
Membrane-Targeting Antivirals

 , , , and
, , , and
Abstract
1. Introduction
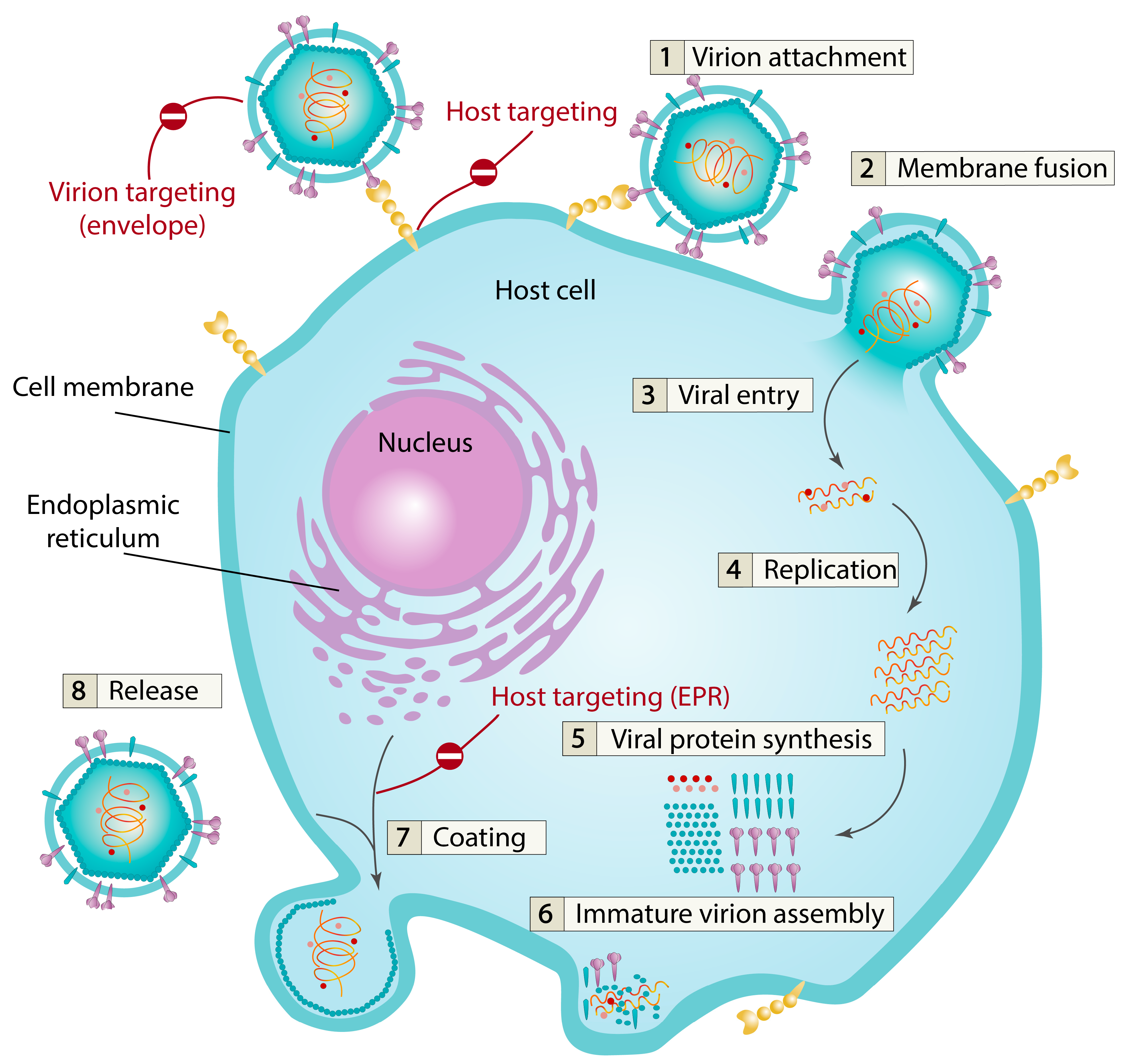
2. Viruses and Viral Membranes
3. Research Methods
- −
- Fluorescence spectroscopy and microscopy—assess membrane permeability and morphological changes;
- −
- Quartz crystal microbalance with dissipation monitoring—evaluate viscoelastic membrane properties;
- −
- Electron spin resonance spectroscopy—monitor membrane organization;
- −
- Electrochemical impedance spectroscopy—study membrane destabilization kinetics.
4. Virion-Targeting Membrane-Active Antivirals
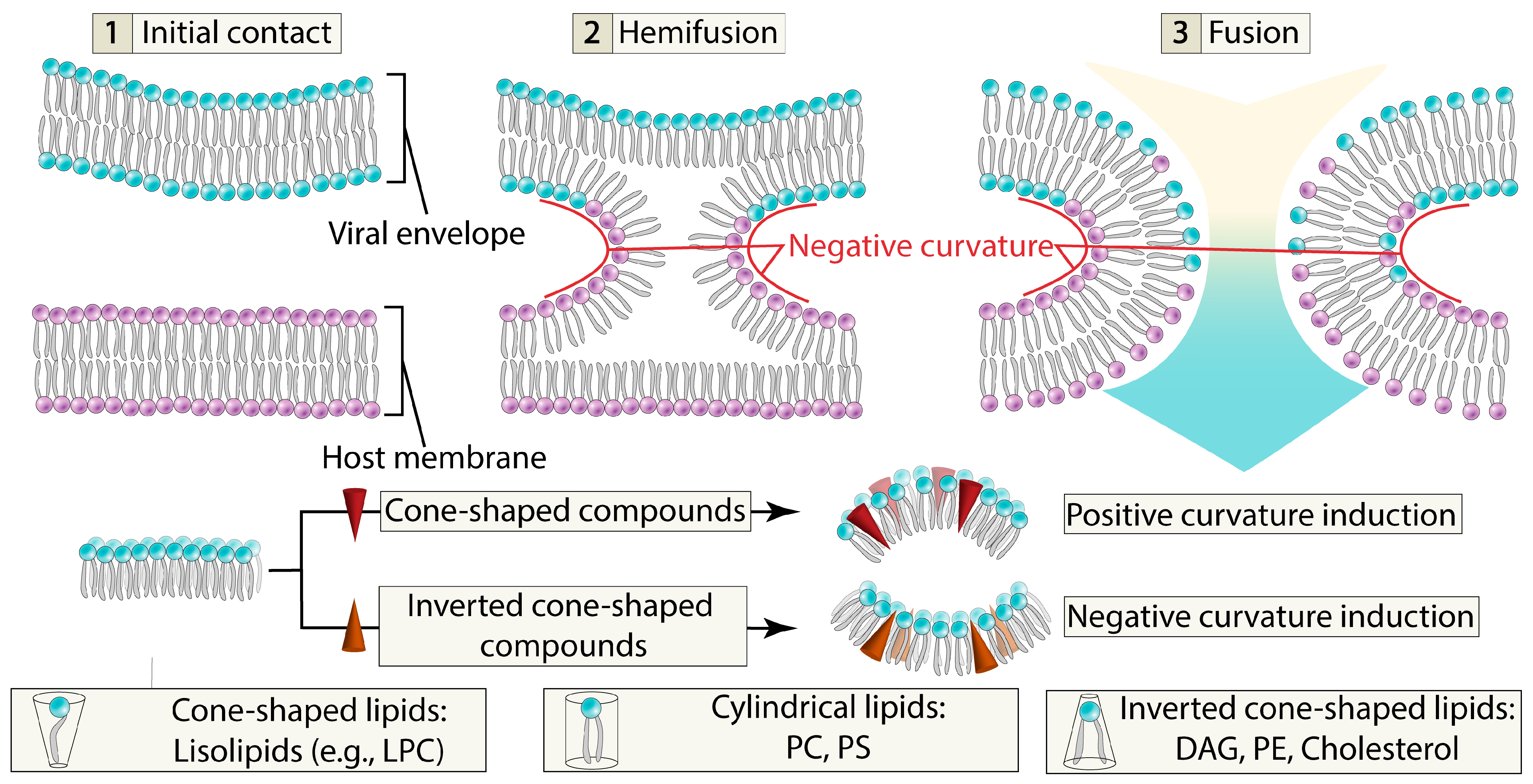
4.1. Membrane Properties Modulation
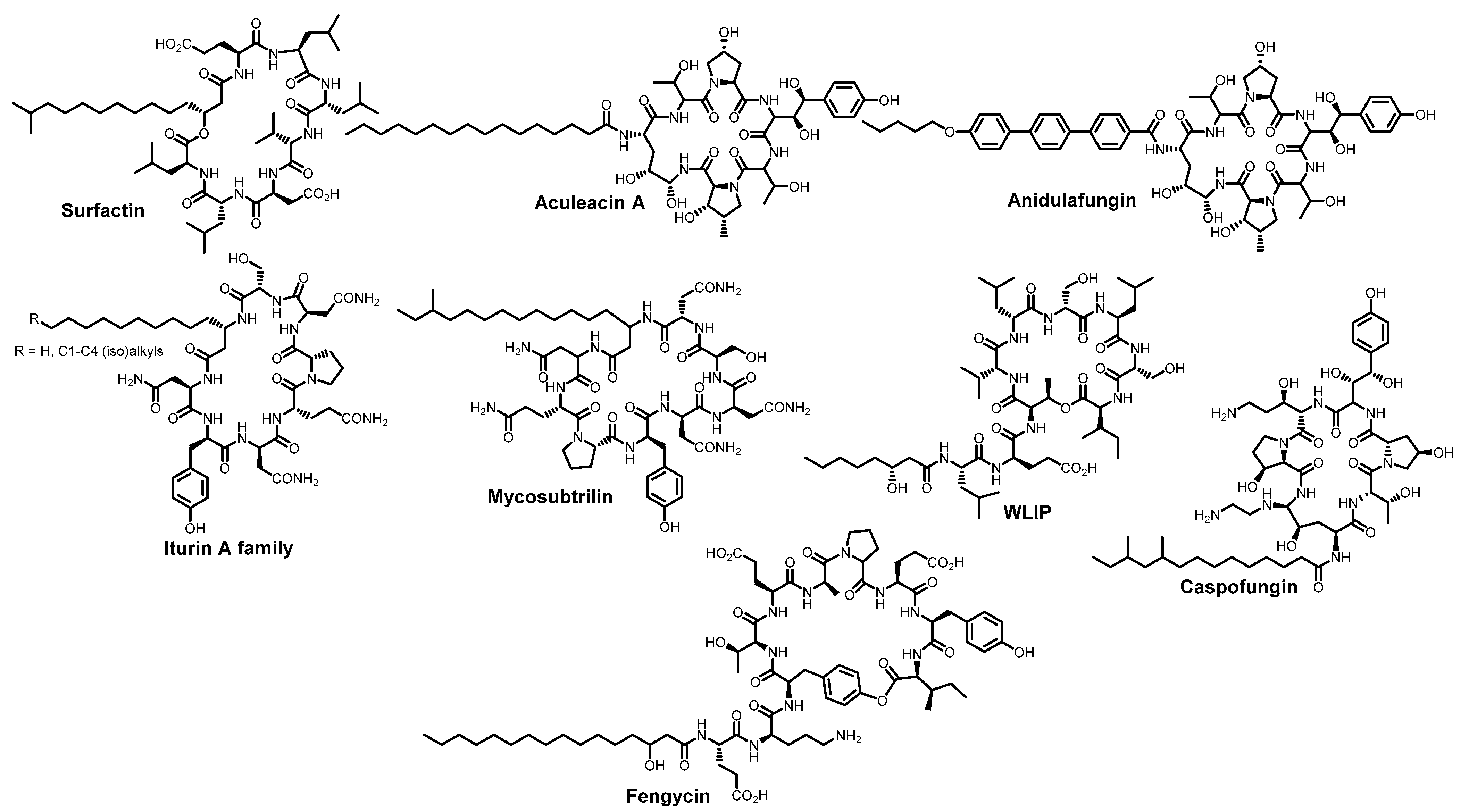
4.2. Membrane-Lytic Compounds
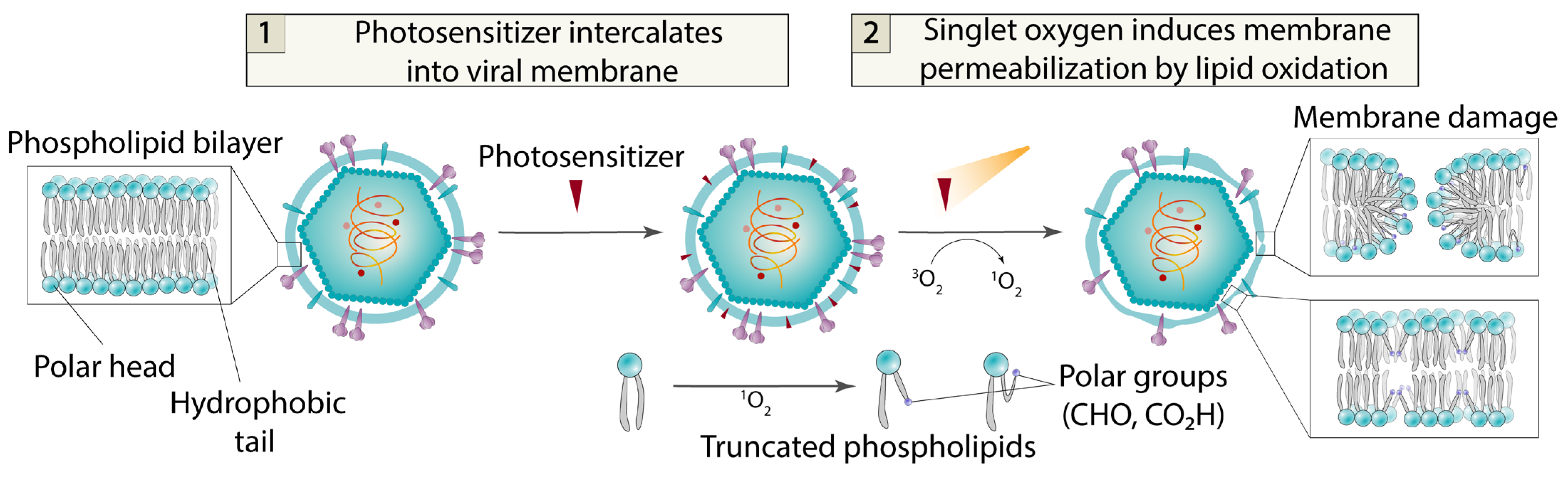
4.3. Photosensitizers
- −
- Exceptionally high antiviral activity, reaching subnanomolar efficacy levels;
- −
- Low cytotoxicity due to the presence of lipid reparation system in the cell;
- −
- Broad-spectrum potential—photosensitizers may theoretically target all enveloped viruses.
