
Circulating Biomarkers for the Early Diagnosis of Alzheimer’s Disease

 , , , and
, , , and
Abstract
1. Introduction
2. Diagnostic Criteria for Alzheimer’s Disease: Historical Perspective

{kind=link}
| Criteria | Applicable Setting | Clinical Presentations | Required Biological Markers | Reference |
|---|---|---|---|---|
| NINCDS–ADRDA (1984) | Research and clinical. | Memory changes and impairment in at least one another cognitive domain. | None. | [21] |
| IWG (2007) | Research. | Amnestic syndrome of a hippocampal type. | CSF biomarkers, MRI atrophy, [18F]-FDG-PET showing glucose hypometabolism, positive Aβ-PET, or AD autosomal dominant mutation. | [17] |
| IWG (2010) | Research. | Amnestic syndrome of a hippocampal type, posterior cortical variant, logopenic variant, or behavioral–frontal variant. | low CSF Aβ42, high phosphorylated tau, or high total tau, or positive amyloid PET. | [18] |
| NIA–AA (2011) | Research and clinical. | Mild cognitive impairment (amnestic or non-amnestic) or dementia. | Amyloid β markers (CSF or PET) or marker of degeneration (CSF tau, phosphorylated tau, [18F]-FDG-PET, and T1-weighted MRI). | [22] |
| IWG (2014) | Research. | Amnestic syndrome of a hippocampal type, posterior cortical variant, logopenic variant, or behavioral– frontal variant. | CSF amyloid β and tau or amyloid PET positive. | [23] |
| IWG–AA (2016) | Research. | None. | Amyloid β marker (CSF or PET) and tau marker (CSF or PET). | [24] |
| NIA–AA (2018) | Research. | None. | Amyloid β marker (CSF or PET) and tau marker (CSF or PET). | [25] |
| IWG (2021) | Research and clinical. | Amnestic variant, posterior cortical atrophy, logopenic variant primary progressive aphasia, behavioral or dysexecutive frontal variant, corticobasal syndrome, semantic and non-fluent variants of primary progressive aphasias. | Amyloid β marker (CSF or PET) and tau marker (CSF or PET). | [19] |
| NIA–AA (2024) | Research. | None. | Amyloid PET, CSF biomarkers and reliable plasma biomarkers (mainly p-tau217) grouped into Core 1 biomarkers, sufficient for diagnosing AD; tau-PET. | [20] |
- -
- For stage A, the proposed fluid biomarkers would be CSF Aβ1-42/Aβ1-40, p-tau181/Aβ1-42, t-tau/Aβ1-42, or accurate plasma assays.
- -
- For stage B, other p-tau forms, such as p-tau205, could be used.
- -
- In stage C, MBTR-tau243 would be altered.
- -
- In stage D other, non-phosphorylated tau fragments could be detected in biological fluids.
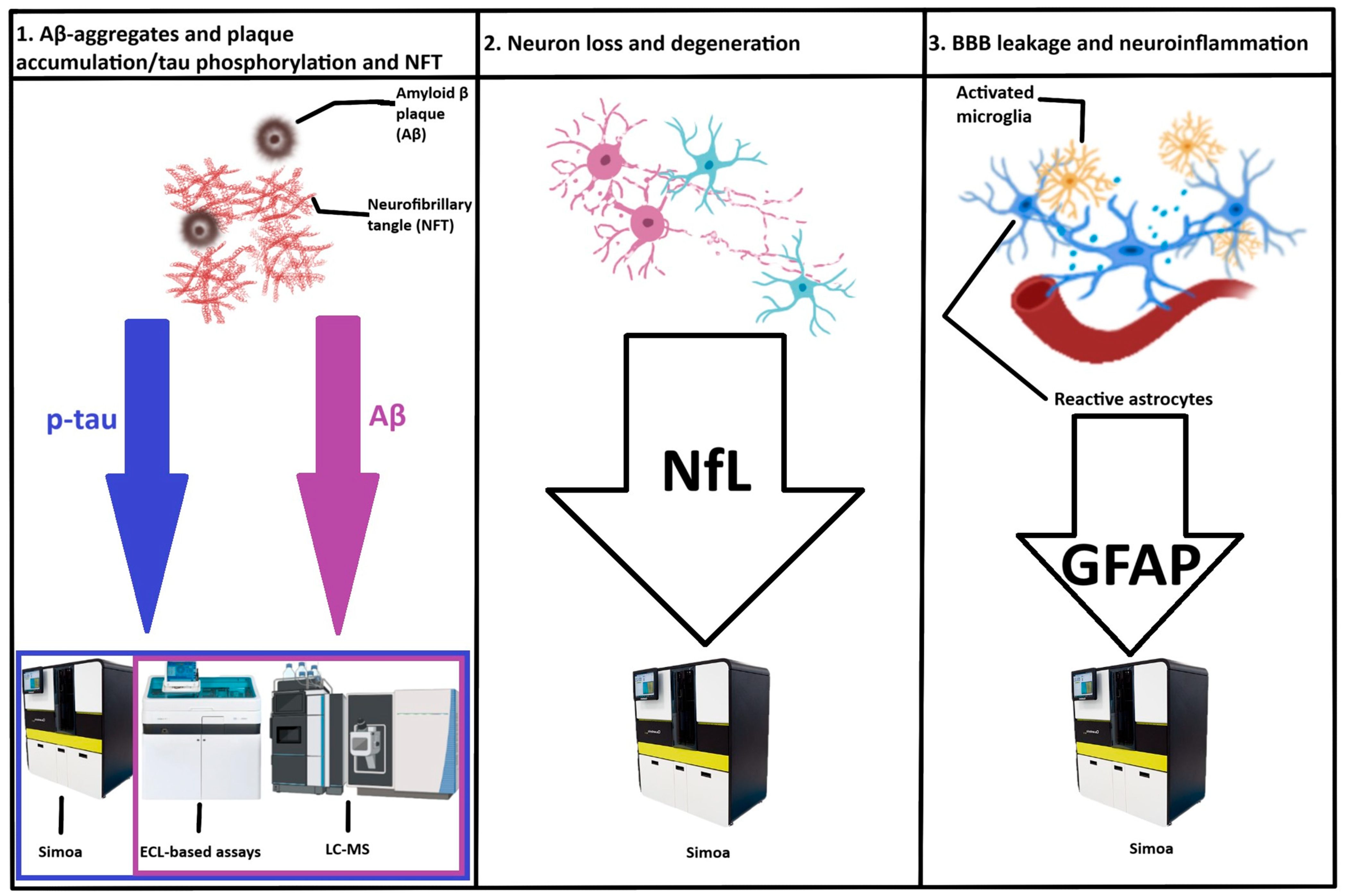
3. Circulating Biomarkers for the Detection and Staging of Alzheimer’s Disease
3.1. Definition of a Biomarker
3.2. Plasma vs. CSF Biomarkers
3.3. Circulating Biomarkers for Amyloid Pathology
3.4. Circulating Biomarkers for Tau Pathology
3.5. Biomarkers of Neurodegeneration
3.6. Biomarkers of Neuroinflammation
3.6.1. Glial Fibrillary Acidic Protein (GFAP)
3.6.2. Triggering Receptor Expressed on Myeloid Cells 2 (TREM2)
3.6.3. Chitinase-3 Like-Protein 1 (CHI3L1)
3.6.4. Monocyte Chemoattractant Protein-1 (MCP-1)
3.6.5. Other Inflammatory Biomarkers
3.7. Novel Blood Biomarkers
3.8. MicroRNAs as Potential Biomarkers for AD
- -
- miR-142.3p, miR-98.5p, and miR-9985 yielded an area under the curve (AUC) of 0.72 for AD.
- -
- miR-590.3p, miR-369.3p, and miR-9985 predicted early mild cognitive impairment with an AUC of 0.71.
- -
- miR-1306, miR-4429, and miR-22.5p characterized late mild cognitive impairment with an AUC of 0.71.
3.9. Studies of Circulating Extracellular Vesicles
4. Critical Appraisal of the Use of Novel Biomarkers for Diagnosing Alzheimer’s Disease
4.1. Selecting Between Available Laboratory Methods
4.2. Establishing Validated Cut-Offs for the Selected Biomarkers
4.2.1. Plasma Amyloid-β
4.2.2. Plasma Phosphorylated Tau
4.2.3. Other Promising Biomarkers
4.3. Issues to Be Addressed Before Proceeding to Clinical Implementation of Biomarkers for Alzheimer’s Disease Diagnosis and Monitoring of Progression
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. The Top 10 Causes of Death [Internet]. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 2 June 2025).
