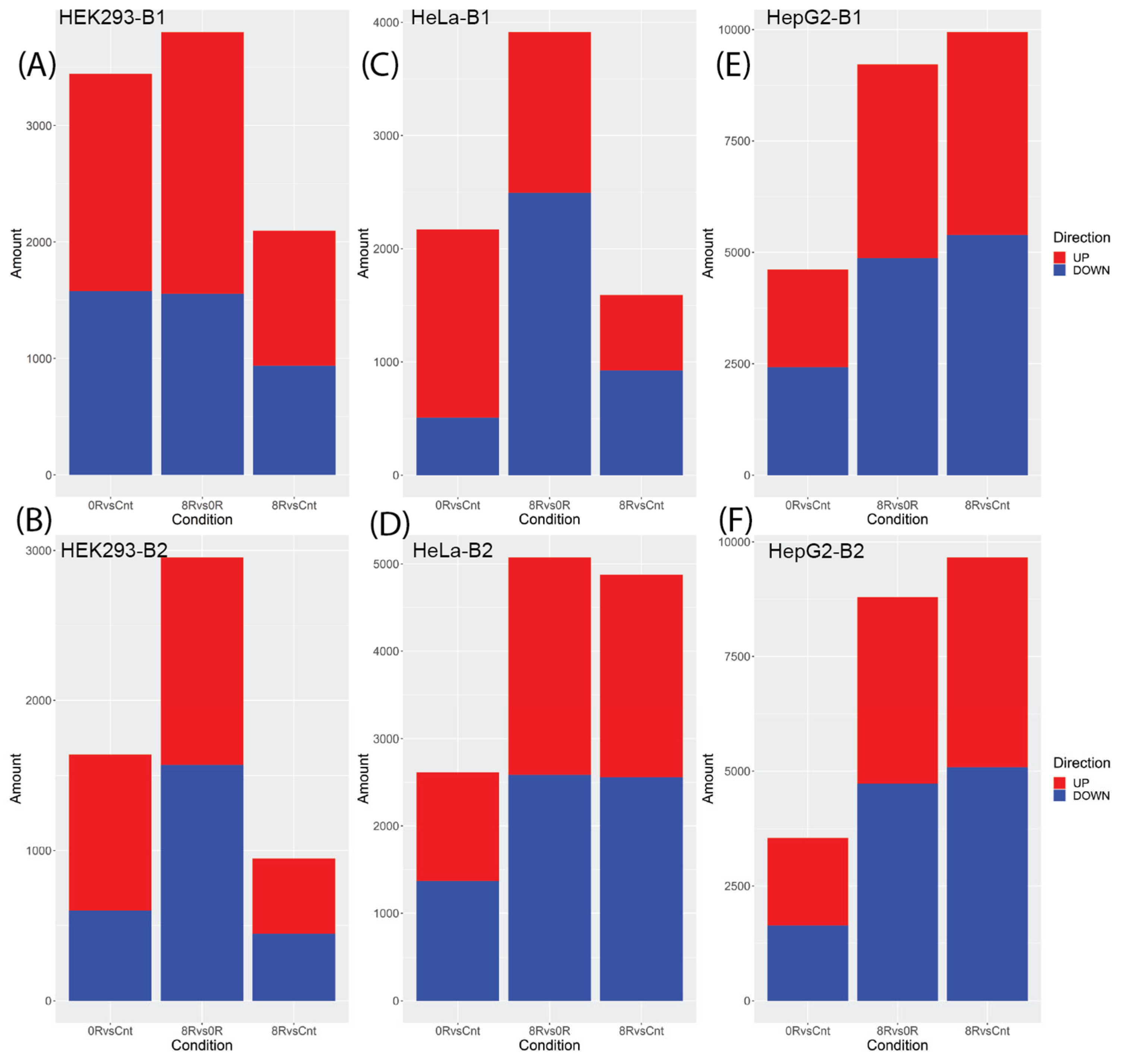
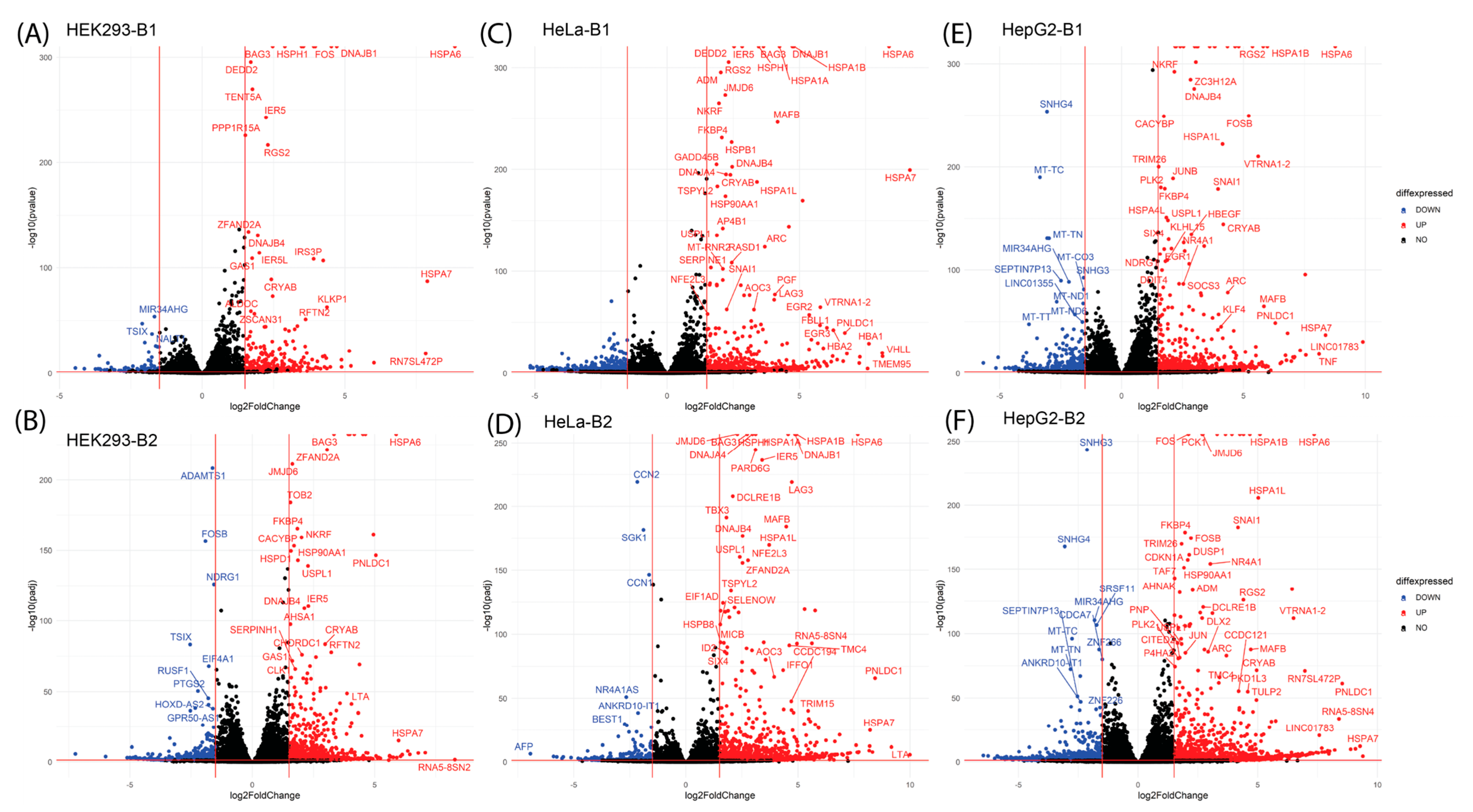
2.2. Differential Gene Expression Analysis
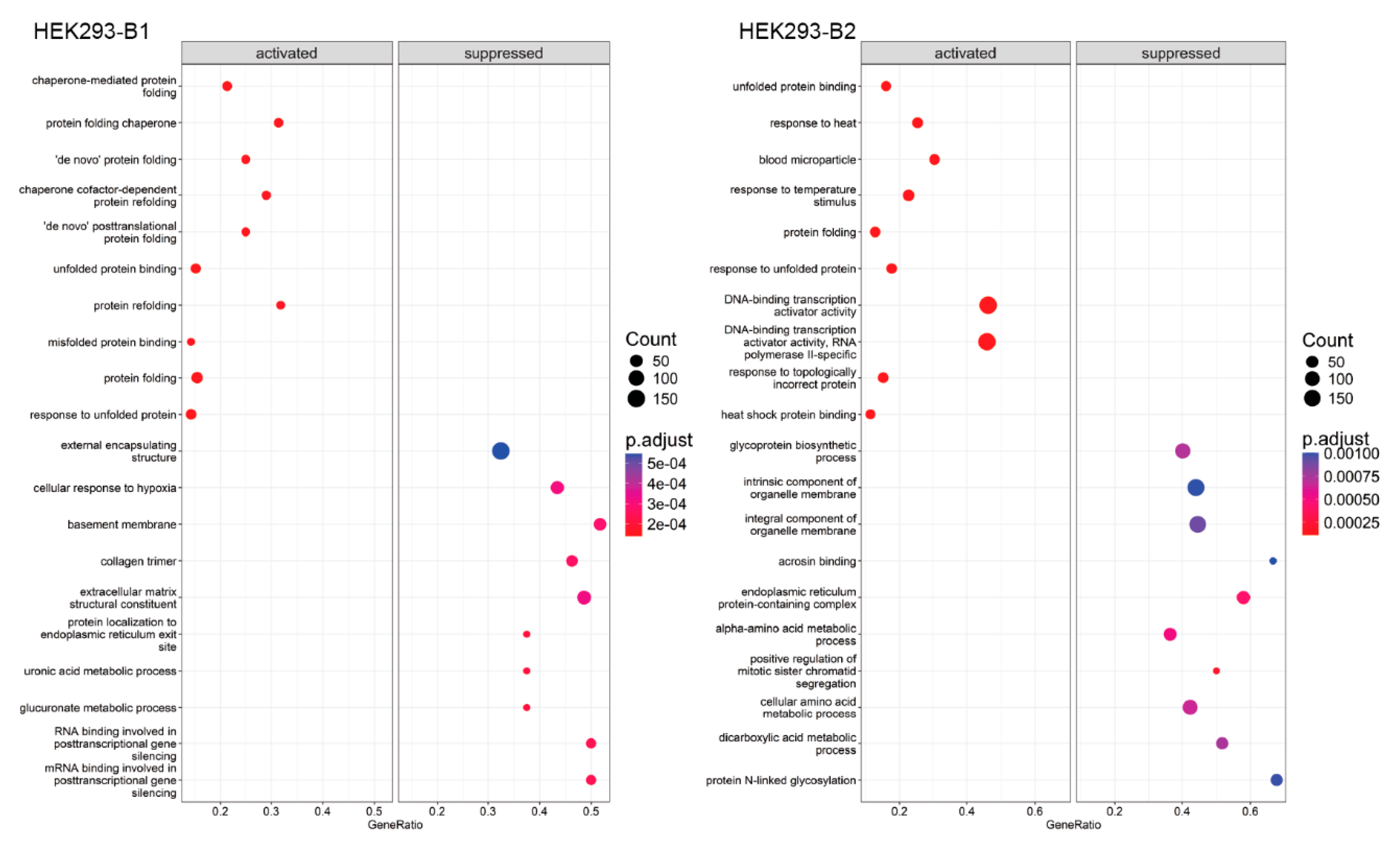
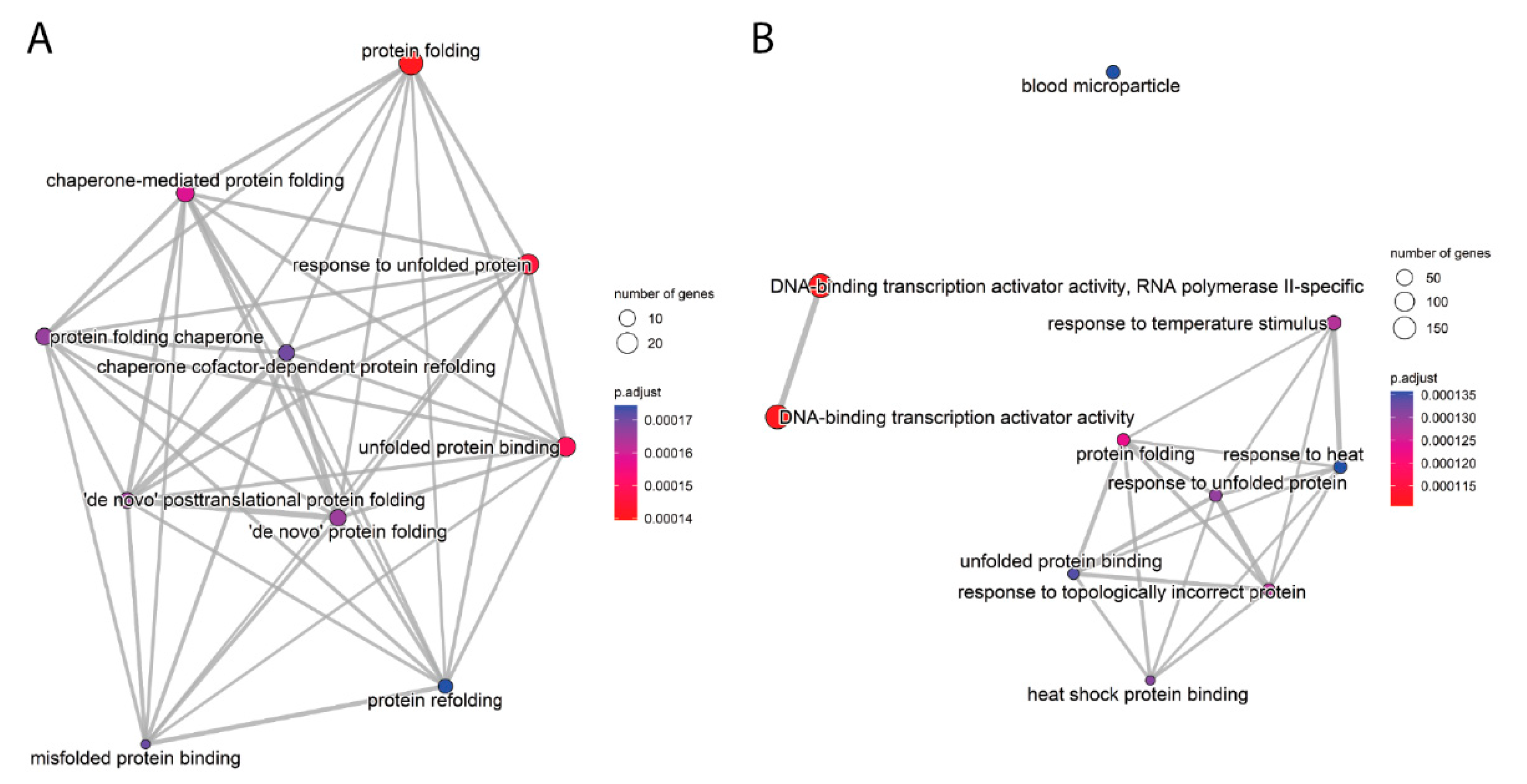
2.3. Functional Enrichment Analysis
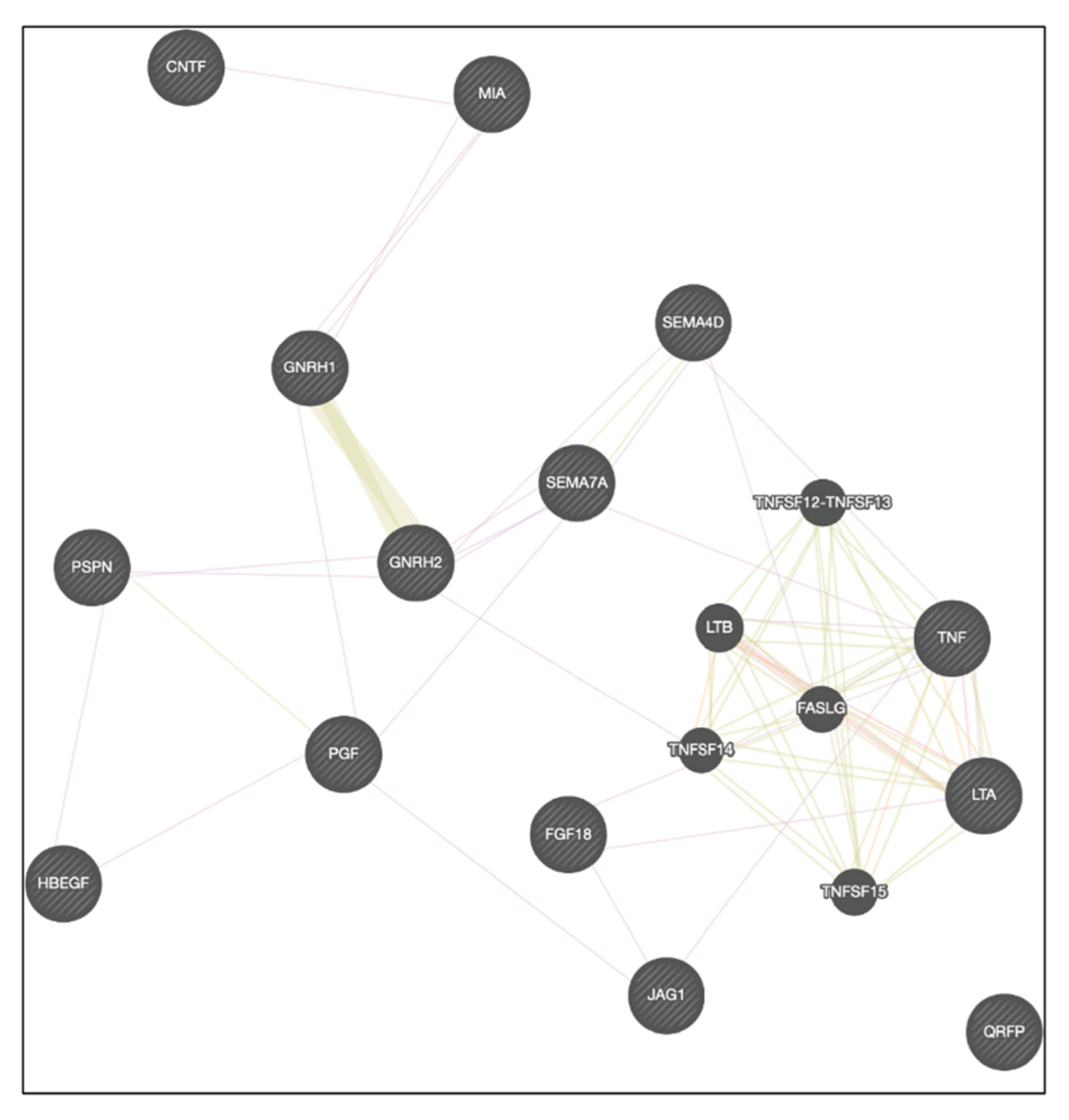
2.4. Analysis of Receptor Ligand Activity (GO:0048018) Genes
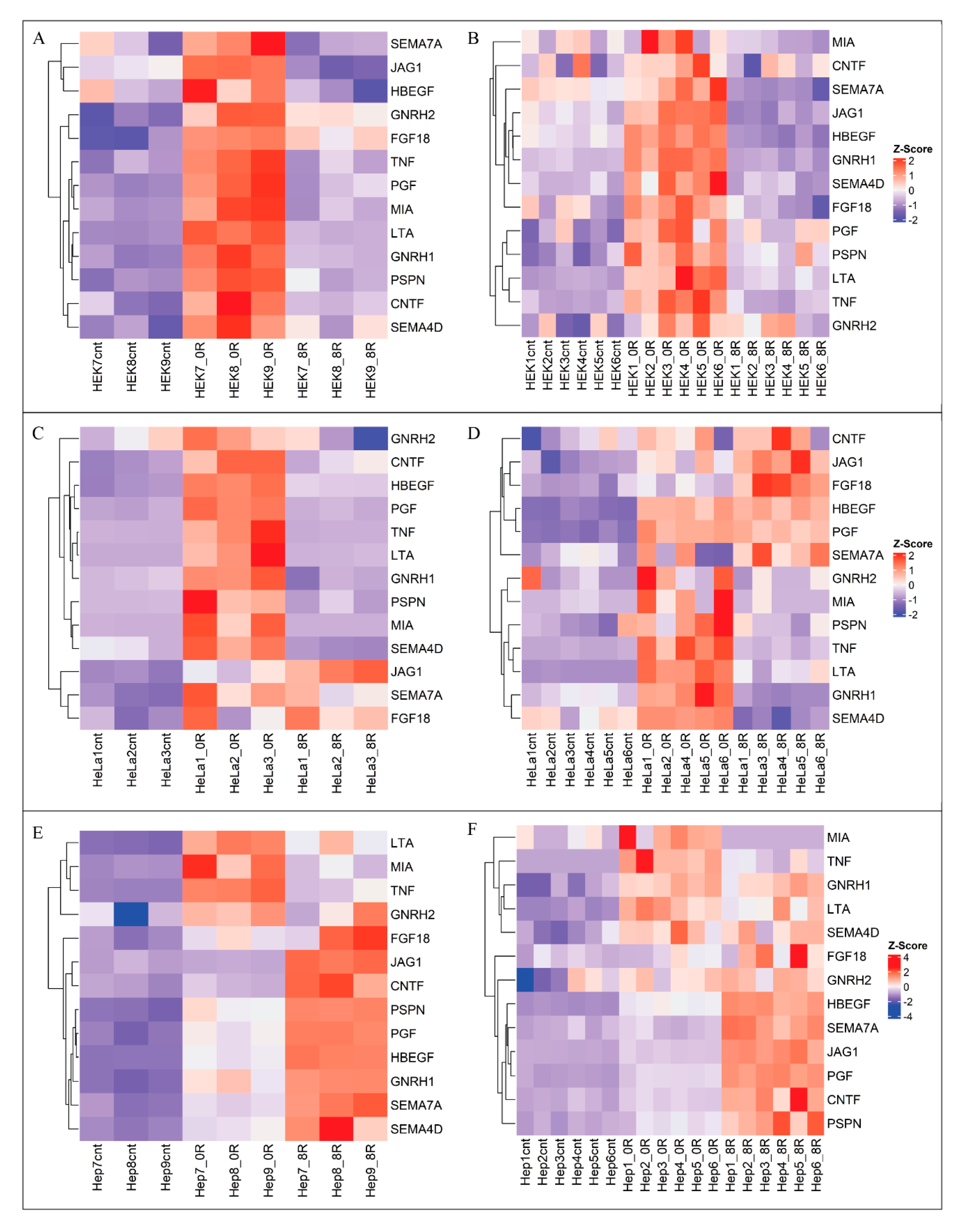
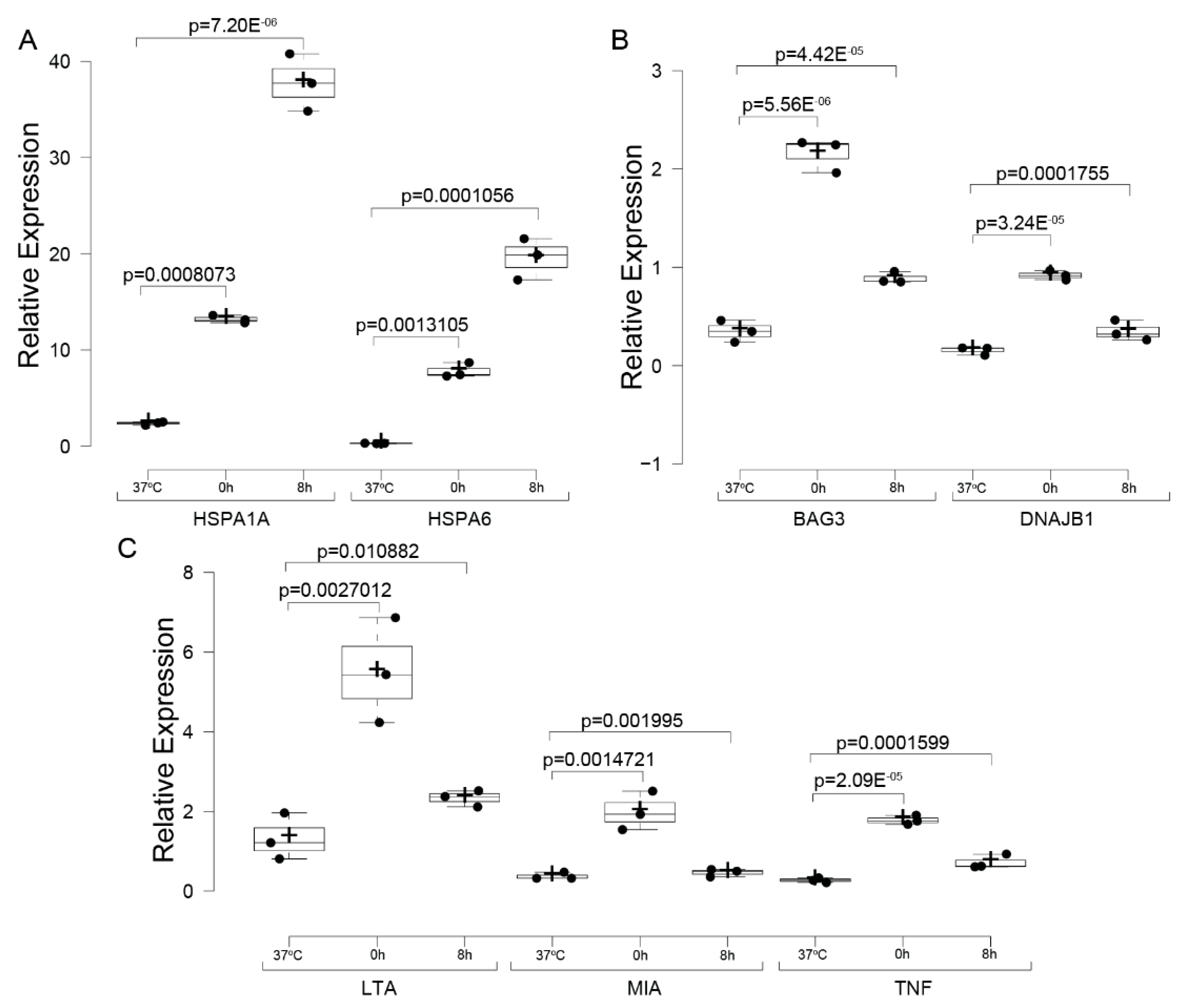
2.5. Gene Expression Assessment via qPCR
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Heat Shock Treatment
4.3. Sample Preparation, cDNA Library Preparation, and Sequencing
4.4. Transcriptomics Analyses (Detailed Methodology in Supplementary Data S1)
4.5. Molecular Validation of Using qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D. Evolution of the cellular stress proteome: From monophyletic origin to ubiquitous function. J. Exp. Biol. 2003, 206, 3119–3124. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Somero, G.N. The cellular stress response and temperature: Function, regulation, and evolution. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 379–397. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, J. Molecular mechanisms underlying stress response and adaptation. Thorac. Cancer 2018, 9, 218–227. [Google Scholar] [CrossRef]