- −
- Primary limitations include the following:
- −
- Light-dependent action with no dark activity mechanism;
- −
- Poor aqueous solubility resulting from the essential non-polar aromatic core required for both singlet oxygen photogeneration and membrane intercalation.
5. Host Cell-Targeted Antiviral Drugs
- These agents do not directly interact with viral proteins, thereby reducing the likelihood of drug-resistance mutations emerging.
- They can target host receptor proteins common to multiple viruses, enabling broad-spectrum activity.
5.1. Host Cell Receptor Proteins
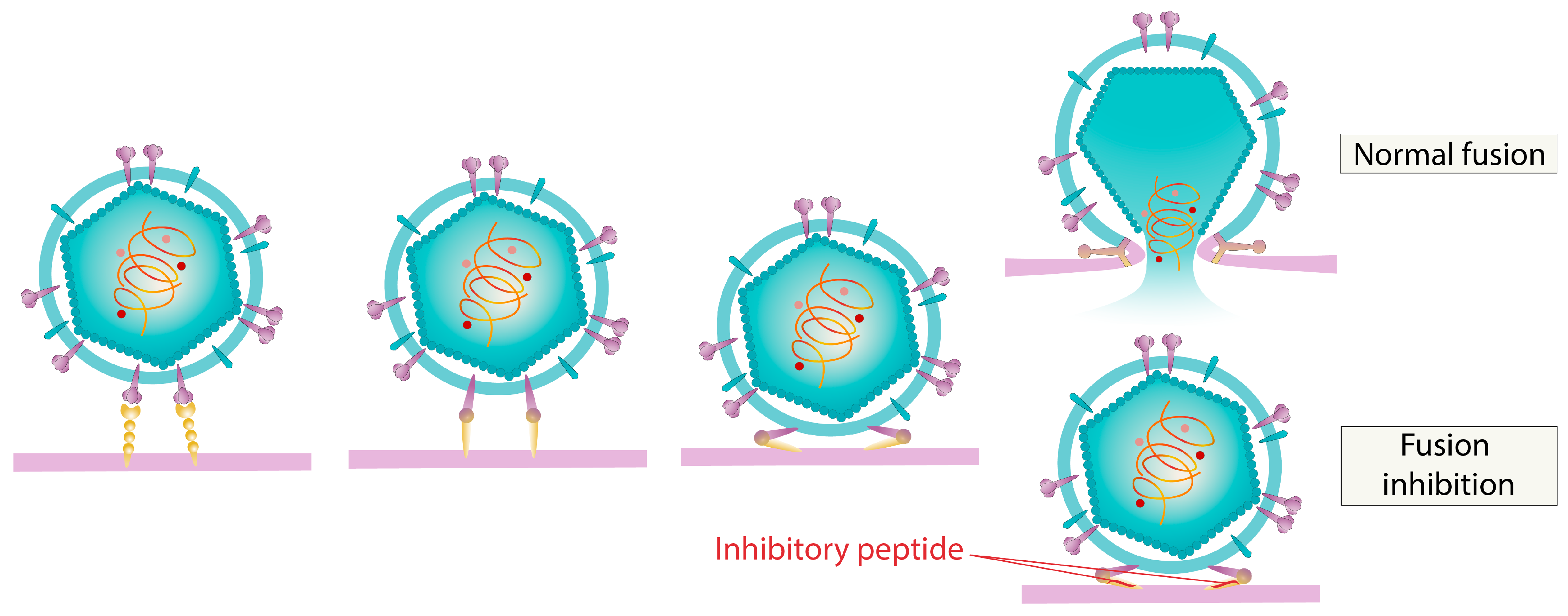
5.2. Inhibition of Virus Coating
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, J. NIH Cancels Research Grants for COVID-19 and Future Pandemics. Science 2025, 388, 16. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Chu, H.; Sridhar, S.; Yuen, K.-Y. COVID-19 Drug Discovery and Treatment Options. Nat. Rev. Microbiol. 2024, 22, 391–407. [Google Scholar] [CrossRef]
- Bekerman, E.; Einav, S. Combating Emerging Viral Threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palù, G.; Loregian, A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Tr. Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef]
- Ianevski, A.; Andersen, P.I.; Merits, A.; Bjørås, M.; Kainov, D. Expanding the Activity Spectrum of Antiviral Agents. Drug Discov. Today 2019, 24, 1224–1228. [Google Scholar] [CrossRef]
- García-Serradilla, M.; Risco, C.; Pacheco, B. Drug Repurposing for New, Efficient, Broad Spectrum Antivirals. Virus Res. 2019, 264, 22–31. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-Based Drug Repurposing for Novel Coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-Ray Screening Identifies Active Site and Allosteric Inhibitors of SARS-CoV-2 Main Protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Von Delft, A.; Hall, M.D.; Kwong, A.D.; Purcell, L.A.; Saikatendu, K.S.; Schmitz, U.; Tallarico, J.A.; Lee, A.A. Accelerating Antiviral Drug Discovery: Lessons from COVID-19. Nat. Rev. Drug Discov. 2023, 22, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Mei, H.; Chen, Y.; Griffin, J.D.; Liu, Q.; Weisberg, E.; Yang, J. Repurposing Clinically Available Drugs and Therapies for Pathogenic Targets to Combat SARS-CoV-2. MedComm 2023, 4, e254. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Efficacy of Repurposed Antiviral Drugs: Lessons from COVID-19. Drug Discov. Today 2022, 27, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Hayden, R.; Hills, T.; Al-Samkari, H.; Casey, J.; Del Sorbo, L.; Lawler, P.R.; Sise, M.E.; Leaf, D.E. Therapeutic Advances in COVID-19. Nat. Rev. Nephrol. 2023, 19, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic Strategies for COVID-19: Progress and Lessons Learned. Nat. Rev. Drug Discov. 2023, 22, 449–475. [Google Scholar] [CrossRef]
- Urquhart, L. FDA New Drug Approvals in Q2 2023. Nat. Rev. Drug Discov. 2023, 22, 614. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Oprea, T.I.; Bauman, J.E.; Bologa, C.G.; Buranda, T.; Chigaev, A.; Edwards, B.S.; Jarvik, J.W.; Gresham, H.D.; Haynes, M.K.; Hjelle, B.; et al. Drug Repurposing from an Academic Perspective. Drug Discov. Today Ther. Strateg. 2011, 8, 61–69. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug Repurposing: A Promising Tool to Accelerate the Drug Discovery Process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Bano, N.; Parveen, S.; Saeed, M.; Siddiqui, S.; Abohassan, M.; Mir, S.S. Drug Repurposing of Selected Antibiotics: An Emerging Approach in Cancer Drug Discovery. ACS Omega 2024, 9, 26762–26779. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Lü, Z.; Dai, X.; Xu, J.; Liu, Z.; Guo, Y.; Gao, Z.; Meng, F. Medicinal Chemistry Strategies toward Broad-Spectrum Antiviral Agents to Prevent next Pandemics. Eur. J. Med. Chem. 2024, 271, 116442. [Google Scholar] [CrossRef]
- Ryu, W.-S. Virus Structure. In Molecular VirolOgy of Human Pathogenic Viruses; Elsevier: Amsterdam, The Netherlands, 2017; pp. 21–29. ISBN 978-0-12-800838-6. [Google Scholar] [CrossRef]
- Simmonds, P.; Adriaenssens, E.M.; Zerbini, F.M.; Abrescia, N.G.A.; Aiewsakun, P.; Alfenas-Zerbini, P.; Bao, Y.; Barylski, J.; Drosten, C.; Duffy, S.; et al. Four Principles to Establish a Universal Virus Taxonomy. PLoS Biol. 2023, 21, e3001922. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e00053-21. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K. Cell Entry of Enveloped Viruses. Curr. Opin. Virol. 2011, 1, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral Membrane Fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Mercer, J.; Lee, J.E.; Saphire, E.O.; Freeman, S.A. SnapShot: Enveloped Virus Entry. Cell 2020, 182, 786–786.e1. [Google Scholar] [CrossRef]
- Kephart, S.M.; Hom, N.; Lee, K.K. Visualizing Intermediate Stages of Viral Membrane Fusion by Cryo-Electron Tomography. Tr. Biochem. Sci. 2024, 49, 916–931. [Google Scholar] [CrossRef]
- Sardar, A.; Dewangan, N.; Panda, B.; Bhowmick, D.; Tarafdar, P.K. Lipid and Lipidation in Membrane Fusion. J. Membr. Biol. 2022, 255, 691–703. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; D’Amelio, N. Biomembrane Lipids: When Physics and Chemistry Join to Shape Biological Activity. Biochimie 2022, 203, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Ripa, I.; Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Membrane Rafts: Portals for Viral Entry. Front. Microbiol. 2021, 12, 631274. [Google Scholar] [CrossRef]
- Prochnow, H.; Rox, K.; Birudukota, N.V.S.; Weichert, L.; Hotop, S.-K.; Klahn, P.; Mohr, K.; Franz, S.; Banda, D.H.; Blockus, S.; et al. Labyrinthopeptins Exert Broad-Spectrum Antiviral Activity through Lipid-Binding-Mediated Virolysis. J. Virol. 2020, 94, e01471-19. [Google Scholar] [CrossRef]
- Blockus, S.; Sake, S.M.; Wetzke, M.; Grethe, C.; Graalmann, T.; Pils, M.; Le Goffic, R.; Galloux, M.; Prochnow, H.; Rox, K.; et al. Labyrinthopeptins as Virolytic Inhibitors of Respiratory Syncytial Virus Cell Entry. Antivir. Res. 2020, 177, 104774. [Google Scholar] [CrossRef]
- Das, A.; Rivera-Serrano, E.E.; Yin, X.; Walker, C.M.; Feng, Z.; Lemon, S.M. Cell Entry and Release of Quasi-Enveloped Human Hepatitis Viruses. Nat. Rev. Microbiol. 2023, 21, 573–589. [Google Scholar] [CrossRef]
- Yang, L.; Li, J.; Li, S.; Dang, W.; Xin, S.; Long, S.; Zhang, W.; Cao, P.; Lu, J. Extracellular Vesicles Regulated by Viruses and Antiviral Strategies. Front. Cell Dev. Biol. 2021, 9, 722020. [Google Scholar] [CrossRef] [PubMed]
- Ketter, E.; Randall, G. Virus Impact on Lipids and Membranes. Annu. Rev. Virol. 2019, 6, 319–340. [Google Scholar] [CrossRef]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-Spectrum Antivirals against Viral Fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef]