- Wimo, A.; Seeher, K.; Cataldi, R.; Cyhlarova, E.; Dielemann, J.L.; Frisell, O.; Guerchet, M.; Jönsson, L.; Malaha, A.K.; Nichols, E.; et al. The worldwide costs of dementia in 2019. Alzheimers Dement. 2023, 19, 2865–2873. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Hendriks, S.; Peetoom, K.; Bakker, C.; van der Flier, W.M.; Papma, J.M.; Koopmans, R.; Verhey, F.R.J.; de Vugt, M.; Köhler, S.; Young-Onset Dementia Epidemiology Study Group. Global Prevalence of Young-Onset Dementia: A Systematic Review and Meta-analysis. JAMA Neurol. 2021, 78, 1080–1090. [Google Scholar] [CrossRef]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef]
- Militaru, M.; Lighezan, D.F.; Tudoran, C.; Tudoran, M.; Militaru, A.G. Factors Influencing the Development and Severity of Cognitive Decline in Patients with Chronic Heart Failure. Medicina 2024, 60, 1859. [Google Scholar] [CrossRef]
- Militaru, M.; Lighezan, D.F.; Tudoran, C.; Zara, F.; Bucur, A.; Militaru, A.G. Relationship Between Depression and Decreased Activity Level and Cognitive Impairment in Patients with Diabetes Mellitus Type 2 and/or Atrial Fibrillation. J. Clin. Med. 2025, 14, 563. [Google Scholar] [CrossRef]
- Militaru, M.; Lighezan, D.F.; Tudoran, C.; Militaru, A.G. Connections between Cognitive Impairment and Atrial Fibrillation in Patients with Diabetes Mellitus Type 2. Biomedicines 2024, 12, 672. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Liu, K.Y.; Costafreda, S.G.; Selbæk, G.; Alladi, S.; Ames, D.; Banerjee, S.; Burns, A.; Brayne, C.; et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet 2024, 404, 572–628. [Google Scholar] [CrossRef]
- Nous, A.; Engelborghs, S.; Smolders, I. Melatonin levels in the Alzheimer’s disease continuum: A systematic review. Alzheimers Res. Ther. 2021, 13, 52. [Google Scholar] [CrossRef]
- Watroba, M.; Szukiewicz, D. Sirtuins promote brain homeostasis, preventing Alzheimer’s disease through targeting neuroinflammation. Front. Physiol. 2022, 13, 962769. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Albert, M.S.; Alonso, A.; Coker, L.H.; Coresh, J.; Davis, S.M.; Deal, J.A.; McKhann, G.M.; Mosley, T.H.; Sharrett, A.R.; et al. Associations Between Midlife Vascular Risk Factors and 25-Year Incident Dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 2017, 74, 1246–1254. [Google Scholar] [CrossRef]
- Pacholko, A.; Iadecola, C. Hypertension, Neurodegeneration, and Cognitive Decline. Hypertension 2024, 81, 991–1007. [Google Scholar] [CrossRef]
- Li, Q.X.; Gao, H.; Guo, Y.X.; Wang, B.Y.; Hua, R.X.; Gao, L.; Shang, H.W.; Lu, X.; Xu, J.D. GLP-1 and Underlying Beneficial Actions in Alzheimer’s Disease, Hypertension, and NASH. Front. Endocrinol. 2021, 12, 721198. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimers Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Bougea, A.; Gourzis, P. Biomarker-Based Precision Therapy for Alzheimer’s Disease: Multidimensional Evidence Leading a New Breakthrough in Personalized Medicine. J. Clin. Med. 2024, 13, 4661. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Cummings, J.L.; Dekosky, S.T.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.; Fox, N.C.; Galasko, D.; et al. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 2024, 20, 5143–5169. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Contributors. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Lee, H.; Hsieh, Y.-C.; Chou, V.; Selkoe, D.J. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer’s disease. Trends Neurosci. 2023, 46, 426–444. [Google Scholar] [CrossRef]
- Lista, S.; Mapstone, M.; Caraci, F.; Emanuele, E.; López-Ortiz, S.; Martín-Hernández, J.; Triaca, V.; Imbimbo, C.; Gabelle, A.; Mielke, M.M.; et al. A critical appraisal of blood-based biomarkers for Alzheimer’s disease. Ageing Res. Rev. 2024, 96, 102290. [Google Scholar] [CrossRef]
- Hampel, H.; Cummings, J.; Blennow, K.; Gao, P.; Jack, C.R.; Vergallo, A. Developing the ATX(N) classification for use across the Alzheimer’s disease continuum. Nat. Rev. Neurol. 2021, 17, 580–589. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef]
- Varesi, A.; Carrara, A.; Pires, V.G.; Floris, V.; Pierella, E.; Savioli, G.; Prasad, S.; Esposito, C.; Ricevuti, G.; Chirumbolo, S.; et al. Blood-Based Biomarkers for Alzheimer’s Disease Diagnosis and Progression: An Overview. Cells 2022, 11, 1367. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Andrews, S.J.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised criteria for the diagnosis and staging of Alzheimer’s disease. Nat. Med. 2024, 30, 2121–2124. [Google Scholar] [CrossRef]