- Camberg, J.L.; Doyle, S.M.; Johnston, D.M.; Wickner, S. Molecular Chaperones. In Brenner’s Encyclopedia of Genetics; Elsevier: Amsterdam, The Netherlands, 2013; pp. 456–460. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- McCallister, C.; Siracusa, M.C.; Shirazi, F.; Chalkia, D.; Nikolaidis, N. Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci. Rep. 2015, 5, 9363. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress. Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Salamanca, H.H.; Duarte, F.M.; Danko, C.G.; Lis, J.T. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol. Cell 2016, 62, 63–78. [Google Scholar] [CrossRef]
- Zhao, P.; Javed, S.; Shi, X.; Wu, B.; Zhang, D.; Xu, S.; Wang, X. Varying architecture of heat shock elements contributes to distinct magnitudes of target gene expression and diverged biological pathways in heat stress response of bread wheat. Front. Genet. 2020, 11, 30. [Google Scholar] [CrossRef]
- Zuo, W.F.; Pang, Q.; Zhu, X.; Yang, Q.Q.; Zhao, Q.; He, G.; Han, B.; Huang, W. Heat shock proteins as hallmarks of cancer: Insights from molecular mechanisms to therapeutic strategies. J. Hematol. Oncol. 2024, 17, 81. [Google Scholar] [CrossRef]
