
Thermodynamics of Molecular Transport Through a Nanochannel: Evidence of Energy–Entropy Compensation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
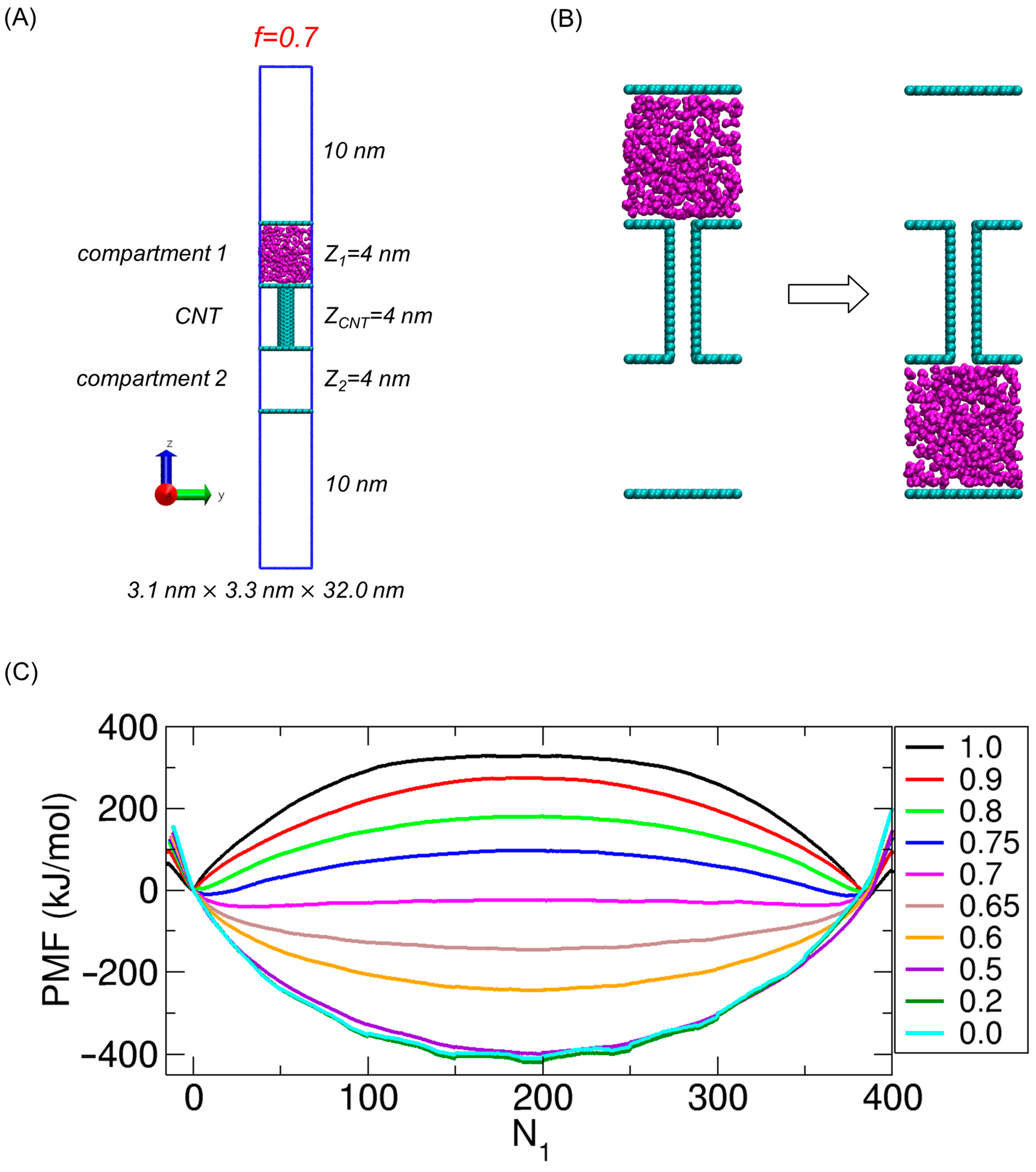
2.1. Model System
2.2. PMF Calculations and Results from Our Previous Work

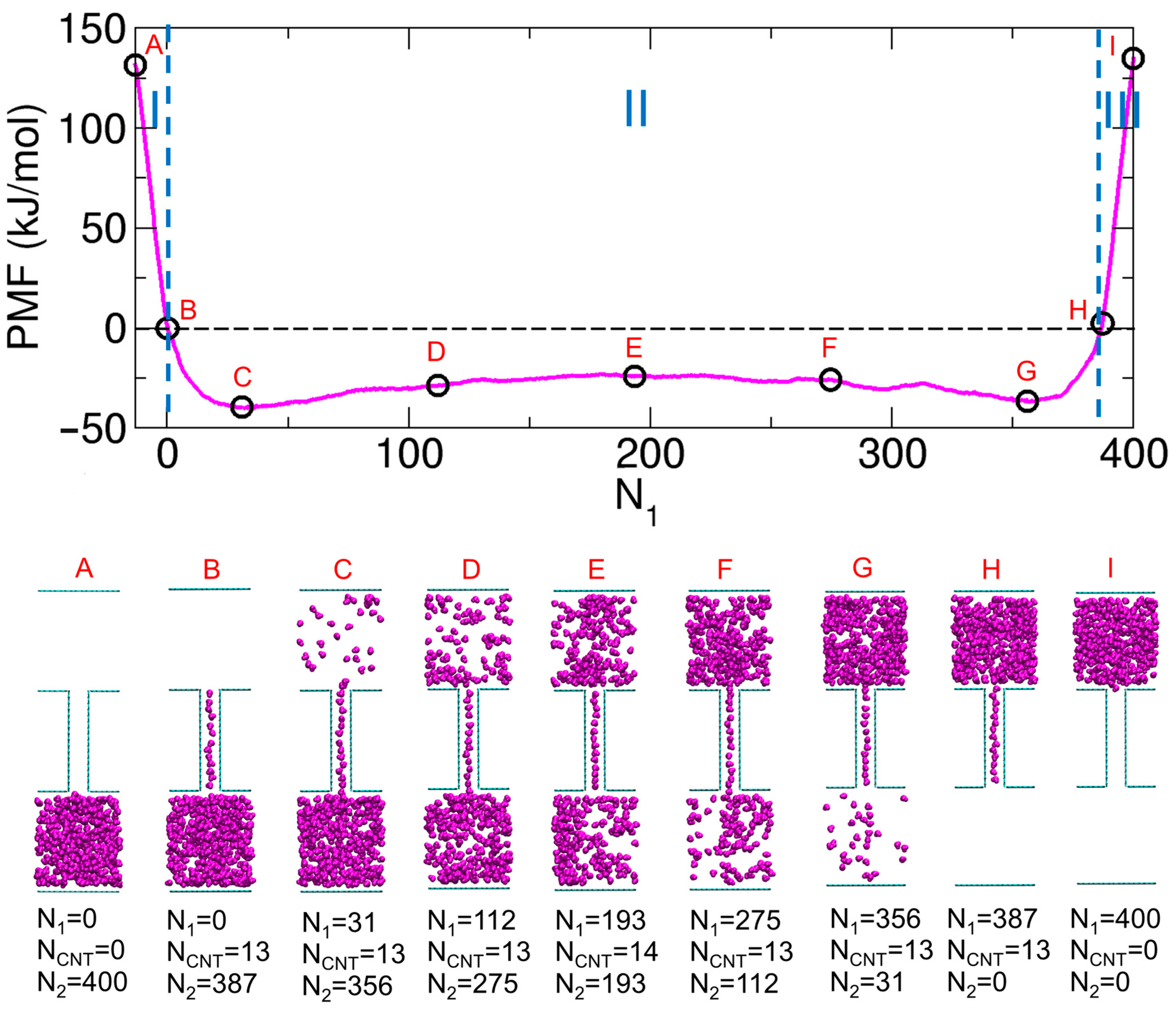
2.3. Thermodynamic Analysis
3. Results and Discussion
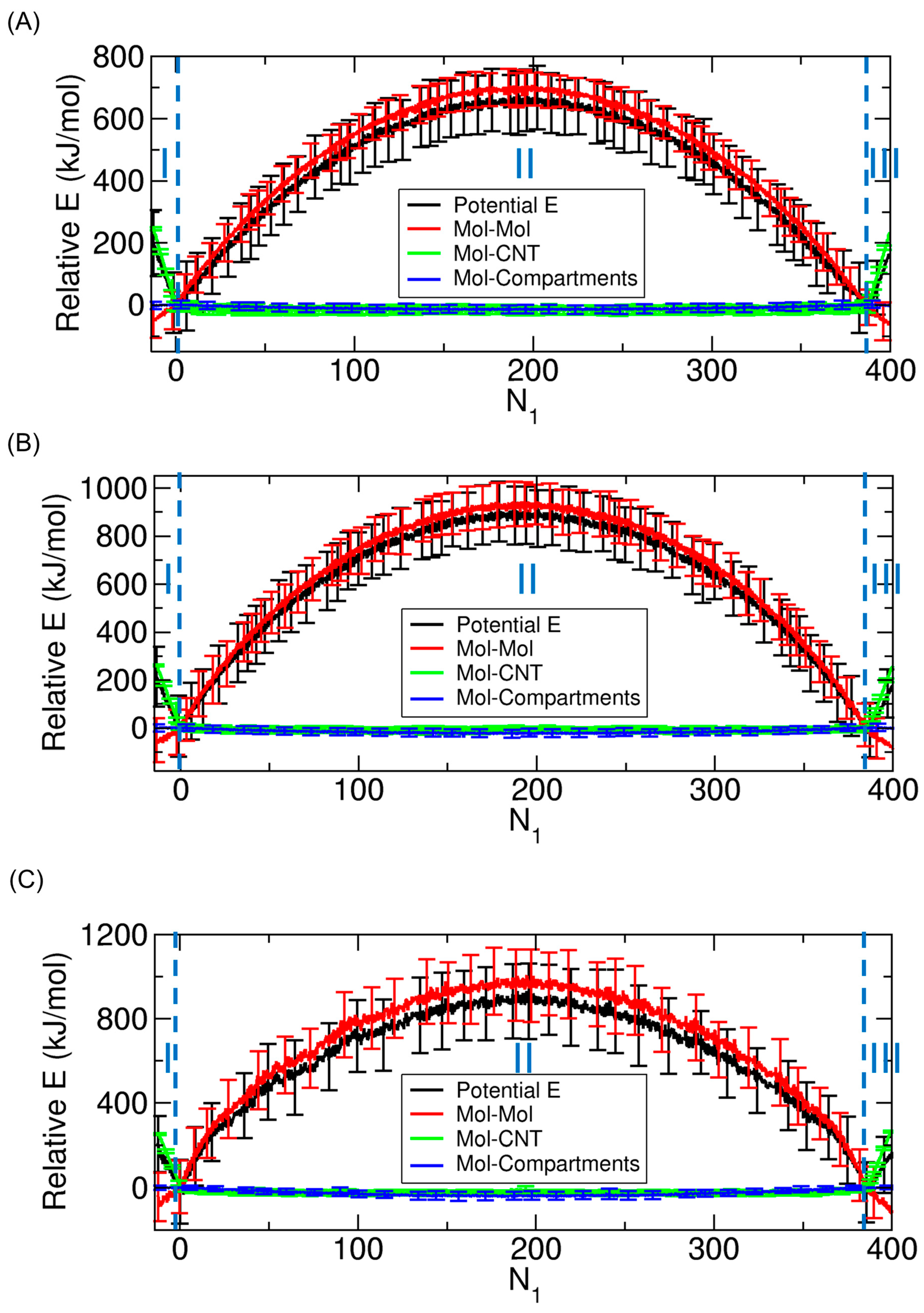
3.1. Total Potential Energy Profiles
3.2. Decomposition of Total Potential Energy by Interaction Type
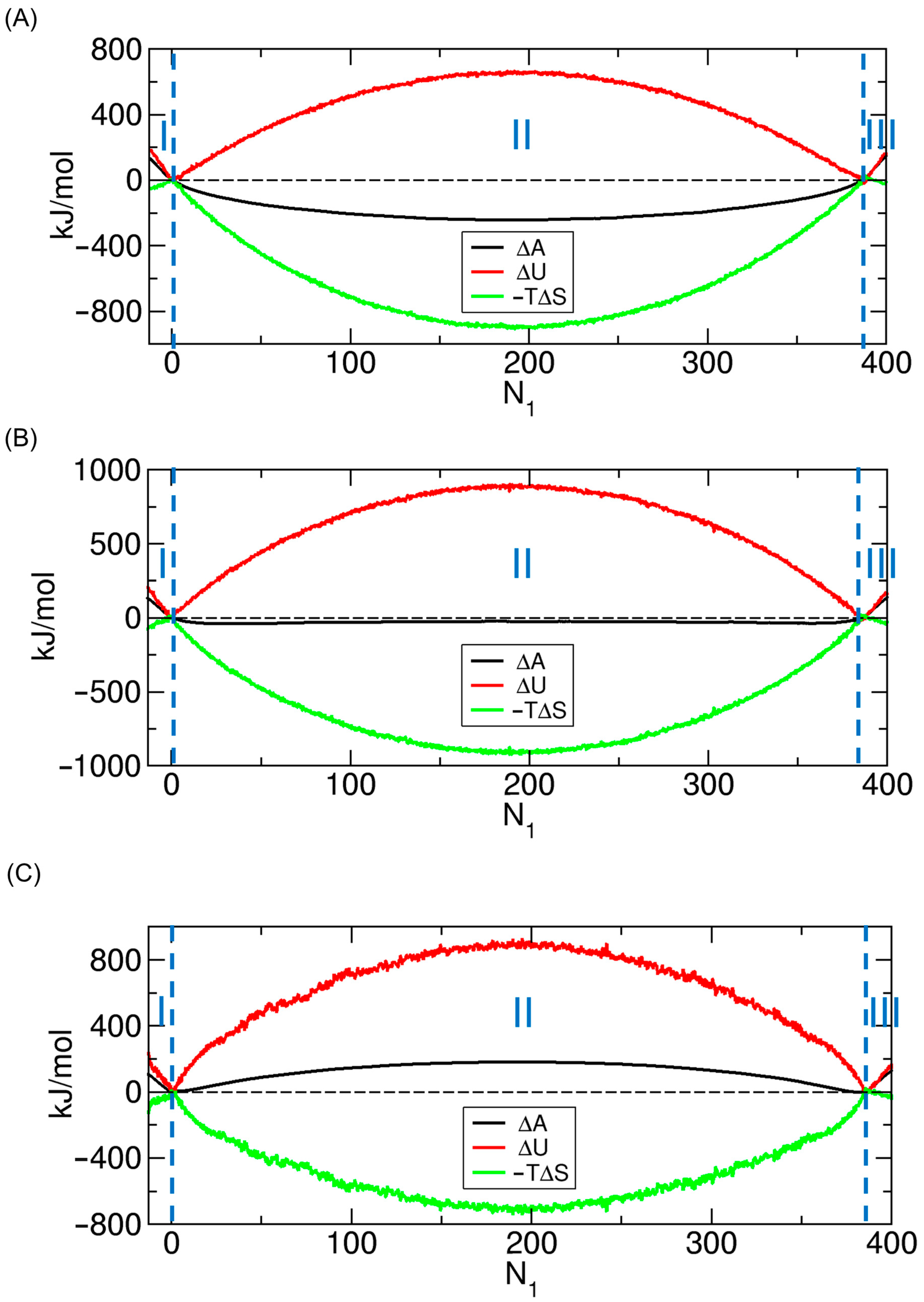
3.3. Decomposition of the PMF into Energetic and Entropic Contributions
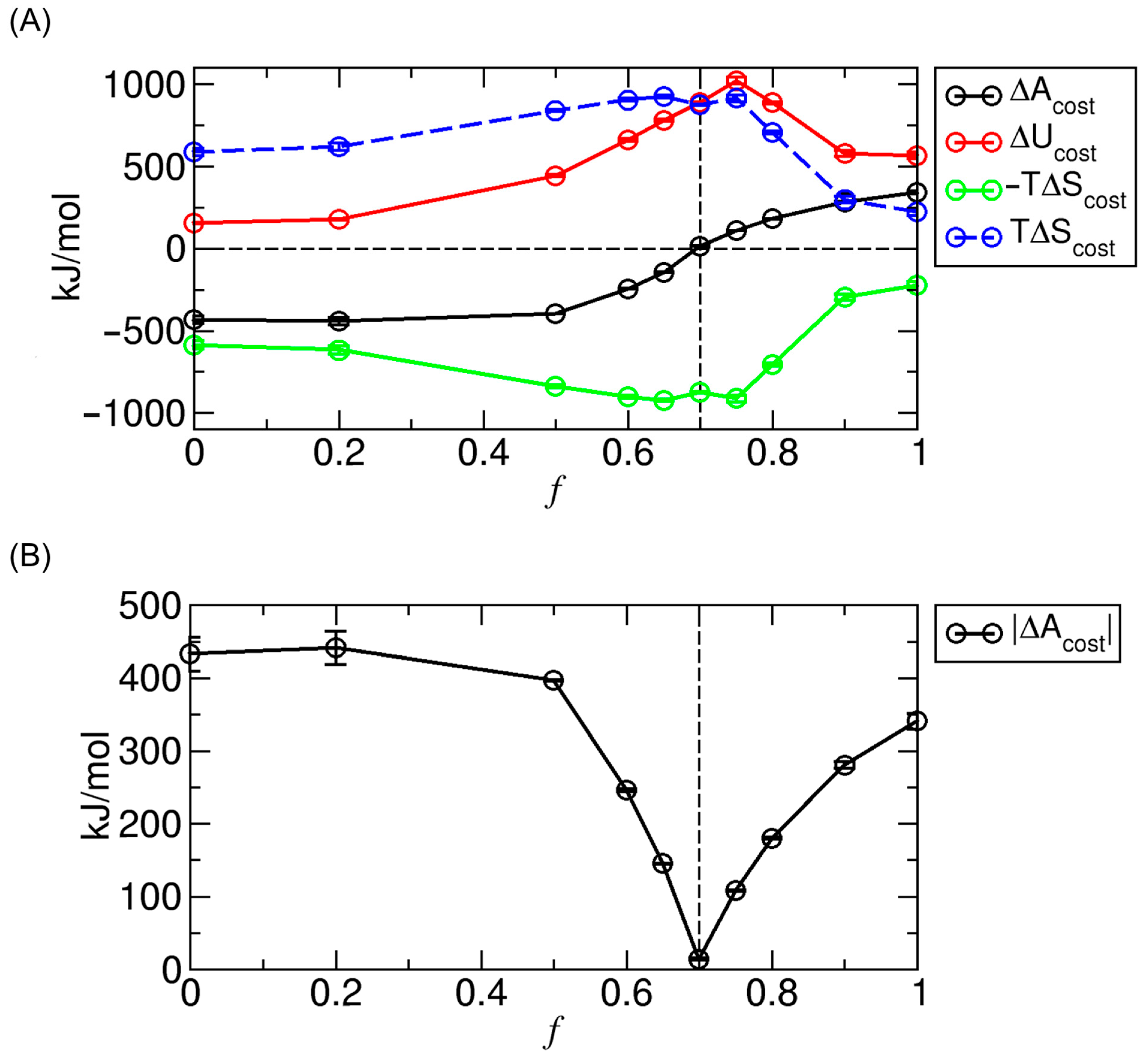
3.4. Free Energy, Energetic, and Entropic Costs of Molecular Transport
3.5. Analysis of Energy–Entropy Compensation Across Interaction Strengths
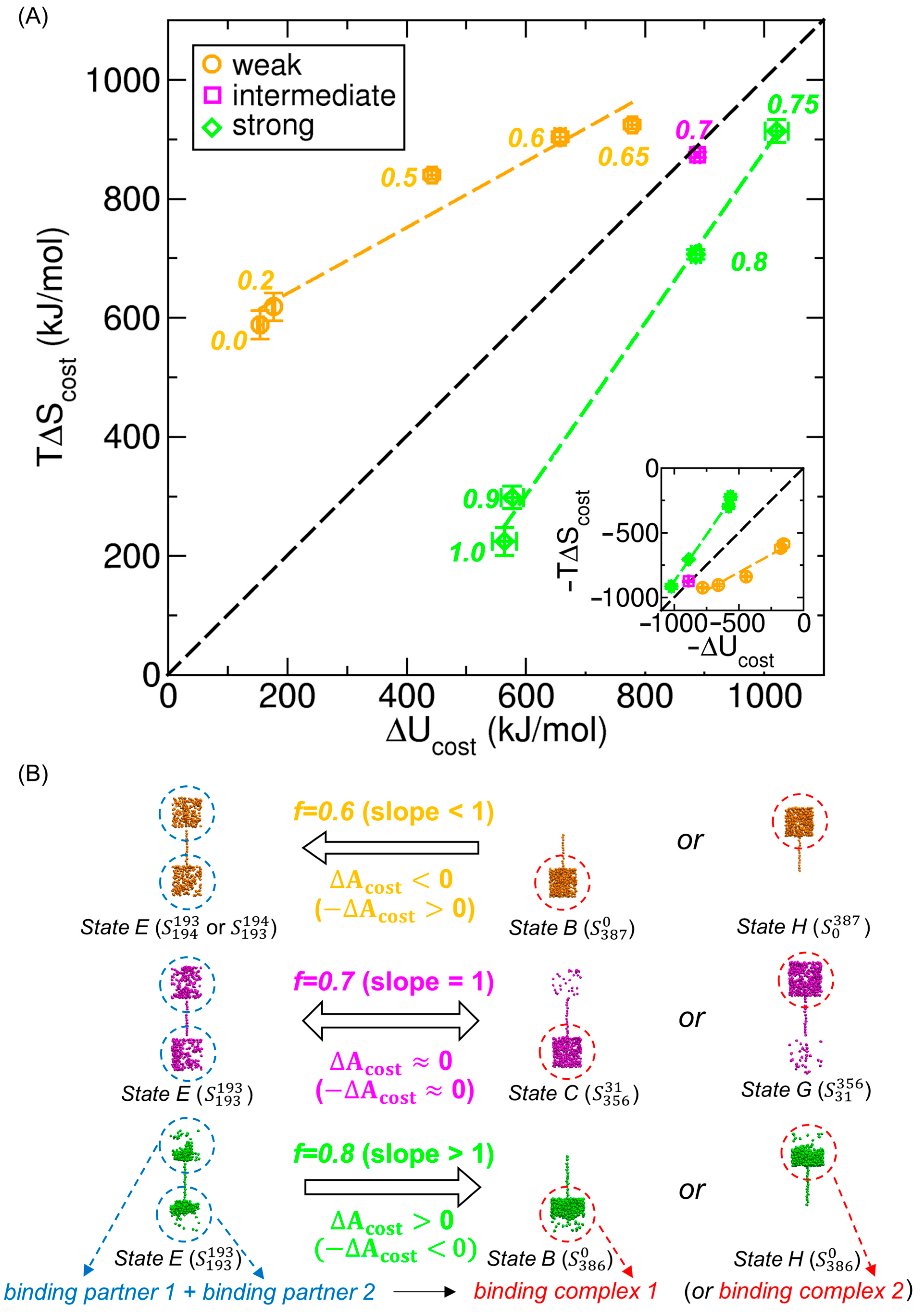
3.6. Extension of Energy–Entropy Compensation Analysis to Molecular Binding
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001; ISBN 0878933212. [Google Scholar]
- Gouaux, E.; MacKinnon, R. Principles of Selective Ion Transport in Channels and Pumps. Science 2005, 310, 1461–1465. [Google Scholar] [CrossRef]
- Jensen, M.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of Voltage Gating in Potassium Channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef]
- Görlich, D.; Kutay, U. Transport between the Cell Nucleus and the Cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar] [CrossRef]
- Nakielny, S.; Dreyfuss, G. Transport of Proteins and RNAs in and out of the Nucleus. Cell 1999, 99, 677–690. [Google Scholar] [CrossRef]
- Stewart, M.; Baker, R.P.; Bayliss, R.; Clayton, L.; Grant, R.P.; Littlewood, T.; Matsuura, Y. Molecular Mechanism of Translocation through Nuclear Pore Complexes during Nuclear Protein Import. FEBS Lett. 2001, 498, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The Nuclear Pore Complex and Nuclear Transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef]
- De Magistris, P. The Great Escape: Mrna Export through the Nuclear Pore Complex. Int. J. Mol. Sci. 2021, 22, 11767. [Google Scholar] [CrossRef] [PubMed]