- Gee, Y.J.; Sea, Y.L.; Lal, S.K. Viral Modulation of Lipid Rafts and Their Potential as Putative Antiviral Targets. Rev. Med. Virol. 2023, 33, e2413. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus Membrane Fusion Mechanism Offers a Potential Target for Antiviral Development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.K.; Jeon, W.-Y.; Sut, T.N.; Cho, N.-J.; Jackman, J.A. Stopping Membrane-Enveloped Viruses with Nanotechnology Strategies: Toward Antiviral Drug Development and Pandemic Preparedness. ACS Nano 2021, 15, 125–148. [Google Scholar] [CrossRef]
- Wisskirchen, K.; Lucifora, J.; Michler, T.; Protzer, U. New Pharmacological Strategies to Fight Enveloped Viruses. Tr. Pharmacol. Sci. 2014, 35, 470–478. [Google Scholar] [CrossRef]
- Cheresiz, S.V.; Ulyanova, E.A.; Pokrovsky, A.G. Enveloped Virus Entry as a Pharmacological Target: Viral Membrane Fusion Machineries and Their Inhibitors. Mol. Biol. 2025, 59. in press. [Google Scholar] [CrossRef]
- Park, S.; Cho, N.-J. Lipid Membrane Interface Viewpoint: From Viral Entry to Antiviral and Vaccine Development. Langmuir 2023, 39, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Zhang, Y.; Tan, C.; Cai, Y.; Zhang, Y.; Chen, J.; Fu, Y.; Liu, G. In Vitro and in Vivo Evaluation of Thapsigargin as an Antiviral Agent against Transmissible Gastroenteritis Virus. Vet. Res. 2024, 55, 97. [Google Scholar] [CrossRef]
- Tarafdar, P.K.; Chakraborty, H.; Dennison, S.M.; Lentz, B.R. Phosphatidylserine Inhibits and Calcium Promotes Model Membrane Fusion. Biophys. J. 2012, 103, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Omer, A.A.M.; Hinkula, J.; Tran, P.-T.-H.; Melik, W.; Zattarin, E.; Aili, D.; Selegård, R.; Bengtsson, T.; Khalaf, H. Plantaricin NC8 αβ Rapidly and Efficiently Inhibits Flaviviruses and SARS-CoV-2 by Disrupting Their Envelopes. PLoS One 2022, 17, e0278419. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Huang, Y.-H.; Rosengren, K.J.; Franquelim, H.G.; Carvalho, F.A.; Johnson, A.; Sonza, S.; Tachedjian, G.; Castanho, M.A.R.B.; Daly, N.L.; et al. Decoding the Membrane Activity of the Cyclotide Kalata B1: The Importance of Phosphatidylethanolamine Phospholipids and Lipid Organization on Hemolytic and Anti-HIV Activities. J. Biol. Chem. 2011, 286, 24231–24241. [Google Scholar] [CrossRef] [PubMed]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef]
- Sardar, A.; Lahiri, A.; Kamble, M.; Mallick, A.I.; Tarafdar, P.K. Translation of Mycobacterium Survival Strategy to Develop a Lipo-peptide Based Fusion Inhibitor. Angew. Chem. Int. Ed. 2021, 60, 6101–6106. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Tr. Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
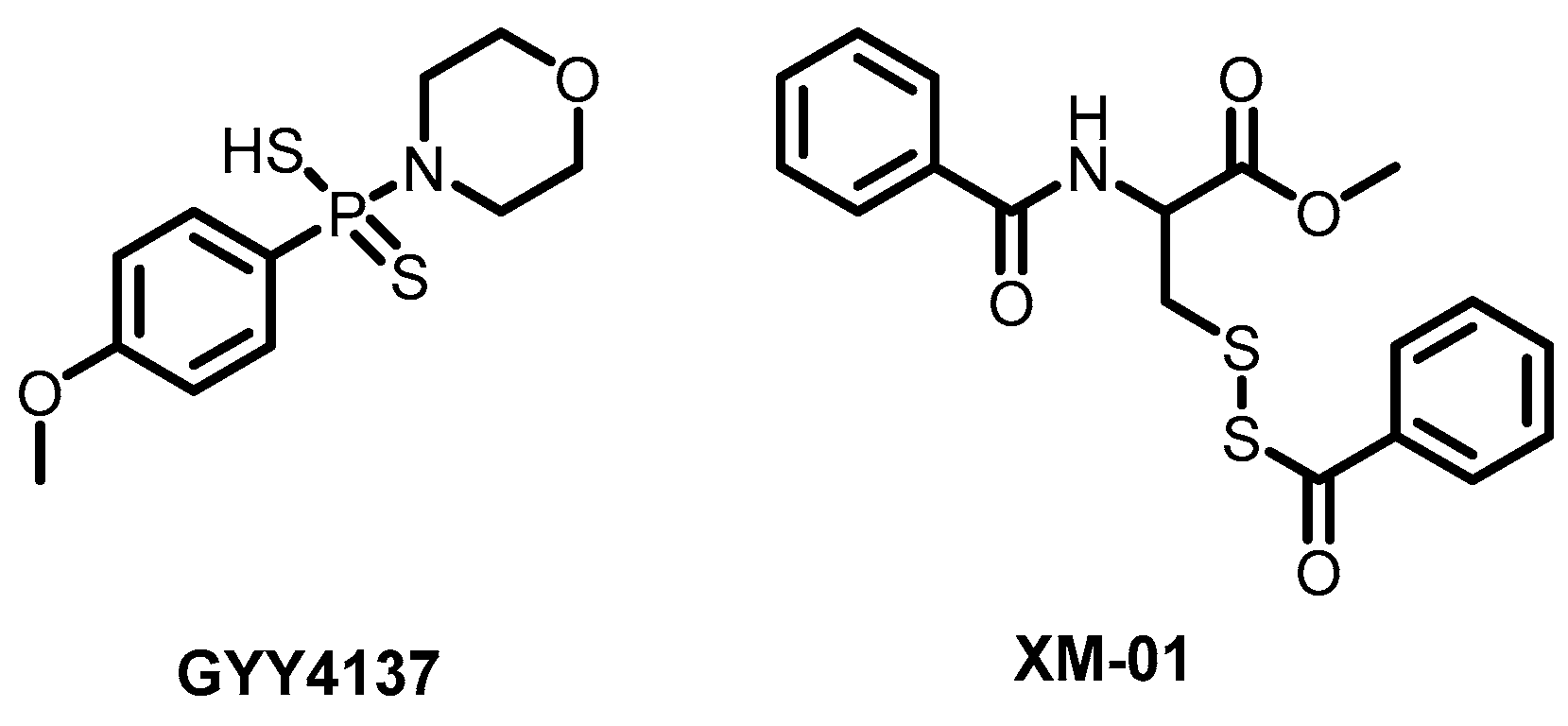
- Haas De Mello, A.H.; Liu, T.; Garofalo, R.P.; Casola, A. Hydrogen Sulfide Donor GYY4137 Rescues NRF2 Activation in Respiratory Syncytial Virus Infection. Antioxidants 2022, 11, 1410. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Brancaleone, V.; Brogi, S.; Gojon, G.; Montanaro, R.; Morales, G.; Testai, L.; Calderone, V. Anti-inflammatory and Antiviral Roles of Hydrogen Sulfide: Rationale for Considering H2S Donors in COVID-19 Therapy. Br. J. Pharmacol. 2020, 177, 4931–4941. [Google Scholar] [CrossRef]
- Pacheco, A.; Buchholz, D.; Monreal, I.; Xu, S.; Imbiakha, B.; Sahler, J.; Jager, M.; Lai, A.; Contreras, E.; Cook, B.; et al. New Class of Broad-Spectrum Antivirals Improves Influenza Virus Vaccine Development. Res. Sq. Prepr. 2022. [Google Scholar] [CrossRef]
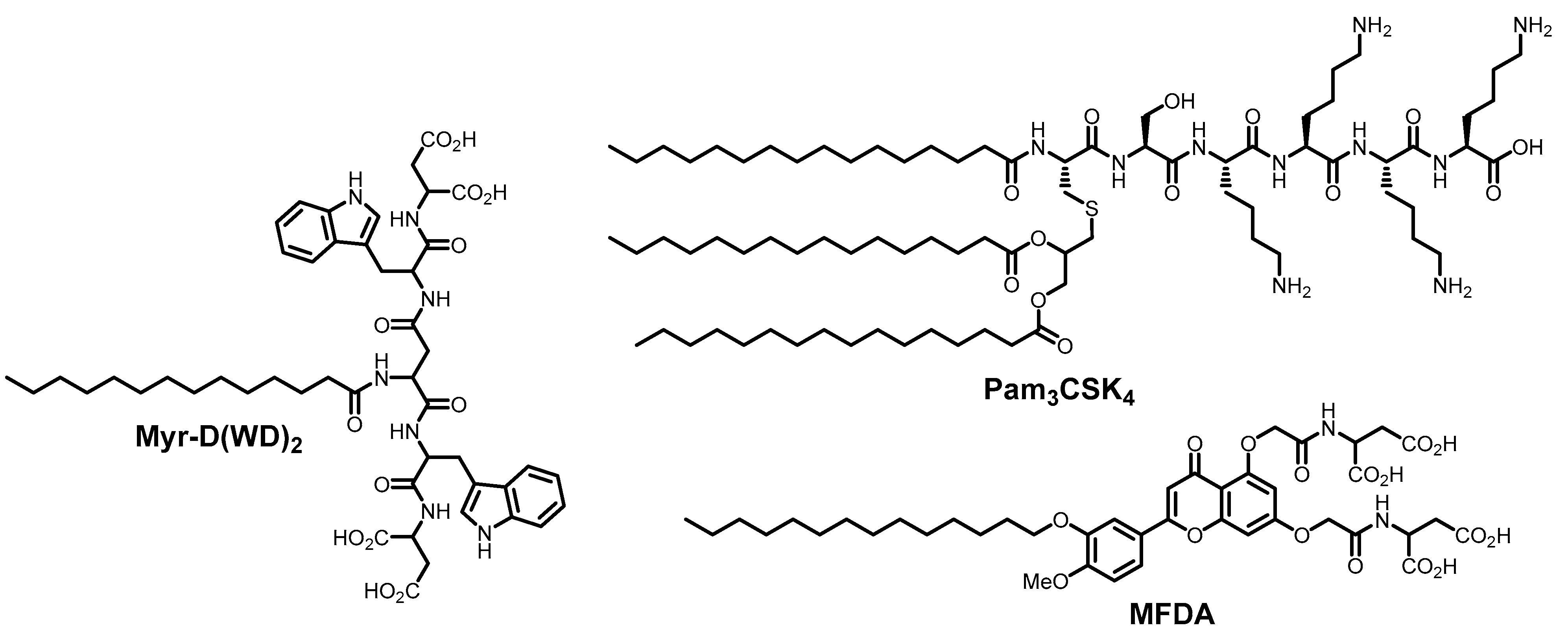
- Sardar, A.; Bhowmick, S.; Kamble, M.; Dewangan, N.; Hazra, B.; Mallick, A.I.; Tarafdar, P.K. De Novo Design of Lipopeptide-Based β-Sheet-Like Self-Assemblies: A Strategy to Develop Fusion Inhibitors as Broad-Spectrum Antivirals. Chem. Eur. J. 2025, 31, e202403039. [Google Scholar] [CrossRef]
- Shekunov, E.V.; Zlodeeva, P.D.; Efimova, S.S.; Muryleva, A.A.; Zarubaev, V.V.; Slita, A.V.; Ostroumova, O.S. Cyclic Lipopeptides as Membrane Fusion Inhibitors against SARS-CoV-2: New Tricks for Old Dogs. Antivir. Res. 2023, 212, 105575. [Google Scholar] [CrossRef]
- Dewangan, N.; Jana, I.D.; Yadav, S.; Sardar, A.; Mallick, A.I.; Mondal, A.; Tarafdar, P.K. Design of Flavonoid-Based Lipid Domains as Fusion Inhibitors to Efficiently Block Coronavirus and Other Enveloped Virus Infection. Small 2025, 21, 2410727. [Google Scholar] [CrossRef]
- Weil, T.; Kirupakaran, A.; Le, M.-H.; Rebmann, P.; Mieres-Perez, J.; Issmail, L.; Conzelmann, C.; Müller, J.A.; Rauch, L.; Gilg, A.; et al. Advanced Molecular Tweezers with Lipid Anchors against SARS-CoV-2 and Other Respiratory Viruses. JACS Au 2022, 2, 2187–2202. [Google Scholar] [CrossRef]
- Mariewskaya, K.A.; Gvozdev, D.A.; Chistov, A.A.; Straková, P.; Huvarová, I.; Svoboda, P.; Kotouček, J.; Ivanov, N.M.; Krasilnikov, M.S.; Zhitlov, M.Y.; et al. Membrane-Targeting Perylenylethynylphenols Inactivate Medically Important Coronaviruses via the Singlet Oxygen Photogeneration Mechanism. Molecules 2023, 28, 6278. [Google Scholar] [CrossRef]
- Griesman, T.; McMillen, C.M.; Negatu, S.G.; Hulahan, J.J.; Whig, K.; Dohnalová, L.; Dittmar, M.; Thaiss, C.A.; Jurado, K.A.; Schultz, D.C.; et al. The Lipopeptide Pam3CSK4 Inhibits Rift Valley Fever Virus Infection and Protects from Encephalitis. PLoS Pathog. 2024, 20, e1012343. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, S.; Wang, Y.; Li, Y.; Wang, X.; Yang, Q. Surfactin Inhibits Membrane Fusion during Invasion of Epithelial Cells by Enveloped Viruses. J. Virol. 2018, 92, e00809-18. [Google Scholar] [CrossRef]
- Johnson, B.A.; Hage, A.; Kalveram, B.; Mears, M.; Plante, J.A.; Rodriguez, S.E.; Ding, Z.; Luo, X.; Bente, D.; Bradrick, S.S.; et al. Peptidoglycan-Associated Cyclic Lipopeptide Disrupts Viral Infectivity. J. Virol. 2019, 93, e01282-19. [Google Scholar] [CrossRef]
- Crovella, S.; De Freitas, L.C.; Zupin, L.; Fontana, F.; Ruscio, M.; Pena, E.P.N.; Pinheiro, I.O.; Calsa Junior, T. Surfactin Bacterial Antiviral Lipopeptide Blocks In Vitro Replication of SARS-CoV-2. App. Microbiol. 2022, 2, 680–687. [Google Scholar] [CrossRef]
- Hoste, A.C.R.; Smeralda, W.; Cugnet, A.; Brostaux, Y.; Deleu, M.; Garigliany, M.; Jacques, P. The Structure of Lipopeptides Impacts Their Antiviral Activity and Mode of Action against SARS-CoV-2 in Vitro. Appl. Environ. Microbiol. 2024, 90, e01036-24. [Google Scholar] [CrossRef]
- Isaia, H.A.; Clerici, N.J.; Brandelli, A. Bacillus Lipopeptides as Versatile Antimicrobial Weapons: Looking toward Antiviral Activity. Crit. Rev. Biotechnol. 2025, 25. in press. [Google Scholar] [CrossRef]
- Chng, C.-P.; Cho, N.-J.; Hsia, K.J.; Huang, C. Role of Membrane Stretch in Adsorption of Antiviral Peptides onto Lipid Membranes and Membrane Pore Formation. Langmuir 2021, 37, 13390–13398. [Google Scholar] [CrossRef]
- Lee, A.G. Lipid Phase Transitions and Phase Diagrams, I. Lipid Phase Transitions. Biochim. Biophys. Acta 1977, 472, 237–281. [Google Scholar] [CrossRef]