- Kunachowicz, D.; Krol-Kulikowska, M.; Raczycka, W.; Sleziak, J.; Blazejewska, M.; Kulbacka, J. Heat shock proteins, a double-edged sword: Significance in cancer progression, chemotherapy resistance and novel therapeutic perspectives. Cancers 2024, 16, 1500. [Google Scholar] [CrossRef]
- Cyran, A.M.; Zhitkovich, A. Heat shock proteins and HSF1 in cancer. Front. Oncol. 2022, 12, 860320. [Google Scholar] [CrossRef]
- Somu, P.; Mohanty, S.; Basavegowda, N.; Yadav, A.K.; Paul, S.; Baek, K.H. The interplay between heat shock proteins and cancer pathogenesis: A novel strategy for cancer therapeutics. Cancers 2024, 16, 638. [Google Scholar] [CrossRef]
- Ruz, C.; Alcantud, J.L.; Vives Montero, F.; Duran, R.; Bandres-Ciga, S. Proteotoxicity and neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5646. [Google Scholar] [CrossRef]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef]
- Flati, T.; Gioiosa, S.; Chillemi, G.; Mele, A.; Oliverio, A.; Mannironi, C.; Rinaldi, A.; Castrignano, T. A gene expression atlas for different kinds of stress in the mouse brain. Sci. Data 2020, 7, 437. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.I.; Whitfield, M.L.; Trinklein, N.D.; Myers, R.M.; Brown, P.O.; Botstein, D. Diverse and specific gene expression responses to stresses in cultured human cells. Mol. Biol. Cell 2004, 15, 2361–2374. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, Y.; Liu, X.; Yan, T.; Wu, L.; Alm, E.; Arkin, A.; Thompson, D.K.; Zhou, J. Global transcriptome analysis of the heat shock response of Shewanella oneidensis. J. Bacteriol. 2004, 186, 7796–7803. [Google Scholar] [CrossRef]
- Xu, P.; Wang, M.; Song, W.M.; Wang, Q.; Yuan, G.C.; Sudmant, P.H.; Zare, H.; Tu, Z.; Orr, M.E.; Zhang, B. The landscape of human tissue and cell type specific expression and co-regulation of senescence genes. Mol. Neurodegener. 2022, 17, 5. [Google Scholar] [CrossRef]
- Pessa, J.C.; Joutsen, J.; Sistonen, L. Transcriptional reprogramming at the intersection of the heat shock response and proteostasis. Mol. Cell 2024, 84, 80–93. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Love, M.I.; Hogenesch, J.B.; Irizarry, R.A. Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nat. Biotechnol. 2016, 34, 1287–1291. [Google Scholar] [CrossRef]
- Vihervaara, A.; Mahat, D.B.; Guertin, M.J.; Chu, T.; Danko, C.G.; Lis, J.T.; Sistonen, L. Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat. Commun. 2017, 8, 255. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; De Smaele, E.; Donadelli, M.; Riganti, C. Hypoxia dictates metabolic rewiring of tumors: Implications for chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Rios, F.; Uriostegui-Arcos, M.; Zurita, M. Transcriptional stress induces the generation of DoGs in cancer cells. Noncoding RNA 2024, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Cayado-Gutierrez, N.; Maccioni, M.; Cuello-Carrion, F.D. Heat shock proteins (HSPs) based anti-cancer vaccines. Curr. Mol. Med. 2012, 12, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 700. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef]
- Kurop, M.K.; Huyen, C.M.; Kelly, J.H.; Blagg, B.S.J. The heat shock response and small molecule regulators. Eur. J. Med. Chem. 2021, 226, 113846. [Google Scholar] [CrossRef]
- Kuo, D.; Ding, J.; Cohn, I.S.; Zhang, F.; Wei, K.; Rao, D.A.; Rozo, C.; Sokhi, U.K.; Shanaj, S.; Oliver, D.J.; et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Transl. Med. 2019, 11, eaau8587. [Google Scholar] [CrossRef]
- Rocourt, D.V.; Mehta, V.B.; Wu, D.; Besner, G.E. Heparin-binding EGF-like growth factor decreases neutrophil-endothelial cell interactions. J. Surg. Res. 2007, 141, 262–266. [Google Scholar] [CrossRef]
- Rampioni Vinciguerra, G.L.; Capece, M.; Reggiani Bonetti, L.; Nigita, G.; Calore, F.; Rentsch, S.; Magistri, P.; Ballarin, R.; Di Benedetto, F.; Distefano, R.; et al. Nutrient restriction-activated Fra-2 