- Eijkel, J.C.T.; van den Berg, A. Nanofluidics: What Is It and What Can We Expect from It? Microfluid. Nanofluidics 2005, 1, 249–267. [Google Scholar] [CrossRef]
- Han, J.; Fu, J.; Schoch, R.B. Molecular Sieving Using Nanofilters: Past, Present and Future. Lab Chip 2007, 8, 23–33. [Google Scholar] [CrossRef]
- Corry, B. Designing Carbon Nanotube Membranes for Efficient Water Desalination. J. Phys. Chem. B 2008, 112, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Napoli, M.; Eijkel, J.C.T.; Pennathur, S. Nanofluidic Technology for Biomolecule Applications: A Critical Review. Lab Chip 2010, 10, 957–985. [Google Scholar] [CrossRef]
- Kwon, Y.; Eun, C. Free Energy Change in the Complete Transport of All Water Molecules through a Carbon Nanotube. Phys. Chem. Chem. Phys. 2023, 25, 7032–7046. [Google Scholar] [CrossRef]
- Eun, C. Free Energy Profile for the Complete Transport of Nonpolar Molecules through a Carbon Nanotube. Int. J. Mol. Sci. 2023, 24, 14565. [Google Scholar] [CrossRef] [PubMed]
- Eun, C. Fluctuation-Induced Extensive Molecular Transport via Modulation of Interaction Strength. Phys. Chem. Chem. Phys. 2025, 27, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water Conduction through the Hydrophobic Channel of a Carbon Nanotube. Nature 2001, 414, 6860. [Google Scholar] [CrossRef]
- Berezhkovskii, A.; Hummer, G. Single-File Transport of Water Molecules through a Carbon Nanotube. Phys. Rev. Lett. 2002, 89, 64503. [Google Scholar] [CrossRef]
- Zhu, F.; Tajkhorshid, E.; Schulten, K. Collective Diffusion Model for Water Permeation through Microscopic Channels. Phys. Rev. Lett. 2004, 93, 224501. [Google Scholar] [CrossRef]
- Joseph, S.; Aluru, N.R. Why Are Carbon Nanotubes Fast Transporters of Water? Nano Lett. 2008, 8, 452–458. [Google Scholar] [CrossRef]
- Thomas, J.A.; McGaughey, A.J.H. Reassessing Fast Water Transport through Carbon Nanotubes. Nano Lett. 2008, 8, 2788–2793. [Google Scholar] [CrossRef] [PubMed]