- Chong, J.R.; Ashton, N.J.; Karikari, T.K.; Tanaka, T.; Schöll, M.; Zetterberg, H.; Blennow, K.; Chen, C.P.; Lai, M.K.P. Blood-based high sensitivity measurements of beta-amyloid and phosphorylated tau as biomarkers of Alzheimer’s disease: A focused review on recent advances. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1231–1241. [Google Scholar] [CrossRef]
- Tikhonova, M.A.; Zhanaeva, S.Y.; Shvaikovskaya, A.A.; Olkov, N.M.; Aftanas, L.I.; Danilenko, K.V. Neurospecific Molecules Measured in Periphery in Humans: How Do They Correlate with the Brain Levels? A Systematic Review. Int. J. Mol. Sci. 2022, 23, 9193. [Google Scholar] [CrossRef]
- Lam, V.; Takechi, R.; Hackett, M.J.; Francis, R.; Bynevelt, M.; Celliers, L.M.; Nesbit, M.; Mamsa, S.; Arfuso, F.; Das, S.; et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease-like neurodegenerative phenotype. PLoS Biol. 2021, 19, e3001358. [Google Scholar] [CrossRef]
- Liu, Z.H.; Wang, Y.J.; Bu, X.L. Alzheimer’s disease: Targeting the peripheral circulation. Mol. Neurodegener. 2023, 18, 3. [Google Scholar] [CrossRef]
- Zetterberg, H. Blood-based biomarkers for Alzheimer’s disease—An update. J. Neurosci. Methods 2019, 319, 2–6. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Durrleman, S.; Younesi, E.; Rojkova, K.; Escott-Price, V.; Corvol, J.-C.; Broich, K.; Dubois, B.; Lista, S. A Precision Medicine Initiative for Alzheimer’s Disease: The Road Ahead to Biomarker-Guided Integrative Disease Modeling. Climacteric 2017, 20, 107–118. [Google Scholar] [CrossRef]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef]
- Constantinides, V.C.; Paraskevas, G.P.; Boufidou, F.; Bourbouli, M.; Pyrgelis, E.-S.; Stefanis, L.; Kapaki, E. CSF Aβ42 and Aβ42/Aβ40 Ratio in Alzheimer’s Disease and Frontotemporal Dementias. Diagnostics 2023, 13, 783. [Google Scholar] [CrossRef]
- Amft, M.; Ortner, M.; Eichenlaub, U.; Goldhardt, O.; Diehl-Schmid, J.; Hedderich, D.M.; Yakushev, I.; Grimmer, T. The cerebrospinal fluid biomarker ratio Aβ42/40 identifies amyloid positron emission tomography positivity better than Aβ42 alone in a heterogeneous memory clinic cohort. Alzheimers Res. Ther. 2022, 14, 60. [Google Scholar] [CrossRef]
- Krawczuk, D.; Kulczyńska-Przybik, A.; Mroczko, B. Clinical Application of Blood Biomarkers in Neurodegenerative Diseases—Present and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 8132. [Google Scholar] [CrossRef]
- Watt, A.D.; Perez, K.A.; Rembach, A.R.; Masters, C.L.; Villemagne, V.L.; Barnham, K.J. Variability in blood-based amyloid-beta assays: The need for consensus on pre-analytical processing. J. Alzheimers Dis. 2012, 30, 323–336. [Google Scholar] [CrossRef]
- Leuzy, A.S.; Mattson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Gage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med. 2022, 14, e14408. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Stomrud, E.; Zetterberg, H.; Karl, J.; Zink, K.; Bittner, T.; Mattson, N.; Eichenlaub, U.; Blennow, K.; et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer’s Disease-Related-β-Amyloid Status. JAMA Neurol. 2019, 76, 1060. [Google Scholar] [CrossRef]
- Shea, D.; Colasurdo, E.; Smith, A.; Paschall, C.; Jayadev, S.; Keene, C.D.; Galasko, D.; Ko, A.; Li, G.; Peskind, E.; et al. SOBA: Development and testing of a soluble oligomer binding assay for detection of amyloidogenic toxic oligomers. Proc. Natl. Acad. Sci. USA 2022, 119, e2213157119. [Google Scholar] [CrossRef]
- Cullen, N.C.; Janelidze, S.; Mattsson-Carlgren, N.; Palmqvist, S.; Bittner, T.; Suridjan, I.; Jethwa, A.; Kollmorgen, G.; Brum, W.S.; Zetterberg, H.; et al. Test-retest variability of plasma biomarkers in Alzheimer’s disease and its effects on clinical prediction models. Alzheimers Dement. 2023, 19, 797–806. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. validation of plasma amyloid-β42/40 for detecting Alzheimer disease amyloid plaques. Neurology 2022, 98, e688–e699. [Google Scholar]
- Hu, H.; Bi, Y.L.; Shen, X.N.; Ma, Y.H.; Ou, Y.N.; Zhang, W.; Ma, L.Z.; Hu, H.Y.; Dong, Q.; Tan, L.; et al. Application of the Amyloid/Tau/Neurodegeneration Framework in Cognitively Intact Adults: The CABLE Study. Ann. Neurol. 2022, 92, 439–450. [Google Scholar] [CrossRef]