- Shekunov, E.V.; Efimova, S.S.; Yudintceva, N.M.; Muryleva, A.A.; Zarubaev, V.V.; Slita, A.V.; Ostroumova, O.S. Plant Alkaloids Inhibit Membrane Fusion Mediated by Calcium and Fragments of MERS-CoV and SARS-CoV/SARS-CoV-2 Fusion Peptides. Biomedicines 2021, 9, 1434. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, X.-W.; Schols, D.; Mori, M.; Botta, B.; Chevigné, A.; Mulinge, M.; Steinmetz, A.; Schmit, J.-C.; Seguin-Devaux, C. Active Components from Cassia abbreviata Prevent HIV-1 Entry by Distinct Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 5052. [Google Scholar] [CrossRef]
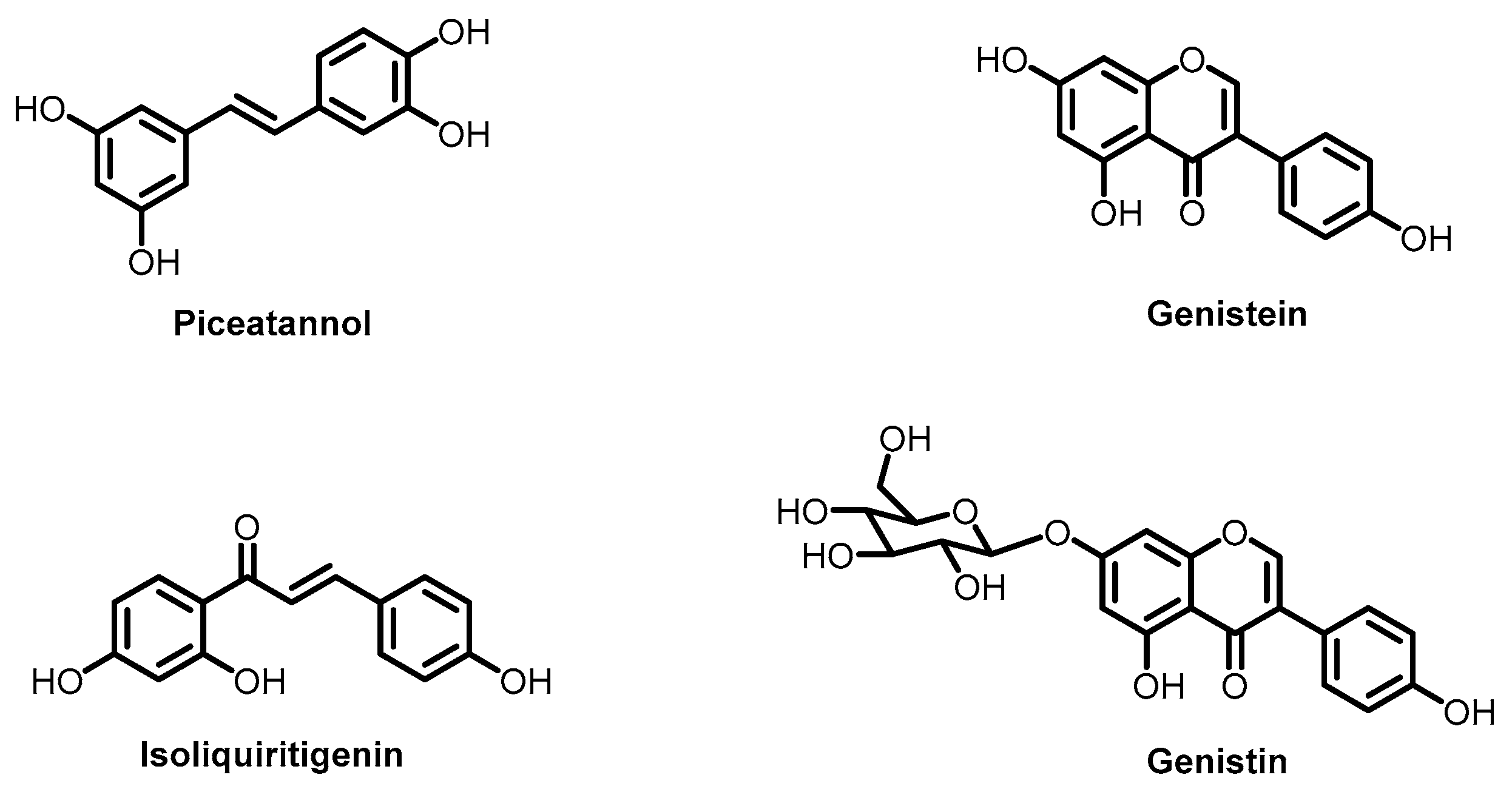
- Zlodeeva, P.D.; Shekunov, E.V.; Ostroumova, O.S.; Efimova, S.S. The Degree of Hydroxylation of Phenolic Rings Determines the Ability of Flavonoids and Stilbenes to Inhibit Calcium-Mediated Membrane Fusion. Nutrients 2023, 15, 1121. [Google Scholar] [CrossRef]
- Shekunov, E.V.; Efimova, S.S.; Kever, L.V.; Ishmanov, T.F.; Ostroumova, O.S. Lipid Selectivity of Membrane Action of the Fragments of Fusion Peptides of Marburg and Ebola Viruses. Int. J. Mol. Sci. 2024, 25, 9901. [Google Scholar] [CrossRef]
- Apaza Ticona, L.; Bermejo, P.; Guerra, J.A.; Abad, M.J.; Beltrán, M.; Martín Lázaro, R.; Alcamí, J.; Bedoya, L.M. Ethanolic Extract of Artemisia campestris subsp. glutinosa (Besser) Batt. Inhibits HIV–1 Replication in Vitro through the Activity of Terpenes and Flavonoids on Viral Entry and NF–κB Pathway. J. Ethnopharmacol. 2020, 263, 113163. [Google Scholar] [CrossRef]
- De Andrea, M.; Ravera, R.; Gioia, D.; Gariglio, M.; Landolfo, S. The Interferon System: An Overview. Eur. J. Paediatr. Neurol. 2002, 6, A41–A46. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croker, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase Inhibits SARS-CoV-2 and Other Coronaviruses by Depleting Membrane Cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef] [PubMed]
- Oeyen, M.; Meyen, E.; Noppen, S.; Claes, S.; Doijen, J.; Vermeire, K.; Süssmuth, R.D.; Schols, D. Labyrinthopeptin A1 Inhibits Dengue and Zika Virus Infection by Interfering with the Viral Phospholipid Membrane. Virology 2021, 562, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.D.; Praest, P.; Thiele, F.; Sadasivam, S.M.; Singethan, K.; Drijfhout, J.W.; Bach, C.; De Boer, S.M.; Lebbink, R.J.; Tao, S.; et al. A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope. Cells 2020, 9, 1989. [Google Scholar] [CrossRef]
- Maiti, B.K. Potential Role of Peptide-Based Antiviral Therapy Against SARS-CoV-2 Infection. ACS Pharmacol. Transl. Sci. 2020, 3, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Molchanova, N.; Herlan, C.; Fortkort, J.; Lin, J.; Figgins, E.; Bopp, N.; Ryan, L.; Chung, D.; Adcock, R.; et al. Potent Antiviral Activity against HSV-1 and SARS-CoV-2 by Antimicrobial Peptoids. Pharmaceuticals 2021, 14, 304. [Google Scholar] [CrossRef]
- Memariani, H.; Memariani, M.; Moravvej, H.; Shahidi-Dadras, M. Melittin: A Venom-Derived Peptide with Promising Anti-Viral Properties. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 5–17. [Google Scholar] [CrossRef]
- Urmi, U.L.; Attard, S.; Vijay, A.K.; Willcox, M.D.P.; Kumar, N.; Islam, S.; Kuppusamy, R. Antiviral Activity of Anthranilamide Peptidomimetics against Herpes Simplex Virus 1 and a Coronavirus. Antibiotics 2023, 12, 1436. [Google Scholar] [CrossRef]
- Tate, P.M.; Mastrodomenico, V.; Cunha, C.; McClure, J.; Barron, A.E.; Diamond, G.; Mounce, B.C.; Kirshenbaum, K. Peptidomimetic Oligomers Targeting Membrane Phosphatidylserine Exhibit Broad Antiviral Activity. ACS Infect. Dis. 2023, 9, 1508–1522. [Google Scholar] [CrossRef]
- Meingast, C.; Heldt, C.L. Arginine-enveloped Virus Inactivation and Potential Mechanisms. Biotechnol. Progr. 2020, 36, e2931. [Google Scholar] [CrossRef]
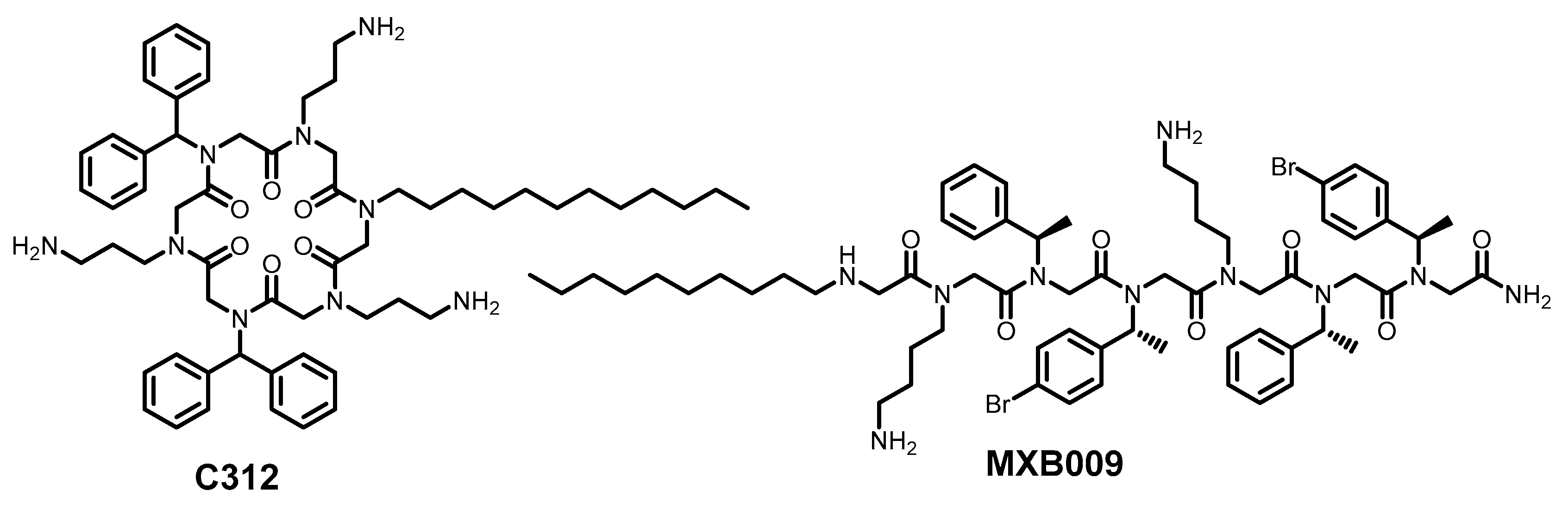
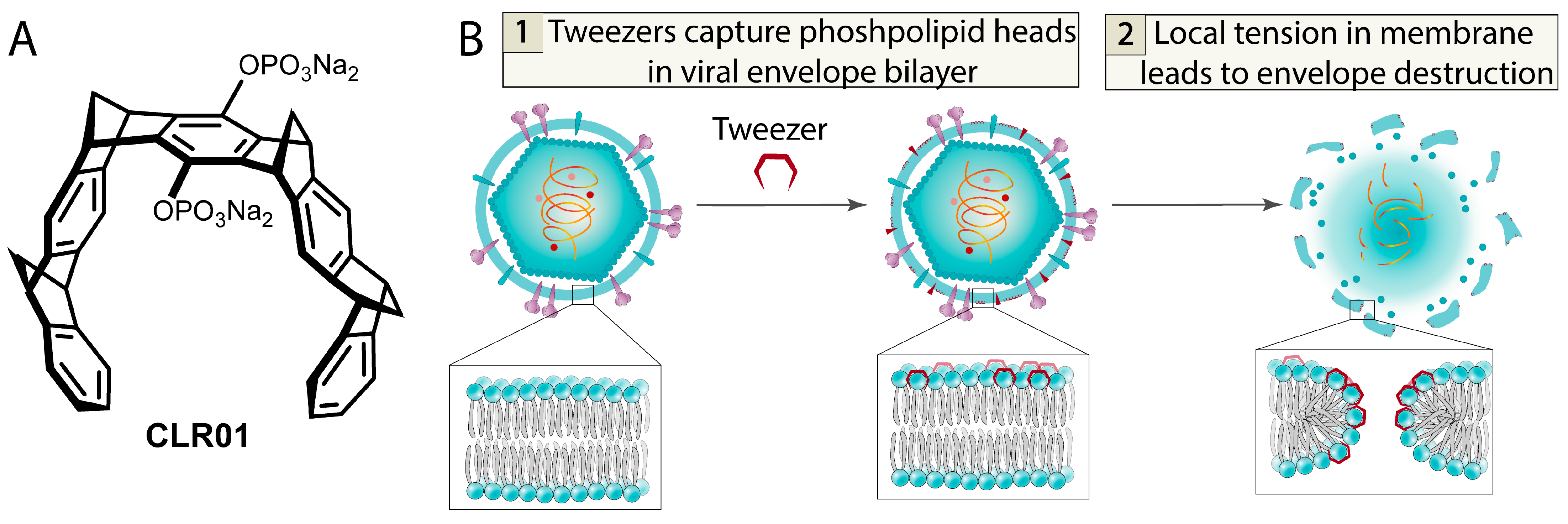
- Weil, T.; Groß, R.; Röcker, A.; Bravo-Rodriguez, K.; Heid, C.; Sowislok, A.; Le, M.-H.; Erwin, N.; Dwivedi, M.; Bart, S.M.; et al. Supramolecular Mechanism of Viral Envelope Disruption by Molecular Tweezers. J. Am. Chem. Soc. 2020, 142, 17024–17038. [Google Scholar] [CrossRef]
- Brenner, S.; Braun, B.; Read, C.; Weil, T.; Walther, P.; Schrader, T.; Münch, J.; Von Einem, J. The Molecular Tweezer CLR01 Inhibits Antibody-Resistant Cell-to-Cell Spread of Human Cytomegalovirus. Viruses 2021, 13, 1685. [Google Scholar] [CrossRef]
- Le, M.-H.; Taghuo, K.E.S.; Schrader, T. Molecular Tweezers—A New Class of Potent Broad-Spectrum Antivirals against Enveloped Viruses. Chem. Commun. 2022, 58, 2954–2966. [Google Scholar] [CrossRef]
- Simon, M.; Veit, M.; Osterrieder, K.; Gradzielski, M. Surfactants–Compounds for Inactivation of SARS-CoV-2 and Other Enveloped Viruses. Curr. Opin. Colloid Interface Sci. 2021, 55, 101479. [Google Scholar] [CrossRef]
- Wainwright, M. Photoinactivation of Viruses. Photochem. Photobiol. Sci. 2004, 3, 406–411. [Google Scholar] [CrossRef]
- Costa, L.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cunha, Â.; Almeida, A. Photodynamic Inactivation of Mammalian Viruses and Bacteriophages. Viruses 2012, 4, 1034–1074. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, L.; Skupin-Mrugalska, P.; Mielcarek, J.; Goslinski, T.; Balzarini, J. Photosensitizers Mediated Photodynamic Inactivation Against Virus Particles. Mini Rev. Med. Chem. 2015, 15, 503–521. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, A.; O’Brien, J.M.; Senge, M.O. Trends and Targets in Antiviral Phototherapy. Photochem. Photobiol. Sci. 2019, 18, 2565–2612. [Google Scholar] [CrossRef]
- Conrado, P.C.V.; Sakita, K.M.; Arita, G.S.; Galinari, C.B.; Gonçalves, R.S.; Lopes, L.D.G.; Lonardoni, M.V.C.; Teixeira, J.J.V.; Bonfim-Mendonça, P.S.; Kioshima, E.S. A Systematic Review of Photodynamic Therapy as an Antiviral Treatment: Potential Guidance for Dealing with SARS-CoV-2. Photodiagn. Photodyn. Ther. 2021, 34, 102221. [Google Scholar] [CrossRef]
- Willis, J.A.; Cheburkanov, V.; Kassab, G.; Soares, J.M.; Blanco, K.C.; Bagnato, V.S.; Yakovlev, V.V. Photodynamic Viral Inactivation: Recent Advances and Potential Applications. Appl. Phys. Rev. 2021, 8, 021315. [Google Scholar] [CrossRef] [PubMed]
- Mariewskaya, K.A.; Tyurin, A.P.; Chistov, A.A.; Korshun, V.A.; Alferova, V.A.; Ustinov, A.V. Photosensitizing Antivirals. Molecules 2021, 26, 3971. [Google Scholar] [CrossRef] [PubMed]
- Kunstek, H.; Vreken, F.; Keita, A.; Hamblin, M.R.; Dumarçay, F.; Varbanov, M. Aspects of Antiviral Strategies Based on Different Phototherapy Approaches: Hit by the Light. Pharmaceuticals 2022, 15, 858. [Google Scholar] [CrossRef]
- Sadraeian, M.; Zhang, L.; Aavani, F.; Biazar, E.; Jin, D. Photodynamic Viral Inactivation Assisted by Photosensitizers. Mater. Today Phys. 2022, 28, 100882. [Google Scholar] [CrossRef]
- Bartosińska, J.; Kowalczuk, D.; Szczepanik-Kułak, P.; Kwaśny, M.; Krasowska, D. A Review of Photodynamic Therapy for the Treatment of Viral Skin Diseases. Antivir. Ther. 2025, 30, 13596535251331728. [Google Scholar] [CrossRef]
- North, J.; Neyndorff, H.; Levy, J.G. New Trends in Photobiology. J. Photochem. Photobiol. B 1993, 17, 99–108. [Google Scholar] [CrossRef]
- Mendonça, D.A.; Cadima-Couto, I.; Buga, C.C.; Arnaut, Z.A.; Schaberle, F.A.; Arnaut, L.G.; Castanho, M.A.R.B.; Cruz-Oliveira, C. Repurposing Anti-Cancer Porphyrin Derivative Drugs to Target SARS-CoV-2 Envelope. Biomed. Pharmacother. 2024, 176, 116768. [Google Scholar] [CrossRef]