- Walther, J.H.; Ritos, K.; Cruz-Chu, E.R.; Megaridis, C.M.; Koumoutsakos, P. Barriers to Superfast Water Transport in Carbon Nanotube Membranes. Nano Lett. 2013, 13, 1910–1914. [Google Scholar] [CrossRef]
- Wang, L.; Dumont, R.S.; Dickson, J.M. Nonequilibrium Molecular Dynamics Simulation of Pressure-Driven Water Transport through Modified CNT Membranes. J. Chem. Phys. 2013, 138, 44101. [Google Scholar] [CrossRef] [PubMed]
- Melillo, M.; Zhu, F.; Snyder, M.A.; Mittal, J. Water Transport through Nanotubes with Varying Interaction Strength between Tube Wall and Water. J. Phys. Chem. Lett. 2011, 2, 2978–2983. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Song, X.; Zhao, T.; Zhao, S.; Liu, H. Confinement Effect on Water Transport in CNT Membranes. Chem. Eng. Sci. 2018, 192, 1252–1259. [Google Scholar] [CrossRef]
- Thomas, J.A.; McGaughey, A.J.; Kuter-Arnebeck, O. Pressure-Driven Water Flow through Carbon Nanotubes: Insights from Molecular Dynamics Simulation. Int. J. Therm. Sci. 2010, 49, 281–289. [Google Scholar] [CrossRef]
- Nicholls, W.D.; Borg, M.K.; Lockerby, D.A.; Reese, J.M. Water Transport through (7,7) Carbon Nanotubes of Different Lengths Using Molecular Dynamics. Microfluid. Nanofluidics 2012, 12, 257–264. [Google Scholar] [CrossRef]
- Nicholls, W.D.; Borg, M.K.; Lockerby, D.A.; Reese, J.M. Water Transport through Carbon Nanotubes with Defects. Mol. Simul. 2012, 38, 781–785. [Google Scholar] [CrossRef]
- Jia, Y.; Lu, X.; Cao, Z.; Yan, T. From a Bulk to Nanoconfined Water Chain: Bridge Water at the Pore of the (6,6) Carbon Nanotube. Phys. Chem. Chem. Phys. 2020, 22, 25747–25759. [Google Scholar] [CrossRef] [PubMed]
- Won, C.Y.; Joseph, S.; Aluru, N.R. Effect of Quantum Partial Charges on the Structure and Dynamics of Water in Single-Walled Carbon Nanotubes. J. Chem. Phys. 2006, 125, 114701. [Google Scholar] [CrossRef]
- Kalra, A.; Garde, S.; Hummer, G. Osmotic Water Transport through Carbon Nanotube Membranes. Proc. Natl. Acad. Sci. USA 2003, 100, 10175–10180. [Google Scholar] [CrossRef]