- Monane, M.; Johnson, K.G.; Snider, B.J.; Turner, R.S.; Drake, J.D.; Maraganore, D.M.; Bicksel, J.L.; Jacobs, D.H.; Ortega, J.L.; Henderson, J.; et al. A blood biomarker test for brain amyloid impacts the clinical evaluation of cognitive impairment. Ann. Clin. Transl. Neurol. 2023, 10, 1738–1748. [Google Scholar] [CrossRef]
- O’Connor, A.; Pannee, J.; Poole, T.; Arber, C.; Portelius, E.; Swift, I.J.; Heslegrave, A.J.; Abel, E.; Willumsen, N.; Rice, H.; et al. Plasma amyloid-β ratios in autosomal dominant Alzheimer’s disease: The influence of genotype. Brain 2021, 144, 2964–2970. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, Y.-Y.; Shen, X.-N.; Chen, S.-D.; Hu, H.; Wang, Z.-T.; Tan, L.; Yu, J.-T. Characterization of Alzheimer’s Tau Biomarker Discordance using Plasma, CSF, and PET. Alzheimer’s Res. Ther. 2021, 13, 93. [Google Scholar] [CrossRef]
- Toombs, J.; Zetterberg, H. In the blood: Biomarkers for amyloid pathology and neurodegeneration in Alzheimer’s disease. Brain Commun. 2020, 2, fcaa054. [Google Scholar] [CrossRef]
- O’Connor, A.; Karikari, T.K.; Poole, T.; Ashton, N.J.; Lantero Rodriguez, J.; Khatun, A.; Swift, I.; Heslegrave, A.J.; Abel, E.; Chung, E.; et al. Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: A longitudinal cohort study. Mol. Psychiatry 2021, 26, 5967–5976. [Google Scholar] [CrossRef]
- Zetterberg, H.; Blennow, K. Blood biomarkers: Democratizing Alzheimer’s diagnostics. Neuron 2020, 106, 881–883. [Google Scholar] [CrossRef]
- Bermudez, C.; Graff-Radford, J.; Syrjanen, J.A.; Stricker, N.H.; Algeciras-Schimnich, A.; Kouri, N.; Kremers, W.K.; Petersen, R.C.; Jack, C.R., Jr.; Knopman, D.S.; et al. Plasma biomarkers for prediction of Alzheimer’s disease neuropathologic change. Acta Neuropathol. 2023, 146, 13–29. [Google Scholar] [CrossRef]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of Plasma Phospho-Tau217 Levels with Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021, 78, 149–156. [Google Scholar] [CrossRef]
- Mielke, M.M.; Dage, J.L.; Frank, R.D.; Algeciras-Schimnich, A.; Knopman, D.S.; Lowe, V.J.; Bu, G.; Vemuri, P.; Graff-Radford, J.; Jack, C.R., Jr.; et al. Performance of plasma phosphorylated tau181 and 217 in the community. Nat. Med. 2022, 28, 1398–1405. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arsian, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau217 Immunoassay for Alzheimer Disase Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef]
- Mattson-Carlgren, N.; Salvadớ, G.; Ashton, N.J.; Tideman, P.; Stomrud, E.; Zetterberg, H.; Ossenkoppele, R.; Betthauser, T.; Cody, K.A.; Jonaitis, E.M.; et al. Prediction of Longitudinal Cognitive Decline in Preclinical Alzheimer Disease Using Plasma Biomarkers. JAMA Neurol. 2023, 80, 360–369. [Google Scholar] [CrossRef]
- Aguillon, D.; Langella, S.; Chen, Y.; Sanchez, J.S.; Su, Y.; Vila-Castelar, C.; Vasquez, D.; Zetterberg, H.; Hansson, O.; Dage, J.L.; et al. Plasma p-tau217 predicts in vivo brain pathology and cognition in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2023, 19, 2585–2594. [Google Scholar] [CrossRef]
- Saloner, R.; VandeVrede, L.; Asken, B.M.; Paolillo, E.W.; Gontrum, E.Q.; Wolf, A.; Lario-Lago, A.; Milà-Alomà, M.; Triana-Baltzer, G.; Kolb, H.C.; et al. Plasma phosphorylated tau-217 exhibits sex-specific prognostication of cognitive decline and brain atrophy in cognitively unimpaired adults. Alzheimers Dement. 2024, 20, 376–387. [Google Scholar] [CrossRef]
- Lehmann, S.; Schraen-Maschke, S.; Vidal, J.S.; Delaby, C.; Buee, L.; Blanc, F.; Paquet, C.; Allinquant, B.; Bombois, S.; Gabelle, A.; et al. Clinical value of plasma ALZpath pTau217 immunoassay for assessing mild cognitive impairment. J. Neurol. Neurosurg. Psychiatry 2024, 95, 1046–1053. [Google Scholar] [CrossRef]
- Ashton, N.J.; Pascoal, T.A.; Karikari, T.K.; Benedet, A.L.; Lantero-Rodriguez, J.; Brinkmalm, G.; Snellman, A.; Schöll, M.; Troakes, C.; Hye, A.; et al. Plasma p-tau231: A new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021, 141, 709–724. [Google Scholar] [CrossRef]
- Horie, K.; Salvadó, G.; Barthélemy, N.R.; Janelidze, S.; Li, Y.; He, Y.; Saef, B.; Chen, C.D.; Jiang, H.; Strandberg, O.; et al. CSF MTBR-tau243 is a specific biomarker of tau tangle pathology in Alzheimer’s disease. Nat. Med. 2023, 29, 1954–1963. [Google Scholar] [CrossRef]