promotes tumor progression via IGF1R in miR-15a downmodulated pancreatic ductal adenocarcinoma. Signal Transduct. Target. Ther. 2024, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Momose, I.; Adachi, H.; Yamazaki, Y.; Sawa, R.; Ohba, S.I.; Kawada, M. Human pancreatic cancer cells under nutrient deprivation are vulnerable to redox system inhibition. J. Biol. Chem. 2020, 295, 16678–16690. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, H.; Resat, H.; Wiley, H.S. Cell surface receptors for signal transduction and ligand transport: A design principles study. PLoS Comput. Biol. 2007, 3, e101. [Google Scholar] [CrossRef]
- Arguello-Miranda, O.; Marchand, A.J.; Kennedy, T.; Russo, M.A.X.; Noh, J. Cell cycle-independent integration of stress signals by Xbp1 promotes Non-G1/G0 quiescence entry. J. Cell Biol. 2022, 221, e202103171. [Google Scholar] [CrossRef]
- Chung, K.M.; Kim, H.; Roque, C.G.; McCurdy, E.P.; Nguyen, T.T.T.; Siegelin, M.D.; Hwang, J.Y.; Hengst, U. A systemic cell stress signal confers neuronal resilience toward oxidative stress in a Hedgehog-dependent manner. Cell Rep. 2022, 41, 111488. [Google Scholar] [CrossRef]
- Kolodkin, A.; Sahin, N.; Phillips, A.; Hood, S.R.; Bruggeman, F.J.; Westerhoff, H.V.; Plant, N. Optimization of stress response through the nuclear receptor-mediated cortisol signalling network. Nat. Commun. 2013, 4, 1792. [Google Scholar] [CrossRef]