- Raghunathan, A.V.; Aluru, N.R. Molecular Understanding of Osmosis in Semipermeable Membranes. Phys. Rev. Lett. 2006, 97, 24501. [Google Scholar] [CrossRef]
- Eun, C. Osmosis-Driven Water Transport through a Nanochannel: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2020, 21, 8030. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Huang, D.; Su, J. Osmotic Water Permeation through a Carbon Nanotube. J. Phys. Chem. Lett. 2020, 11, 940–944. [Google Scholar] [CrossRef]
- Gong, X.; Li, J.; Lu, H.; Wan, R.; Li, J.; Hu, J.; Fang, H. A Charge-Driven Molecular Water Pump. Nat. Nanotechnol. 2007, 2, 709. [Google Scholar] [CrossRef]
- Garate, J.A.; English, N.J.; MacElroy, J.M.D. Static and Alternating Electric Field and Distance-Dependent Effects on Carbon Nanotube-Assisted Water Self-Diffusion across Lipid Membranes. J. Chem. Phys. 2009, 131, 114508. [Google Scholar] [CrossRef] [PubMed]
- Garate, J.A.; English, N.J.; MacElroy, J.M.D. Carbon Nanotube Assisted Water Self-Diffusion across Lipid Membranes in the Absence and Presence of Electric Fields. Mol. Simul. 2009, 35, 3–12. [Google Scholar] [CrossRef]
- Su, J.; Guo, H. Control of Unidirectional Transport of Single-File Water Molecules through Carbon Nanotubes in an Electric Field. ACS Nano 2011, 5, 351–359. [Google Scholar] [CrossRef]
- Kayal, A.; Chandra, A. Wetting and Dewetting of Narrow Hydrophobic Channels by Orthogonal Electric Fields: Structure, Free Energy, and Dynamics for Different Water Models. J. Chem. Phys. 2015, 143, 224308. [Google Scholar] [CrossRef]
- Chen, Q.; Meng, L.; Li, Q.; Wang, D.; Guo, W.; Shuai, Z.; Jiang, L. Water Transport and Purification in Nanochannels Controlled by Asymmetric Wettability. Small 2011, 7, 2225–2231. [Google Scholar] [CrossRef]
- Beu, T.A. Molecular Dynamics Simulations of Ion Transport through Carbon Nanotubes. I. Influence of Geometry, Ion Specificity, and Many-Body Interactions. J. Chem. Phys. 2010, 132, 164513. [Google Scholar] [CrossRef] [PubMed]