- Horie, K.; Salvadó, G.; Koppisetti, R.K.; Janelidze, S.; Barthélemy, N.R.; He, Y.; Sato, C.; Gordon, B.A.; Jiang, H.; Benzinger, T.L.S.; et al. Plasma MTBR-tau243 biomarker identifies tau tangle pathology in Alzheimer’s disease. Nat. Med. 2025, 31, 2044–2053. [Google Scholar] [CrossRef]
- Chhatwal, J.P.; Schultz, A.P.; Dang, Y.; Ostaszewski, B.; Liu, L.; Yang, H.S.; Johnson, K.A.; Sperling, R.A.; Selkoe, D.J. Plasma N-terminal tau fragment levels predict future cognitive decline and neurodegeneration in healthy elderly individuals. Nat. Commun. 2020, 11, 6024. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Olsson, B.; Portelius, E.; Cullen, N.C.; Sandelius, Å.; Zetterberg, H.; Andreasson, U.; Höglund, K.; Irwin, D.J.; Grossman, M.; Weintraub, D.; et al. Association of Cerebrospinal Fluid Neurofilament Light Protein Levels with Cognition in Patients with Dementia, Motor Neuron Disease, and Movement Disorders. JAMA Neurol. 2019, 76, 318–325. [Google Scholar] [CrossRef]
- Jung, Y.; Damoiseaux, J.S. The potential of blood neurofilament light as a marker of neurodegeneration for Alzheimer’s disease. Brain 2024, 147, 12–25. [Google Scholar] [CrossRef]
- Alcolea, D.; Beeri, M.S.; Rojas, J.C.; Gardner, R.C.; Lleó, A. Blood Biomarkers in Neurodegenerative Diseases: Implications for the Clinical Neurologist. Neurology 2023, 101, 172–180. [Google Scholar] [CrossRef]
- Vrillon, A.; Ashton, N.J.; Karikari, T.K.; Götze, K.; Cognat, E.; Dumurgier, J.; Lilamand, M.; Zetterberg, H.; Blennow, K.; Paquet, C. Comparison of CSF and plasma NfL and pNfH for Alzheimer’s disease diagnosis: A memory clinic study. J. Neurol. 2024, 271, 1297–1310. [Google Scholar] [CrossRef]
- Gaetani, L.; Parnetti, L.; Calabresi, P.; Di Filippo, M. Tracing Neurological Diseases in the Presymptomatic Phase: Insights from Neurofilament Light Chain. Front. Neurosci. 2021, 15, 672954. [Google Scholar] [CrossRef]
- De Meyer, S.; Blujdea, E.R.; Schaeverbeke, J.; Reinartz, M.; Luckett, E.S.; Adamczuk, K.; Van Laere, K.; Dupont, P.; Teunissen, C.E.; Vandenberghe, R.; et al. Longitudinal associations of serum biomarkers with early cognitive, amyloid and grey matter changes. Brain 2024, 147, 936–948. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Doecke, J.D.; Thota, R.; Villemagne, V.L.; Doré, V.; Singh, A.K.; Wang, P.; Rainey-Smith, S.; Fowler, C.; et al. Plasma Aβ42/40 ratio, p-tau181, GFAP, and NfL across the Alzheimer’s disease continuum: A cross-sectional and longitudinal study in the AIBL cohort. Alzheimers Dement. 2023, 19, 1117–1134. [Google Scholar] [CrossRef]
- Nabizadeh, F.; Balabandian, M.; Rostami, M.R.; Kankam, S.B.; Ranjbaran, F.; Pourhamzeh, M.; Alzheimer’s Disease Neuroimaging Initiative (ADNI). Plasma neurofilament light levels correlate with white matter damage prior to Alzheimer’s disease: Results from ADNI. Aging Clin. Exp. Res. 2022, 34, 2363–2372. [Google Scholar] [CrossRef]
- Hofmann, A.; Haesler, L.M.; Preische, O.; Gräber-Sultan, S.; Obermüller, U.; Vöglein, J.; Levin, J.; Laske, C.; Fitzpatrick, C.D.; Levin, R.; et al. Refinement of Neurofilament light Dynamics in CSF and Blood for familial Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, e078802. [Google Scholar] [CrossRef]
- Jurcau, M.C.; Jurcau, A.; Diaconu, R.G.; Hogea, V.O.; Nunkoo, V.S. A Systematic Review of Sporadic Creutzfeldt-Jakob Disease: Pathogenesis, Diagnosis, and Therapeutic Attempts. Neurol. Int. 2024, 16, 1039–1065. [Google Scholar] [CrossRef]
- Kang, Y.; Feng, Z.; Zhang, Q.; Liu, M.; Li, Y.; Yang, H.; Zheng, L.; Cheng, C.; Zhou, W.; Lou, D.; et al. Identification of circulating biomarkers for cognitive decline in a large community-based population in Chongqing China. Alzheimers Dement. 2025, 21, e14443. [Google Scholar] [CrossRef]
- Vila-Castelar, C.; Chen, Y.; Langella, S.; Lopera, F.; Zetterberg, H.; Hansson, O.; Dage, J.L.; Janelidzde, S.; Su, Y.; Chen, K.; et al. Sex differences in blood biomarkers and cognitive performance in individuals with autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2023, 19, 4127–4138. [Google Scholar] [CrossRef]
- Fitzgerald, K.C.; Sotirchos, E.S.; Smith, M.D.; Lord, H.N.; DuVal, A.; Mowry, E.M.; Calabresi, P.A. Contributors to Serum NfL Levels in People without Neurologic Disease. Ann. Neurol. 2022, 92, 688–698. [Google Scholar] [CrossRef]
- Jurcau, M.C.; Jurcau, A.; Cristian, A.; Hogea, V.O.; Diaconu, R.G.; Nunkoo, V.S. Inflammaging and Brain Aging. Int. J. Mol. Sci. 2024, 25, 10535. [Google Scholar] [CrossRef]
- Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. [Google Scholar] [CrossRef]
- Bellaver, B.; Ferrari-Souza, J.P.; Uglione da Ros, L.; Carter, S.F.; Rodriguez-Vieitez, E.; Nordberg, A.; Pellerin, L.; Rosa-Neto, P.; Leffa, D.T.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer Disease: A Systematic Review and Meta-analysis. Neurology 2021, 96, e2944–e2955. [Google Scholar] [CrossRef]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J.; et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol. 2021, 78, 1471–1483. [Google Scholar] [CrossRef]
- Stocker, H.; Beyer, L.; Perna, L.; Rujescu, D.; Holleczek, B.; Beyreuther, K.; Stockmann, J.; Schöttker, B.; Gerwert, K.; Brenner, H. Association of plasma biomarkers, p-tau181, glial fibrillary acidic protein, and neurofilament light, with intermediate and long-term clinical Alzheimer’s disease risk: Results from a prospective cohort followed over 17 years. Alzheimers Dement. 2023, 19, 25–35. [Google Scholar] [CrossRef]
- Snellman, A.; Ekblad, L.L.; Ashton, N.J.; Karikari, T.K.; Lantero-Rodriguez, J.; Pietilä, E.; Koivumäki, M.; Helin, S.; Karrasch, M.; Zetterberg, H.; et al. Head-to-head comparison of plasma p-tau181, p-tau231 and glial fibrillary acidic protein in clinically unimpaired elderly with three levels of APOE4-related risk for Alzheimer’s disease. Neurobiol. Dis. 2023, 183, 106175. [Google Scholar] [CrossRef]
- Park, M.-K.; Ahn, J.; Kim, Y.-J.; Lee, J.-W.; Lee, J.-C.; Hwang, S.-J.; Kim, K.-C. Predicting Longitudinal Cognitive Decline and Alzheimer’s Conversion in Mild Cognitive Impairment Patients Based on Plasma Biomarkers. Cells 2024, 13, 1085. [Google Scholar] [CrossRef]
- Bellaver, B.; Povala, G.; Ferreira, P.C.L.; Ferrari-Souza, J.P.; Leffa, D.T.; Lussier, F.Z.; Benedet, A.L.; Ashton, N.J.; Triana-Baltzer, G.; Kolb, H.C.; et al. Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer’s disease. Nat. Med. 2023, 29, 1775–1781. [Google Scholar] [CrossRef]
- Asken, B.M.; Elahi, F.M.; La Joie, R.; Strom, A.; Staffaroni, A.M.; Lindbergh, C.A.; Apple, A.C.; You, M.; Weiner-Light, S.; Brathaban, N.; et al. Plasma Glial Fibrillary Acidic Protein Levels Differ Along the Spectra of Amyloid Burden and Clinical Disease Stage. J. Alzheimers Dis. 2020, 78, 265–276. [Google Scholar] [CrossRef]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; Steinacker, P.; Huss, A.M.; Neugebauer, H.; von Arnim, C.A.F.; Diehl-Schmid, J.; Grimmer, T.; Kornhuber, J.; et al. Glial Fibrillary Acidic Protein in Serum is Increased in Alzheimer’s Disease and Correlates with Cognitive Impairment. J. Alzheimers Dis. 2019, 67, 481–488. [Google Scholar] [CrossRef]
- Chouliaras, L.; Thomas, A.; Malpetti, M.; Donaghy, P.; Kane, J.; Mak, E.; Savulich, G.; Prats-Sedano, M.A.; Heslegrave, A.J.; Zetterberg, H.; et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer’s disease, frontotemporal dementia and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 2022, 93, 651–658. [Google Scholar] [CrossRef]
- Morenas-Rodríguez, E.; Li, Y.; Nuscher, B.; Franzmeier, N.; Xiong, C.; Suárez-Calvet, M.; Fagan, A.M.; Schultz, S.; Gordon, B.A.; Benzinger, T.L.S.; et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: A longitudinal observational study. Lancet Neurol. 2022, 21, 329–341. [Google Scholar] [CrossRef]
- La Rosa, F.; Agostini, S.; Piancone, F.; Marventano, I.; Hernis, A.; Fenoglio, C.; Galimberti, D.; Scarpini, E.; Saresella, M.; Clerici, M. TREM2 Expression and Amyloid-Beta Phagocytosis in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8626. [Google Scholar] [CrossRef]
- Hu, N.; Tan, M.-S.; Yu, J.-T.; Sun, L.; Tan, L.; Wang, Y.-L.; Jiang, T.; Tan, L. Increased Expression of TREM2 in Peripheral Blood of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2013, 38, 497–501. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, Y.; Qi, C.; Liu, A.; Zhao, Y. Association Study of Serum Soluble TREM2 with Vascular Dementia in Chinese Han Population. Int. J. Neurosci. 2020, 130, 708–712. [Google Scholar] [CrossRef]
- Hok-A-Hin, Y.S.; Del Campo, M.; Boiten, W.A.; Stoops, E.; Vanhooren, M.; Lemstra, A.W.; van der Flier, W.M.; Teunissen, C.E. Neuroinflammatory CSF biomarkers MIF, sTREM1, and sTREM2 show dynamic expression profiles in Alzheimer’s disease. J. Neuroinflammation 2023, 20, 107. [Google Scholar] [CrossRef]