- Plastiras, O.-E.; Bouquet, P.; Raczkiewicz, I.; Belouzard, S.; Martin De Fourchambault, E.; Dhainaut, J.; Dacquin, J.-P.; Goffard, A.; Volkringer, C. Virucidal Activity of Porphyrin-Based Metal-Organic Frameworks against Highly Pathogenic Coronaviruses and Hepatitis C Virus. Mater. Today Bio 2024, 28, 101165. [Google Scholar] [CrossRef]
- Holoubek, J.; Salát, J.; Kotouček, J.; Kastl, T.; Vancová, M.; Huvarová, I.; Bednář, P.; Bednářová, K.; Růžek, D.; Renčiuk, D.; et al. Antiviral Activity of Porphyrins and Porphyrin-like Compounds against Tick-Borne Encephalitis Virus: Blockage of the Viral Entry/Fusion Machinery by Photosensitization-Mediated Destruction of the Viral Envelope. Antiv. Res. 2024, 221, 105767. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Cokarić Brdovčak, M.; Djaković, L.; Bertović, I.; Knežević, K.; Lončarić, M.; Jurak Begonja, A.; Malatesti, N. Photodynamic Inhibition of Herpes Simplex Virus 1 Infection by Tricationic Amphiphilic Porphyrin with a Long Alkyl Chain. Pharmaceutics 2023, 15, 956. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-M.; Wu, B.; Huang, G.-G.; Feng, C.-L.; Wang, X.-H.; Wang, H.-Y.; Wu, Y.-W.; Tang, W. Hemin Protects against Zika Virus Infection by Disrupting Virus-Endosome Fusion. Antivir. Res. 2022, 203, 105347. [Google Scholar] [CrossRef]
- Meunier, T.; Desmarets, L.; Bordage, S.; Bamba, M.; Hervouet, K.; Rouillé, Y.; François, N.; Decossas, M.; Sencio, V.; Trottein, F.; et al. A Photoactivable Natural Product with Broad Antiviral Activity against Enveloped Viruses, Including Highly Pathogenic Coronaviruses. Antimicrob. Agents Chemother. 2022, 66, e015821. [Google Scholar] [CrossRef]
- Lu, S.; Pan, X.; Chen, D.; Xie, X.; Wu, Y.; Shang, W.; Jiang, X.; Sun, Y.; Fan, S.; He, J. Broad-Spectrum Antivirals of Protoporphyrins Inhibit the Entry of Highly Pathogenic Emerging Viruses. Bioorg. Chem. 2021, 107, 104619. [Google Scholar] [CrossRef]
- Chen, D.; Lu, S.; Yang, G.; Pan, X.; Fan, S.; Xie, X.; Chen, Q.; Li, F.; Li, Z.; Wu, S.; et al. The Seafood Musculus senhousei Shows Anti-Influenza A Virus Activity by Targeting Virion Envelope Lipids. Biochem. Pharmacol. 2020, 177, 113982. [Google Scholar] [CrossRef]
- Ries, A.S.; Cargnelutti, J.F.; Basso, G.; Acunha, T.V.; Iglesias, B.A.; Flores, E.F.; Weiblen, R. Water-Soluble Tetra-Cationic Porphyrins Display Virucidal Activity against Bovine adenovirus and Bovine alphaherpesvirus 1. Photodiagn. Photodyn. Ther. 2020, 31, 101947. [Google Scholar] [CrossRef]
- Sengupta, D.; Timilsina, U.; Mazumder, Z.H.; Mukherjee, A.; Ghimire, D.; Markandey, M.; Upadhyaya, K.; Sharma, D.; Mishra, N.; Jha, T.; et al. Dual Activity of Amphiphilic Zn(II) Nitroporphyrin Derivatives as HIV-1 Entry Inhibitors and in Cancer Photodynamic Therapy. Eur. J. Med. Chem. 2019, 174, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.; Bartolomeu, M.; Vieira, C.; Gomes, A.T.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Almeida, A. Photoinactivation of Phage Phi6 as a SARS-CoV-2 Model in Wastewater: Evidence of Efficacy and Safety. Microorganisms 2022, 10, 659. [Google Scholar] [CrossRef]
- Savelyeva, I.O.; Zhdanova, K.A.; Gradova, M.A.; Gradov, O.V.; Bragina, N.A. Cationic Porphyrins as Antimicrobial and Antiviral Agents in Photodynamic Therapy. Curr. Iss. Mol. Biol. 2023, 45, 9793–9822. [Google Scholar] [CrossRef] [PubMed]
- Rywkin, S.; Ben-Hur, E.; Malik, Z.; Prince, A.M.; Li, Y.-S.; Kenney, M.E.; Oleinick, N.L.; Horowitz, B. New Phthalocyanines for Photodynamic Virus Inactivation in Red Blood Cell Concentrates. Photochem. Photobiol. 1994, 60, 165–170. [Google Scholar] [CrossRef]
- Da Silva Santos, P.S.; Da Fonseca Orcina, B.; Machado, R.R.G.; Vilhena, F.V.; Da Costa Alves, L.M.; Zangrando, M.S.R.; De Oliveira, R.C.; Soares, M.Q.S.; Simão, A.N.C.; Pietro, E.C.I.N.; et al. Beneficial Effects of a Mouthwash Containing an Antiviral Phthalocyanine Derivative on the Length of Hospital Stay for COVID-19: Randomised Trial. Sci. Rep. 2021, 11, 19937. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, F.V.; Orcina, B.D.F.; Lemos, L.; Less, J.C.F.; Pinto, I.; Santos, P.S.D.S. Elimination of SARS-CoV-2 in Nasopharynx and Oropharynx after Use of an Adjuvant Gargling and Rinsing Protocol with an Antiseptic Mouthwash. Einstein 2021, 19, eCE6999. [Google Scholar] [CrossRef]
- Efimov, A.; Dagallier, C.; Frochot, C.; Myrzakhmetov, B.; Arnoux, P.; Heinonen, T.; Mannerström, M.; Toimela, T.; Ahmed, Z.; Audibert, J.F.; et al. LASU: An Efficient and Stable Phthalocyanine Dye with Tolerable Safety Profile for Self-Disinfecting Anti-COVID Textiles Activated by Ambient Light. Photodiagn. Photodyn. Ther. 2024, 45, 103978. [Google Scholar] [CrossRef]
- Vilhelmova-Ilieva, N.; Mantareva, V.; Braikova, D.; Iliev, I. Photodynamic Inactivation of Human Herpes Virus in Vitro with Ga(III) and Zn(II) Phthalocyanines. Viruses 2024, 16, 1937. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.R.; Parks, R.J. Curcumin as an Antiviral Agent. Viruses 2020, 12, 1242. [Google Scholar] [CrossRef]
- Keil, S.D.; Ragan, I.; Yonemura, S.; Hartson, L.; Dart, N.K.; Bowen, R. Inactivation of Severe Acute Respiratory Syndrome Coronavirus 2 in Plasma and Platelet Products Using a Riboflavin and Ultraviolet Light-based Photochemical Treatment. Vox Sanguinis 2020, 115, 495–501. [Google Scholar] [CrossRef]
- Ragan, I.; Hartson, L.; Pidcoke, H.; Bowen, R.; Goodrich, R. Pathogen Reduction of SARS-CoV-2 Virus in Plasma and Whole Blood Using Riboflavin and UV Light. PLoS One 2020, 15, e0233947. [Google Scholar] [CrossRef]
- Farah, N.; Chin, V.K.; Chong, P.P.; Lim, W.F.; Lim, C.W.; Basir, R.; Chang, S.K.; Lee, T.Y. Riboflavin as a Promising Antimicrobial Agent? A Multi-Perspective Review. Curr. Res. Microb. Sci. 2022, 3, 100111. [Google Scholar] [CrossRef]
- Crocker, L.B.; Lee, J.H.; Mital, S.; Mills, G.C.; Schack, S.; Bistrović-Popov, A.; Franck, C.O.; Mela, I.; Kaminski, C.F.; Christie, G.; et al. Tuning Riboflavin Derivatives for Photodynamic Inactivation of Pathogens. Sci. Rep. 2022, 12, 6580. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, Q.; Tian, L.; Huang, Z.; Tang, Y.; Wen, Y.; Yu, F.; Yan, X.; Zhao, Y.; Wu, Z.; et al. Advancements and Future Prospects in Hypocrellins Production and Modification for Photodynamic Therapy. Fermentation 2024, 10, 559. [Google Scholar] [CrossRef]
- Deng, H.; Liu, X.; Xie, J.; Yin, R.; Huang, N.; Gu, Y.; Zhao, J. Quantitative and Site-Directed Chemical Modification of Hypocrellins toward Direct Drug Delivery and Effective Photodynamic Activity. J. Med. Chem. 2012, 55, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Alferova, V.A.; Mikhnovets, I.E.; Chistov, A.A.; Korshun, V.A.; Tyurin, A.P.; Ustinov, A.V. Perylene as a Controversial Antiviral Scaffold. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2022; Volume 58, pp. 93–156. ISBN 978-0-323-98893-3. [Google Scholar]