- Garate, J.A.; Perez-Acle, T.; Oostenbrink, C. On the Thermodynamics of Carbon Nanotube Single-File Water Loading: Free Energy, Energy and Entropy Calculations. Phys. Chem. Chem. Phys. 2014, 16, 5119–5128. [Google Scholar] [CrossRef] [PubMed]
- Alexiadis, A.; Kassinos, S. Molecular Simulation of Water in Carbon Nanotubes. Chem. Rev. 2008, 108, 5014–5034. [Google Scholar] [CrossRef] [PubMed]
- Rasaiah, J.C.; Garde, S.; Hummer, G. Water in Nonpolar Confinement: From Nanotubes to Proteins and Beyond. Annu. Rev. Phys. Chem. 2008, 59, 713–740. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.I.; Rao, S.; Sansom, M.S.P. Water in Nanopores and Biological Channels: A Molecular Simulation Perspective. Chem. Rev. 2020, 120, 10298–10335. [Google Scholar] [CrossRef] [PubMed]
- Chatzichristos, A.; Hassan, J. Current Understanding of Water Properties inside Carbon Nanotubes. Nanomater 2022, 12, 174. [Google Scholar] [CrossRef]
- Leffler, J.E. The Enthalpy-Entropy Relationship and Its Implications for Organic Chemistry. J. Org. Chem. 1955, 20, 1202–1231. [Google Scholar] [CrossRef]
- Krug, R.R.; Hunter, W.G.; Grieger, R.A. Enthalpy-Entropy Compensation. 1. Some Fundamental Statistical Problems Associated with the Analysis of van’t Hoff and Arrhenius Data. J. Phys. Chem. 1976, 80, 2335–2341. [Google Scholar] [CrossRef]
- Houk, K.N.; Leach, A.G.; Kim, S.P.; Zhang, X. Binding Affinities of Host–Guest, Protein–Ligand, and Protein–Transition-State Complexes. Angew. Chemie Int. Ed. 2003, 42, 4872–4897. [Google Scholar] [CrossRef]
- Kuroki, R.; Nitta, K.; Yutani, K. Thermodynamic Changes in the Binding of Ca2+ to a Mutant Human Lysozyme (D86/92): Enthalpy-Entropy Compensation Observed upon Ca2+ Binding to Proteins. J. Biol. Chem. 1992, 267, 24297–24301. [Google Scholar] [CrossRef]
- Dunitz, J.D. Win Some, Lose Some: Enthalpy-Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef]
- Qian, H.; Hopfield, J.J. Entropy-Enthalpy Compensation: Perturbation and Relaxation in Thermodynamic Systems. J. Chem. Phys. 1996, 105, 9292–9298. [Google Scholar] [CrossRef]
- Sharp, K. Entropy—Enthalpy Compensation: Fact or Artifact? Protein Sci. 2001, 10, 661–667. [Google Scholar] [CrossRef]