- Vergallo, A.; Lista, S.; Lemercier, P.; Chiesa, P.A.; Zetterberg, H.; Blennow, K.; Potier, M.C.; Habert, M.O.; Baldacci, F.; Cavedo, E.; et al. Association of plasma YKL-40 with brain amyloid-β levels, memory performance, and sex in subjective memory complainers. Neurobiol. Aging 2020, 96, 22–32. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4781–4789. [Google Scholar] [CrossRef]
- Wilczyńska, K.; Maciejczyk, M.; Zalewska, A.; Waszkiewicz, N. Serum Amyloid Biomarkers, Tau Protein and YKL-40 Utility in Detection, Differential Diagnosing, and Monitoring of Dementia. Front. Psychiatry 2021, 12, 725511. [Google Scholar] [CrossRef]
- Thomas, A.; Guo, J.; Reyes-Dumeyer, D.; Sanchez, D.; Scarmeas, N.; Manly, J.J.; Brickman, A.M.; Lantigua, R.A.; Mayeux, R.; Gu, Y. Inflammatory biomarkers profiles and cognition among older adults. Sci. Rep. 2025, 15, 2265. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral Inflammatory Markers in Alzheimer’s Disease: A Systematic Review and Meta-Analysis of 175 Studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s Disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Foley, K.E.; Winder, Z.; Sudduth, T.L.; Martin, B.J.; Nelson, P.T.; Jicha, G.A.; Harp, J.P.; Weekman, E.M.; Wilcock, D.M. Alzheimer’s disease and inflammatory biomarkers positively correlate in plasma in the UK-ADRC cohort. Alzheimers Dement. 2024, 20, 1374–1386. [Google Scholar] [CrossRef]
- Liang, C.-S.; Tsai, C.-L.; Lin, G.-Y.; Lee, J.-T.; Lin, Y.-K.; Chu, C.-S.; Sung, Y.-F.; Tsai, C.-K.; Yeh, T.-C.; Chu, H.-T.; et al. Better Identification of Cognitive Decline with Interleukin-2 than with Amyloid and Tau Protein Biomarkers in Amnestic Mild Cognitive Impairment. Front. Aging Neurosci. 2021, 13, 670115. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef]
- Ji, D.; Chen, W.Z.; Zhang, L.; Zhang, Z.H.; Chen, L.J. Gut microbiota, circulating cytokines and dementia: A Mendelian randomization study. J. Neuroinflammation 2024, 21, 2. [Google Scholar] [CrossRef]
- Hampel, H.; Nisticò, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, X.; Ip, F.C.; Chan, P.; Chen, Y.; Lai, N.C.H.; Cheung, K.; Lo, R.M.N.; Tong, E.P.S.; Wong, B.W.Y.; et al. Large-scale plasma proteomic profiling identifies a high-performance biomarker panel for Alzheimer’s disease screening and staging. Alzheimers Dement. 2022, 18, 88–102. [Google Scholar] [CrossRef]
- Lacar, B.; Ferdosi, S.; Alavi, A.; Stukalov, A.; Venkataraman, G.R.; de Geus, M.; Dodge, H.; Wu, C.Y.; Kivisakk, P.; Das, S.; et al. Identification of Novel Biomarkers for Alzheimer’s Disease and Related Dementias Using Unbiased Plasma Proteomics. bioRxiv 2024. [Google Scholar] [CrossRef]
- Huang, Y.-L.; Chang, W.-J.; Huang, C.-H.; Lin, C.-H.; Peng, L.-N.; Chung, C.-P.; Chen, L.-K.; Lee, W.-J. Proteo-metabolomic insights for early dual physical and cognitive impairments: A search for biomarkers of healthy aging based on muscle-brain crosstalk. Aging Cell 2025, 24, e14407. [Google Scholar] [CrossRef]
- Jurcau, A.; Jurcau, M.C. Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy. Biomedicines 2022, 10, 1895. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Wang, L.; Shui, X.; Diao, Y.; Chen, D.; Zhou, Y.; Lee, T.H. Potential Implications of miRNAs in the Pathogenesis, Diagnosis, and therapeutics of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 16259. [Google Scholar] [CrossRef]
- Denk, J.; Oberhauser, F.; Kornhuber, J.; Wiltfang, J.; Fassbender, K.; Schroeter, M.L.; Volk, A.E.; Diehl-Schmid, J.; Prudlo, J.; Danek, A.; et al. Specific serum and CSF microRNA profiles distinguish sporadic behavioural variant of frontotemporal dementia compared with Alzheimer patients and cognitively healthy controls. PLoS ONE 2018, 13, e0197329. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells 2021, 10, 113. [Google Scholar] [CrossRef]
- Kenny, A.; McArdle, H.; Calero, M.; Rabano, A.; Madden, S.F.; Adamson, K.; Forster, R.; Spain, E.; Prehn, J.H.M.; Henshall, D.C.; et al. Elevated Plasma microRNA-206 Levels Predict Cognitive Decline and Progression to Dementia from Mild Cognitive Impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef]
- Ansari, A.; Maffioletti, E.; Milanesi, E.; Marizzoni, M.; Frisoni, G.B.; Blin, O.; Richardson, J.C.; Bordet, R.; Forloni, G.; Gennarelli, M.; et al. miR-146a and miR-181a are involved in the progression of mild cognitive impairment to Alzheimer’s disease. Neurobiol. Aging 2019, 82, 102–109. [Google Scholar] [CrossRef]