- Delcanale, P.; Uriati, E.; Mariangeli, M.; Mussini, A.; Moreno, A.; Lelli, D.; Cavanna, L.; Bianchini, P.; Diaspro, A.; Abbruzzetti, S.; et al. The Interaction of Hypericin with SARS-CoV-2 Reveals a Multimodal Antiviral Activity. ACS Appl. Mater. Interfaces 2022, 14, 14025–14032. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, Y.; Xu, C.; Gao, J.; Feng, Y.; Wu, Q. Disinfection of Influenza a Viruses by Hypocrellin A-Mediated Photodynamic Inactivation. Photodiagn. Photodyn. Ther. 2023, 43, 103674. [Google Scholar] [CrossRef] [PubMed]
- Orlov, A.A.; Chistov, A.A.; Kozlovskaya, L.I.; Ustinov, A.V.; Korshun, V.A.; Karganova, G.G.; Osolodkin, D.I. Rigid Amphipathic Nucleosides Suppress Reproduction of the Tick-Borne Encephalitis Virus. Med. Chem. Commun. 2016, 7, 495–499. [Google Scholar] [CrossRef]
- Aralov, A.V.; Proskurin, G.V.; Orlov, A.A.; Kozlovskaya, L.I.; Chistov, A.A.; Kutyakov, S.V.; Karganova, G.G.; Palyulin, V.A.; Osolodkin, D.I.; Korshun, V.A. Perylenyltriazoles Inhibit Reproduction of Enveloped Viruses. Eur. J. Med. Chem. 2017, 138, 293–299. [Google Scholar] [CrossRef]
- Speerstra, S.; Chistov, A.A.; Proskurin, G.V.; Aralov, A.V.; Ulashchik, E.A.; Streshnev, P.P.; Shmanai, V.V.; Korshun, V.A.; Schang, L.M. Antivirals Acting on Viral Envelopes via Biophysical Mechanisms of Action. Antivir. Res. 2018, 149, 164–173. [Google Scholar] [CrossRef]
- Chistov, A.A.; Orlov, A.A.; Streshnev, P.P.; Slesarchuk, N.A.; Aparin, I.O.; Rathi, B.; Brylev, V.A.; Kutyakov, S.V.; Mikhura, I.V.; Ustinov, A.V.; et al. Compounds Based on 5-(Perylen-3-ylethynyl)Uracil Scaffold: High Activity against Tick-Borne Encephalitis Virus and Non-Specific Activity against Enterovirus, A. Eur. J. Med. Chem. 2019, 171, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Slesarchuk, N.A.; Khvatov, E.V.; Chistov, A.A.; Proskurin, G.V.; Nikitin, T.D.; Lazarevich, A.I.; Ulanovskaya, A.A.; Ulashchik, E.A.; Orlov, A.A.; Jegorov, A.V.; et al. Simplistic Perylene-Related Compounds as Inhibitors of Tick-Borne Encephalitis Virus Reproduction. Bio. Med. Chem. Lett. 2020, 30, 127100. [Google Scholar] [CrossRef]
- Mikhnovets, I.E.; Holoubek, J.; Panina, I.S.; Kotouček, J.; Gvozdev, D.A.; Chumakov, S.P.; Krasilnikov, M.S.; Zhitlov, M.Y.; Gulyak, E.L.; Chistov, A.A.; et al. Alkyl Derivatives of Perylene Photosensitizing Antivirals: Towards Understanding the Influence of Lipophilicity. Int. J. Mol. Sci. 2023, 24, 16483. [Google Scholar] [CrossRef]
- Straková, P.; Bednář, P.; Kotouček, J.; Holoubek, J.; Fořtová, A.; Svoboda, P.; Štefánik, M.; Huvarová, I.; Šimečková, P.; Mašek, J.; et al. Antiviral Activity of Singlet Oxygen-Photogenerating Perylene Compounds against SARS-CoV-2: Interaction with the Viral Envelope and Photodynamic Virion Inactivation. Virus Res. 2023, 334, 199158. [Google Scholar] [CrossRef]
- Krasilnikov, M.S.; Mazur, R.V.; Chumakov, S.P.; Denisov, V.S.; Goldenberg, E.A.; Nikolaenko, Y.I.; Bersenev, E.A.; Nikitin, T.D.; Orinicheva, P.S.; Brylev, V.A.; et al. Donor-Acceptor (Perylenethienyl)ethylenes as Singlet Oxygen-Photogenerating Viral Inhibitors. ChemBioChem 2025, 26, e202401019. [Google Scholar] [CrossRef] [PubMed]
- Maryewski, X.A.; Krasilnikov, M.S.; Straková, P.; Holoubek, J.; Frčková, T.; Panina, I.S.; Krylov, N.A.; Gvozdev, D.A.; Denisov, V.S.; Semenov, A.N.; et al. Membrane-Active Singlet Oxygen Photogenerators as a Paradigm for Broad-Spectrum Antivirals: The Case of Halogenated (BOron)-DIPYrromethenes. ACS Appl. Mater. Interfaces 2025, 17, 4502–4528. [Google Scholar] [CrossRef]
- Carpenter, B.; Situ, X.; Scholle, F.; Bartelmess, J.; Weare, W.; Ghiladi, R. Antiviral, Antifungal and Antibacterial Activities of a BODIPY-Based Photosensitizer. Molecules 2015, 20, 10604–10621. [Google Scholar] [CrossRef] [PubMed]
- Stoll, K.R.; Scholle, F.; Zhu, J.; Zhang, X.; Ghiladi, R.A. BODIPY-Embedded Electrospun Materials in Antimicrobial Photodynamic Inactivation. Photochem. Photobiol. Sci. 2019, 18, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Schneider, J.E.; Dittmer, D.P. Methylene Blue Photoinactivation of RNA Viruses. Antivir. Res. 2004, 61, 141–151. [Google Scholar] [CrossRef]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A Mechanistic Paradigm for Broad-Spectrum Antivirals That Target Virus-Cell Fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef]
- Rubekina, A.A.; Kamzeeva, P.N.; Alferova, V.A.; Shustova, E.Y.; Kolpakova, E.S.; Yakovchuk, E.V.; Karpova, E.V.; Borodulina, M.O.; Belyaev, E.S.; Khrulev, A.A.; et al. Hydrophobic Rose Bengal Derivatives Exhibit Submicromolar-to-Subnanomolar Activity against Enveloped Viruses. Biomolecules 2022, 12, 1609. [Google Scholar] [CrossRef]
- Cagno, V.; Medaglia, C.; Cerny, A.; Cerny, T.; Zwygart, A.C.-A.; Cerny, E.; Tapparel, C. Methylene Blue Has a Potent Antiviral Activity against SARS-CoV-2 and H1N1 Influenza Virus in the Absence of UV-Activation in Vitro. Sci. Rep. 2021, 11, 14295. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, M.; Ding, Q.; Zhang, Z.; Gong, W.; Yuan, Y.; Miao, X.; Ma, H.; Hong, X.; Hu, W.; et al. Highly Twisted Conformation Thiopyrylium Photosensitizers for In Vivo Near Infrared-II Imaging and Rapid Inactivation of Coronavirus. Angew. Chem. Int. Ed. 2023, 62, e202214875. [Google Scholar] [CrossRef]
- Zaharieva, M.M.; Foka, P.; Karamichali, E.; Kroumov, A.D.; Philipov, S.; Ilieva, Y.; Kim, T.C.; Podlesniy, P.; Manasiev, Y.; Kussovski, V.; et al. Photodynamic Inactivation of Bovine Coronavirus with the Photosensitizer Toluidine Blue O. Viruses 2024, 16, 48. [Google Scholar] [CrossRef]
- Wu, M.; Gu, M.; Leung, J.; Li, X.; Yuan, Y.; Shen, C.; Wang, L.; Zhao, E.; Chen, S. A Membrane-Targeting Photosensitizer with Aggregation-Induced Emission Characteristics for Highly Efficient Photodynamic Combat of Human Coronaviruses. Small 2021, 17, 2101770. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, E.Y.; Liu, Y.; Jin, H.; Liu, X.; Wu, X.; Zhang, M.; Feng, N.; Huang, P.; Zhang, H.; et al. Molecular Engineering of AIE Photosensitizers for Inactivation of Rabies Virus. Small 2023, 19, 2303542. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Kraus, G.A. Photosensitization Is Required for Inactivation of Equine Infectious Anemia Virus by Hypericin. Photochem. Photobiol. 1991, 53, 169–174. [Google Scholar] [CrossRef]
- Bacellar, I.O.L.; Oliveira, M.C.; Dantas, L.S.; Costa, E.B.; Junqueira, H.C.; Martins, W.K.; Durantini, A.M.; Cosa, G.; Di Mascio, P.; Wainwright, M.; et al. Photosensitized Membrane Permeabilization Requires Contact-Dependent Reactions between Photosensitizer and Lipids. J. Am. Chem. Soc. 2018, 140, 9606–9615. [Google Scholar] [CrossRef]
- Bacellar, I.O.L.; Baptista, M.S. Mechanisms of Photosensitized Lipid Oxidation and Membrane Permeabilization. ACS Omega 2019, 4, 21636–21646. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxygen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef]
- Jori, G.; Brown, S.B. Photosensitized Inactivation of Microorganisms. Photochem. Photobiol. Sci. 2004, 3, 403–405. [Google Scholar] [CrossRef]