- Williams, D.H.; Stephens, E.; O’Brien, D.P.; Zhou, M. Understanding Noncovalent Interactions: Ligand Binding Energy and Catalytic Efficiency from Ligand-Induced Reductions in Motion within Receptors and Enzymes. Angew. Chemie-Int. Ed. 2004, 43, 6596–6616. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L. Entropy-Enthalpy Compensation: Role and Ramifications in Biomolecular Ligand Recognition and Design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef]
- Fox, J.M.; Zhao, M.; Fink, M.J.; Kang, K.; Whitesides, G.M. The Molecular Origin of Enthalpy/Entropy Compensation in Biomolecular Recognition. Annu. Rev. Biophys. 2018, 47, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, C.; Holm, R. Exploring the Origins of Enthalpy-Entropy Compensation by Calorimetric Studies of Cyclodextrin Complexes. J. Phys. Chem. B 2019, 123, 6686–6693. [Google Scholar] [CrossRef]
- Peccati, F.; Jiménez-Osés, G. Enthalpy-Entropy Compensation in Biomolecular Recognition: A Computational Perspective. ACS Omega 2021, 6, 11122–11130. [Google Scholar] [CrossRef]
- Milne, J.S.; Xu, Y.; Mayne, L.C.; Englander, S.W. Experimental Study of the Protein Folding Landscape: Unfolding Reactions in Cytochrome C. J. Mol. Biol. 1999, 290, 811–822. [Google Scholar] [CrossRef]
- Liu, L.; Yang, C.; Guo, Q.X. A Study on the Enthalpy-Entropy Compensation in Protein Unfolding. Biophys. Chem. 2000, 84, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ben-Naim, A.; Marcus, Y. Solvation Thermodynamics of Nonionic Solutes. J. Chem. Phys. 1984, 81, 2016–2027. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.X. Isokinetic Relationship, Isoequilibrium Relationship, and Enthalpy-Entropy Compensation. Chem. Rev. 2001, 101, 673–695. [Google Scholar] [CrossRef]
- Moulik, S.P.; Naskar, B.; Rakshit, A.K. Current Status of Enthalpy–Entropy Compensation Phenomenon. Curr. Sci. 2019, 117, 1286–1291. [Google Scholar] [CrossRef]
- Linert, W.; Jameson, R.F. The Isokinetic Relationship. Chem. Soc. Rev. 1989, 18, 477–505. [Google Scholar] [CrossRef]