- Phillips, N.L.H.; Roth, T.L. Animal models and their contribution to our understanding of the relationship between environments, epigenetic modifications, and behavior. Genes 2019, 10, 47. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nordberg, H.; Shabalov, I.; Aerts, A.; Cantor, M.; Goodstein, D.; Kuo, A.; Minovitsky, S.; Nikitin, R.; Ohm, R.A.; et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012, 40, D26–D32. [Google Scholar] [CrossRef]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 91. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.; Wildenhain, J.; Rappsilber, J.; Tyers, M. BoxPlotR: A web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0R vs. Control | 8R vs. Cnt | 8R vs. 0R | |||
|---|---|---|---|---|---|
| ID | Name | ID | Name | ID | Name |
| GO:0048018 | Receptor Ligand Activity | GO:0048018 | Receptor Ligand Activity | GO:0048018 | Receptor Ligand Activity |
| GO:0030545 | Signaling receptor activator activity | hsa04080 | Neuroactive ligand-receptor interaction | hsa04080 | Neuroactive ligand-receptor interaction |
| GO:0044183 | Protein folding chaperone | HSA-500792 | GPCR ligand binding | HSA-500792 | GPCR ligand binding |
| HSA-373076 | Class A/1 (Rhodopsin-like receptors) | HSA-373076 | Class A/1 (Rhodopsin-like receptors) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinschmidt, A.; Solano, L.; Chavez, Y.; Hulsy, W.D.; Nikolaidis, N. Transcriptomics Unveil Canonical and Non-Canonical Heat Shock-Induced Pathways in Human Cell Lines. Int. J. Mol. Sci. 2025, 26, 1057. https://doi.org/10.3390/ijms26031057
Reinschmidt A, Solano L, Chavez Y, Hulsy WD, Nikolaidis N. Transcriptomics Unveil Canonical and Non-Canonical Heat Shock-Induced Pathways in Human Cell Lines. International Journal of Molecular Sciences. 2025; 26(3):1057. https://doi.org/10.3390/ijms26031057
Chicago/Turabian StyleReinschmidt, Andrew, Luis Solano, Yonny Chavez, William Drew Hulsy, and Nikolas Nikolaidis. 2025. "Transcriptomics Unveil Canonical and Non-Canonical Heat Shock-Induced Pathways in Human Cell Lines" International Journal of Molecular Sciences 26, no. 3: 1057. https://doi.org/10.3390/ijms26031057
APA StyleReinschmidt, A., Solano, L., Chavez, Y., Hulsy, W. D., & Nikolaidis, N. (2025). Transcriptomics Unveil Canonical and Non-Canonical Heat Shock-Induced Pathways in Human Cell Lines. International Journal of Molecular Sciences, 26(3), 1057. https://doi.org/10.3390/ijms26031057