- Xie, M.; Derks, M.G.N.; Koch, E.H.W.; Van Boven, C.B.; Janlad, M.; Bagheri, B.; Xu, Z.; Kovryzhenko, D.; Van Walree, C.A.; Sobota, A.; et al. Structure and pH Dependence of Membranolytic Mechanisms by Truncated Oxidized Phospholipids. J. Am. Chem. Soc. 2025, 147, 9175–9189. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Hollmann, A.; Lee, J.; Santos, N.C.; Jung, M.E.; Lee, B. The Rigid Amphipathic Fusion Inhibitor dUY11 Acts through Photosensitization of Viruses. J. Virol. 2014, 88, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Castanho, M.A.R.B.; Lee, B.; Santos, N.C. Singlet Oxygen Effects on Lipid Membranes: Implications for the Mechanism of Action of Broad-Spectrum Viral Fusion Inhibitors. Biochem. J. 2014, 459, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Gonçalves, S.; Augusto, M.T.; Castanho, M.A.R.B.; Lee, B.; Santos, N.C. Effects of Singlet Oxygen Generated by a Broad-Spectrum Viral Fusion Inhibitor on Membrane Nanoarchitecture. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1163–1167. [Google Scholar] [CrossRef]
- Zeng, L.; Wang, M.-D.; Ming, S.-L.; Li, G.-L.; Yu, P.-W.; Qi, Y.-L.; Jiang, D.-W.; Yang, G.-Y.; Wang, J.; Chu, B.-B. An Effective Inactivant Based on Singlet Oxygen-Mediated Lipid Oxidation Implicates a New Paradigm for Broad-Spectrum Antivirals. Redox Biol. 2020, 36, 101601. [Google Scholar] [CrossRef]
- Chistov, A.A.; Chumakov, S.P.; Mikhnovets, I.E.; Nikitin, T.D.; Slesarchuk, N.A.; Uvarova, V.I.; Rubekina, A.A.; Nikolaeva, Y.V.; Radchenko, E.V.; Khvatov, E.V.; et al. 5-(Perylen-3-ylethynyl)Uracil as an Antiviral Scaffold: Potent Suppression of Enveloped Virus Reproduction by 3-Methyl Derivatives in Vitro. Antivir. Res. 2023, 209, 105508. [Google Scholar] [CrossRef]
- Wang, J.; Liu, G.; Leung, K.; Loffroy, R.; Lu, P.-X.; Wáng, Y.X. Opportunities and Challenges of Fluorescent Carbon Dots in Translational Optical Imaging. Curr. Pharm. Design 2015, 21, 5401–5416. [Google Scholar] [CrossRef]
- Sengupta, D.; Rai, M.; Hoque Mazumdar, Z.; Sharma, D.; Malabika Singha, K.; Pandey, P.; Gaur, R. Two Cationic Meso-Thiophenium Porphyrins and Their Zinc-Complexes as Anti-HIV-1 and Antibacterial Agents under Non-Photodynamic Therapy (PDT) Conditions. Bioorg. Med. Chem. Lett. 2022, 65, 128699. [Google Scholar] [CrossRef] [PubMed]
- Ardebili, A.; Pouriayevali, M.H.; Aleshikh, S.; Zahani, M.; Ajorloo, M.; Izanloo, A.; Siyadatpanah, A.; Razavi Nikoo, H.; Wilairatana, P.; Coutinho, H.D.M. Antiviral Therapeutic Potential of Curcumin: An Update. Molecules 2021, 26, 6994. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hovan, A.; Jutková, A.; Kruglik, S.G.; Jancura, D.; Miskovsky, P.; Bánó, G. Phosphorescence Kinetics of Singlet Oxygen Produced by Photosensitization in Spherical Nanoparticles. Part II. The Case of Hypericin-Loaded Low-Density Lipoprotein Particles. J. Phys. Chem. B 2018, 122, 5154–5160. [Google Scholar] [CrossRef]
- Gattuso, H.; Marazzi, M.; Dehez, F.; Monari, A. Deciphering the Photosensitization Mechanisms of Hypericin towards Biological Membranes. Phys. Chem. Chem. Phys. 2017, 19, 23187–23193. [Google Scholar] [CrossRef]
- Mariangeli, M.; Moreno, A.; Delcanale, P.; Abbruzzetti, S.; Diaspro, A.; Viappiani, C.; Bianchini, P. Insights on the Mechanical Properties of SARS-CoV-2 Particles and the Effects of the Photosensitizer Hypericin. Int. J. Mol. Sci. 2024, 25, 8724. [Google Scholar] [CrossRef]
- Akasov, R.A.; Chepikova, O.E.; Pallaeva, T.N.; Gorokhovets, N.V.; Siniavin, A.E.; Gushchin, V.A.; Savvateeva, L.V.; Vinokurov, I.A.; Khochenkov, D.A.; Zamyatnin, A.A.; et al. Evaluation of Molecular Mechanisms of Riboflavin Anti-COVID-19 Action Reveals Anti-Inflammatory Efficacy Rather than Antiviral Activity. Biochim. Biophys. Acta 2024, 1868, 130582. [Google Scholar] [CrossRef]
- Correa Sierra, C.B.; Schang, L.M. On the Sensitivity of the Virion Envelope to Lipid Peroxidation. Microbiol. Spectr. 2022, 10, e0300922. [Google Scholar] [CrossRef] [PubMed]
- Szabó, D.; Crowe, A.; Mamotte, C.; Strappe, P. Natural Products as a Source of Coronavirus Entry Inhibitors. Front. Cell. Infect. Microbiol. 2024, 14, 1353971. [Google Scholar] [CrossRef]
- Agarwal, G.; Gabrani, R. Antiviral Peptides: Identification and Validation. Int. J. Pept. Res. Ther. 2021, 27, 149–168. [Google Scholar] [CrossRef]
- Wu, R.; Wang, L.; Kuo, H.-C.D.; Shannar, A.; Peter, R.; Chou, P.J.; Li, S.; Hudlikar, R.; Liu, X.; Liu, Z.; et al. An Update on Current Therapeutic Drugs Treating COVID-19. Curr. Pharmacol. Rep. 2020, 6, 56–70. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and Peptide-Based Inhibitors of SARS-CoV-2 Entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J. Membr. Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Hu, S.; Gao, J. Discovering Drugs to Treat Coronavirus Disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gao, Z.; Feng, X.; Cheng, L.; Liu, N.; Liu, C.; Han, S.; Yang, Q.; Zou, Q.; Chong, H.; et al. Development of Potent Pan-coronavirus Fusion Inhibitors with a New Design Strategy. MedComm 2024, 5, e666. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of Potent Membrane Fusion Inhibitors against SARS-CoV-2, an Emerging Coronavirus with High Fusogenic Activity. J. Virol. 2020, 94, e00635-20. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; Kreutzberger, A.J.B.; Ojha, R.; Kuivanen, S.; Couoh-Cardel, S.; Muratcioglu, S.; Eisen, T.J.; White, K.I.; Held, R.G.; et al. Nanomolar Inhibition of SARS-CoV-2 Infection by an Unmodified Peptide Targeting the Prehairpin Intermediate of the Spike Protein. Proc. Natl. Acad. Sci. USA 2022, 119, e2210990119. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Xia, S.; Tu, J.; Zheng, L.; Wang, Q.; Jiang, S.; Wang, C. Design of MERS-CoV Entry Inhibitory Short Peptides Based on Helix-Stabilizing Strategies. Bioorg. Med. Chem. Lett. 2024, 97, 129569. [Google Scholar] [CrossRef]
- Tsai, P.-H.; Sun, J.-R.; Chien, Y.; Chan, M.S.; Khor, W.; Yang, H.-C.; Huang, C.-H.; Hsiung, C.-N.; Hwa, T.-Y.; Lin, Y.-Y.; et al. Modifications of Lipid Pathways Restrict SARS-CoV-2 Propagation in Human Induced Pluripotent Stem Cell-Derived 3D Airway Organoids. J. Adv. Res. 2024, 60, 127–140. [Google Scholar] [CrossRef]
- Bird, G.H.; Patten, J.J.; Zavadoski, W.; Barucci, N.; Godes, M.; Moyer, B.M.; Owen, C.D.; DaSilva-Jardine, P.; Neuberg, D.S.; Bowen, R.A.; et al. A Stapled Lipopeptide Platform for Preventing and Treating Highly Pathogenic Viruses of Pandemic Potential. Nat. Commun. 2024, 15, 274. [Google Scholar] [CrossRef] [PubMed]
- Shaban, M.S.; Mayr-Buro, C.; Meier-Soelch, J.; Albert, B.V.; Schmitz, M.L.; Ziebuhr, J.; Kracht, M. Thapsigargin: Key to New Host-Directed Coronavirus Antivirals? Tr. Pharmacol. Sci. 2022, 43, 557–568. [Google Scholar] [CrossRef]
- Al Otaibi, A.; Al Shaikh Mubarak, S.; Al Hejji, F.; Almasaud, A.; Al Jami, H.; Iqbal, J.; Al Qarni, A.; Harbi, N.K.A.; Bakillah, A. Thapsigargin and Tunicamycin Block SARS-CoV-2 Entry into Host Cells via Differential Modulation of Unfolded Protein Response (UPR), AKT Signaling, and Apoptosis. Cells 2024, 13, 769. [Google Scholar] [CrossRef]