- Lafont, V.; Armstrong, A.A.; Ohtaka, H.; Kiso, Y.; Mario Amzel, L.; Freire, E. Compensating Enthalpic and Entropic Changes Hinder Binding Affinity Optimization. Chem. Biol. Drug Des. 2007, 69, 413–422. [Google Scholar] [CrossRef]
- Exner, O. Entropy-Enthalpy Compensation and Anticompensation: Solvation and Ligand Binding. Chem. Commun. 2000, 17, 1655–1656. [Google Scholar] [CrossRef]
- Graziano, G. Case Study of Enthalpy-Entropy Noncompensation. J. Chem. Phys. 2004, 120, 4467–4471. [Google Scholar] [CrossRef]
- Ford, D.M. Enthalpy-Entropy Compensation Is Not a General Feature of Weak Association. J. Am. Chem. Soc. 2005, 127, 16167–16170. [Google Scholar] [CrossRef] [PubMed]
- Ryde, U. A Fundamental View of Enthalpy-Entropy Compensation. Medchemcomm 2014, 5, 1324–1336. [Google Scholar] [CrossRef]
- Pan, A.; Kar, T.; Rakshit, A.K.; Moulik, S.P. Enthalpy-Entropy Compensation (EEC) Effect: Decisive Role of Free Energy. J. Phys. Chem. B 2016, 120, 10531–10539. [Google Scholar] [CrossRef]
- Grunwald, E.; Steel, C. Solvent Reorganization and Thermodynamic Enthalpy—Entropy Compensation. J. Am. Chem. Soc. 1995, 117, 5687–5692. [Google Scholar] [CrossRef]
- Lee, B.; Graziano, G. A Two-State Model of Hydrophobic Hydration That Produces Compensating Enthalpy and Entropy Changes. J. Am. Chem. Soc. 1996, 118, 5163–5168. [Google Scholar] [CrossRef]
- Krug, R.R.; Hunter, W.G.; Grieger, R.A. Statistical Interpretation of Enthalpy-Entropy Compensation. Nature 1976, 261, 566–567. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Enthalpy-Entropy Compensation: A Phantom Phenomenon. J. Biosci. 2002, 27, 121–126. [Google Scholar] [CrossRef]
- Khrapunov, S. The Enthalpy-Entropy Compensation Phenomenon. Limitations for the Use of Some Basic Thermodynamic Equations. Curr. Protein Pept. Sci. 2018, 19, 1088–1091. [Google Scholar] [CrossRef]
- Lumry, R.; Rajender, S. Enthalpy–Entropy Compensation Phenomena in Water Solutions of Proteins and Small Molecules: A Ubiquitous Properly of Water. Biopolymers 1970, 9, 1125–1227. [Google Scholar] [CrossRef]