- Sasaki, J.; Sato, A.; Sasaki, M.; Okabe, I.; Kodama, K.; Otsuguro, S.; Yasuda, K.; Kojima, H.; Orba, Y.; Sawa, H.; et al. X-206 Exhibits Broad-Spectrum Anti-β-Coronavirus Activity, Covering SARS-CoV-2 Variants and Drug-Resistant Isolates. Antiv. Res. 2024, 232, 106039. [Google Scholar] [CrossRef]
- Jäntti, M.H.; Jackson, S.N.; Kuhn, J.; Parkkinen, I.; Sree, S.; Hinkle, J.J.; Jokitalo, E.; Deterding, L.J.; Harvey, B.K. Palmitate and Thapsigargin Have Contrasting Effects on ER Membrane Lipid Composition and ER Proteostasis in Neuronal Cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159219. [Google Scholar] [CrossRef]
- Al-Beltagi, S.; Preda, C.A.; Goulding, L.V.; James, J.; Pu, J.; Skinner, P.; Jiang, Z.; Wang, B.L.; Yang, J.; Banyard, A.C.; et al. Thapsigargin Is a Broad-Spectrum Inhibitor of Major Human Respiratory Viruses: Coronavirus, Respiratory Syncytial Virus and Influenza A Virus. Viruses 2021, 13, 234. [Google Scholar] [CrossRef]
- Al-Beltagi, S.; Goulding, L.V.; Chang, D.K.E.; Mellits, K.H.; Hayes, C.J.; Gershkovich, P.; Coleman, C.M.; Chang, K.-C. Emergent SARS-CoV-2 Variants: Comparative Replication Dynamics and High Sensitivity to Thapsigargin. Virulence 2021, 12, 2946–2956. [Google Scholar] [CrossRef]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-Level Inhibition of Coronavirus Replication by Chemical ER Stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- Goulding, L.V.; Yang, J.; Jiang, Z.; Zhang, H.; Lea, D.; Emes, R.D.; Dottorini, T.; Pu, J.; Liu, J.; Chang, K.-C. Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response That Blocks Influenza A Virus Replication. Viruses 2020, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-C.; Yu, W.-Y.; Huang, S.-H.; Lai, M.M.C. Ubiquitination of the Cytoplasmic Domain of Influenza A Virus M2 Protein Is Crucial for Production of Infectious Virus Particles. J. Virol. 2018, 92, e01972-17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, M.; Wang, Q.; Li, M.; Bai, J.; Dai, Q.; Zhang, Y.; Yan, M.; Li, X.; Chen, J.; et al. Two Novel Compounds Inhibit Flavivirus Infection in Vitro and in Vivo by Targeting Lipid Metabolism. J. Virol. 2024, 98, e0063524. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Emerging Mechanisms of Drug-Induced Phospholipidosis. Biol. Chem. 2019, 401, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; O’Meara, M.J.; Monel, B.; Vallet, T.; White, K.M.; Zhang, Z.; Alon, A.; et al. Drug-Induced Phospholipidosis Confounds Drug Repurposing for SARS-CoV-2. Science 2021, 373, 541–547. [Google Scholar] [CrossRef]
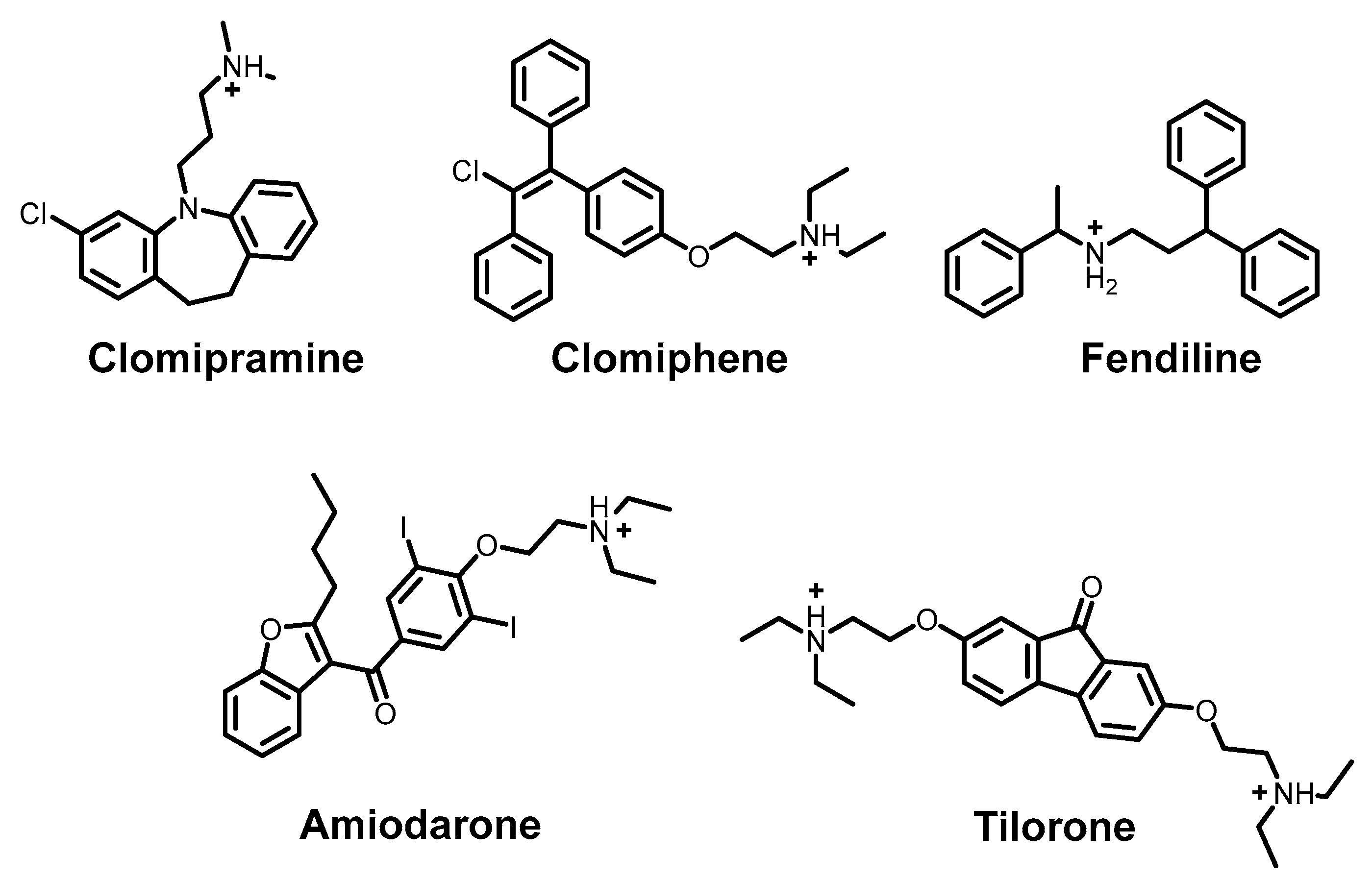
- Liu, Y.; Wang, X.; Li, Q.; Zhu, S.; Zhu, W.; Chen, H.; Si, Y.; Zhu, B.; Cao, S.; Zhao, Z.; et al. Screening a Neurotransmitter-Receptor-Related Inhibitor Library Identifies Clomipramine HCl as a Potential Antiviral Compound against Japanese Encephalitis Virus. Infect. Med. 2024, 3, 100130. [Google Scholar] [CrossRef]
- Ghasemnejad-Berenji, M.; Pashapour, S.; Ghasemnejad-Berenji, H. Therapeutic Potential for Clomiphene, a Selective Estrogen Receptor Modulator, in the Treatment of COVID-19. Med. Hyp. 2020, 145, 110354. [Google Scholar] [CrossRef]
- Husby, M.L.; Stahelin, R. Repurposing Fendiline as a Novel Anti-viral Therapeutic. FASEB J. 2018, 32, 671.9. [Google Scholar] [CrossRef]
- Aimo, A.; Baritussio, A.; Emdin, M.; Tascini, C. Amiodarone as a Possible Therapy for Coronavirus Infection. Eur. J. Prevent. Cardiol. 2021, 28, e16–e18. [Google Scholar] [CrossRef]
- Ekins, S.; Lingerfelt, M.A.; Comer, J.E.; Freiberg, A.N.; Mirsalis, J.C.; O’Loughlin, K.; Harutyunyan, A.; McFarlane, C.; Green, C.E.; Madrid, P.B. Efficacy of Tilorone Dihydrochloride against Ebola Virus Infection. Antimicrob. Agents Chemother. 2018, 62, e01711-17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibition Stage | Compound | Class | Mechanism | Research Stage |
|---|---|---|---|---|
| Attachment | Palivizumab | Antibody | Blocking the binding of the virus to cell receptors | Approved and in use |
| AEEA-16 | Peptidomimetic | Preclinical trials | ||
| Fusion | Maraviroc | Small molecule combining various functions | Blocking fusion proteins | Approved and in use |
| LJ-oo1 | Photosensitizer | Membrane damage | Preclinical trials | |
| CLR01 | Molecular tweezers | Membrane destruction | ||
| Entrance | Amantadine | Cage hydrocarbon amine | Ion channel blocker | Approved and in use |
| Transcription/replication | Remdesivir | Nucleoside | RNA polymerase inhibitors | Approved and in use |
| Favipiravir | Nucleoside analogue | |||
| Lopinavir | Peptidomimetic | Protease inhibitors | ||
| Nirmatrelvir | Peptidomimetic | |||
| Reverse transcriptase inhibitors | Lamivudine | Nucleoside | DNA chain break | Approved and in use |
| Efavirenz | Benzoxazinone | Allosteric inhibition of enzymes | ||
| Virion assembly | Thapsigargin | Terpene | Activation of EPR stress | Preclinical trials |
| Tunicamycin | Nucleoside | |||
| Virion release | Oseltamivir | Small molecule combining various functions | Neuraminidase inhibition | Approved and in use |
| Baloxavir | Small molecule combining various functions | Hemagglutinin inhibition | ||
| Immunomodulators | Interferon-α | Protein | Activation of immunity | Approved and in use |
| Virus | Type | Genetic Material |
|---|---|---|
| Tick-borne encephalitis virus (TBEV) | Enveloped | (+)ssRNA |
| Herpes simplex virus (HSV) | Enveloped | dsDNA |
| Vesicular stomatitis virus (VSV) | Enveloped | (−)ssRNA |
| Influenza virus A (IVA) | Enveloped | (+)ssRNA |
| Zika virus (ZIKV) | Enveloped | (+)ssRNA |
| Human immunodeficiency virus (HIV) | Enveloped | (+)ssRNA |
| Rift Valley fever virus (RVFV) | Enveloped | (−)ssRNA |
| Hepatitis A virus (HAV) | Quasi-enveloped | (+)ssRNA |
| Hepatitis E virus (HEV) | Quasi-enveloped | (+)ssRNA |
| Human papilloma virus (HPV) | Non-enveloped | dsDNA |
| Rotavirus A (RVA) | Non-enveloped | dsRNA |
| Human rhinovirus A (HRV-A) | Non-enveloped | (+)ssRNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasilnikov, M.S.; Denisov, V.S.; Korshun, V.A.; Ustinov, A.V.; Alferova, V.A. Membrane-Targeting Antivirals. Int. J. Mol. Sci. 2025, 26, 7276. https://doi.org/10.3390/ijms26157276
Krasilnikov MS, Denisov VS, Korshun VA, Ustinov AV, Alferova VA. Membrane-Targeting Antivirals. International Journal of Molecular Sciences. 2025; 26(15):7276. https://doi.org/10.3390/ijms26157276
Chicago/Turabian StyleKrasilnikov, Maxim S., Vladislav S. Denisov, Vladimir A. Korshun, Alexey V. Ustinov, and Vera A. Alferova. 2025. "Membrane-Targeting Antivirals" International Journal of Molecular Sciences 26, no. 15: 7276. https://doi.org/10.3390/ijms26157276
APA StyleKrasilnikov, M. S., Denisov, V. S., Korshun, V. A., Ustinov, A. V., & Alferova, V. A. (2025). Membrane-Targeting Antivirals. International Journal of Molecular Sciences, 26(15), 7276. https://doi.org/10.3390/ijms26157276