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy–Entropy Compensation: The Role of Solvation. Eur. Biophys. J. 2017, 46, 301–308. [Google Scholar] [CrossRef]
- Gilbert, A.S. Entropy-Enthalpy Compensation in the Fusion of Organic Molecules: Implications for Walden’s Rule and Molecular Freedom in the Liquid State. Thermochim. Acta 1999, 339, 131–142. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 72, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bayly, C.I.; Merz, K.M.; Ferguson, D.M.; Cornell, W.D.; Fox, T.; Caldwell, J.W.; Kollman, P.A.; Cieplak, P.; Gould, I.R.; Spellmeyer, D.C. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber Die Anwendung Des Satzes Vom Virial in Der Kinetischen Theorie Der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- Berthelot, D. Sur Le Mélange Des Gaz. Compt. Rendus 1898, 126, 15. [Google Scholar]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Hub, J.S.; De Groot, B.L.; Van Der Spoel, D. G-Whams-a Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Gill, S.J. Stability of Protein Structure and Hydrophobic Interaction. Adv. Protein Chem. 1988, 39, 191–234. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eun, C. Thermodynamics of Molecular Transport Through a Nanochannel: Evidence of Energy–Entropy Compensation. Int. J. Mol. Sci. 2025, 26, 7277. https://doi.org/10.3390/ijms26157277
Eun C. Thermodynamics of Molecular Transport Through a Nanochannel: Evidence of Energy–Entropy Compensation. International Journal of Molecular Sciences. 2025; 26(15):7277. https://doi.org/10.3390/ijms26157277
Chicago/Turabian StyleEun, Changsun. 2025. "Thermodynamics of Molecular Transport Through a Nanochannel: Evidence of Energy–Entropy Compensation" International Journal of Molecular Sciences 26, no. 15: 7277. https://doi.org/10.3390/ijms26157277
APA StyleEun, C. (2025). Thermodynamics of Molecular Transport Through a Nanochannel: Evidence of Energy–Entropy Compensation. International Journal of Molecular Sciences, 26(15), 7277. https://doi.org/10.3390/ijms26157277