- Liu, S.; Park, T.; Krüger, D.M.; Pena-Centeno, T.; Burkhardt, S.; Schutz, A.-L.; Huang, Y.-N.; Rosewood, T.; Chaudhuri, S.; Cho, M.Y.; et al. Plasma miRNAs across the Alzheimer’s disease continuum: Relationship to central biomarkers. Alzheimer’s Dement. 2024, 20, 7698–7714. [Google Scholar] [CrossRef]
- Krüger, D.M.; Pena-Centeno, T.; Liu, S.; Park, T.; Kaurani, L.; Pradhan, R.; Huang, Y.-N.; Risacher, S.L.; Burkhardt, S.; Schütz, A.-L.; et al. The plasma miRNAome in ADNI: Signatures to aid the detection of at-risk individuals. Alzheimer’s Dement. 2024, 20, 7479–7494. [Google Scholar] [CrossRef]
- Gupta, N.; Jadhav, S.; Tan, K.L.; Saw, G.; Mallilankaraman, K.B.; Dheen, S.T. miR-142-3p regulates BDNF expression in activated rodent microglia through its target CMAK2A. Front. Cell. Neurosci. 2020, 14, 132. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, H.; Si, Y.; Wu, N.; Cao, H.; Mei, B.; Meng, B. miR-125b promotes tau phosphorylation by targeting the neural cell adhesion molecule in neuropathological progression. Neurobiol. Aging 2019, 73, 41–49. [Google Scholar] [CrossRef]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef]
- Yin, C.; Liufu, C.; Ye, S.; Zhu, T.; Jiang, J.; Wang, M.; Zhou, L.; Yao, L.; Wang, Y.; Shi, B. Tumor-derived exosomal KPNA2 activates fibroblasts and interacts with KIFC1 to promote bladder cancer progression, a process inhibited by miR-26b-5p. Cell Mol. Biol. Lett. 2025, 30, 20. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, S.; Duan, N.; Li, X.; Wen, J. MicroRNA-26b-5p alleviates cerebral ischemia-reperfusion injury in rats via inhibiting the N-myc/PTEN axis by downregulating KLF10 expression. Hum. Exp. Toxicol. 2021, 40, 1250–1262. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Eren, E.; Hunt, J.F.V.; Shardell, M.; Chawla, S.; Tran, J.; Gu, J.; Vogt, N.M.; Johnson, S.C.; Bendlin, B.B.; Kapogiannis, D. Extracellular vesicle biomarkers of Alzheimer’s disease associated with sub-clinical cognitive decline in late middle age. Alzheimers Dement. 2020, 16, 1293–1304. [Google Scholar] [CrossRef]
- Nistor-Cseppentö, D.C.; Jurcău, M.C.; Jurcău, A.; Andronie-Cioară, F.L.; Marcu, F. Stem Cell- and Cell-Based Therapies for Ischemic Stroke. Bioengineering 2022, 9, 717. [Google Scholar] [CrossRef]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef]
- Delgado-Peraza, F.; Nogueras-Ortiz, C.J.; Volpert, O.; Liu, D.; Goetzl, E.J.; Mattson, M.P.; Greig, N.H.; Eitan, E.; Kapogiannis, D. Neuronal and Astrocytic Extracellular Vesicle Biomarkers in Blood Reflect Brain Pathology in Mouse Models of Alzheimer’s Disease. Cells 2021, 10, 993. [Google Scholar] [CrossRef]
- Cai, H.; Pang, Y.; Wang, Q.; Qin, W.; Wei, C.; Li, Y.; Li, T.; Li, F.; Wang, Q.; Li, Y.; et al. Proteomic profiling of circulating plasma exosomes reveals novel biomarkers of Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 181. [Google Scholar] [CrossRef]
- Kalra, H.; Adda, C.G.; Liem, M.; Ang, C.S.; Mechler, A.; Simpson, R.J.; Hulett, M.D.; Mathivanan, S. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 2013, 13, 3354–3364. [Google Scholar] [CrossRef]
- Eitan, E.; Thornton-Wells, T.; Elgart, K.; Erden, E.; Gershun, E.; Levine, A.; Volpert, O.; Azadeh, M.; Smith, D.G.; Kapogiannis, D. Synaptic proteins in neuron-derived extracellular vesicles as biomarkers for Alzheimer’s disease: Novel methodology and clinical proof of concept. Extracell. Vesicles Circ. Nucleic Acids. 2023, 4, 133–150. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957. [Google Scholar] [CrossRef]
- Nakamura, M.; Li, Y.; Choi, B.R.; Matas-Rico, E.; Troncoso, J.; Takahashi, C.; Sockanathan, S. GDE2-RECK controls ADAM10 α-secretase-mediated cleavage of amyloid precursor protein. Sci. Transl. Med. 2021, 13, eabe6178. [Google Scholar] [CrossRef]
- McMillan, N.; Kirschen, G.W.; Desai, S.; Xia, E.; Tsirka, S.E.; Aguirre, A. ADAM10 facilitates rapid neural stem cell cycling and proper positioning within the subventricular zone niche via JAMC/RAP1Gap signaling. Neural Regen. Res. 2022, 17, 2472–2483. [Google Scholar]
- Liu, W.-L.; Lin, H.-W.; Lin, M.-R.; Yu, Y.; Liu, H.-H.; Dai, Y.-L.; Chen, L.-W.; Jia, W.-W.; He, X.-J.; Li, X.-L.; et al. Emerging blood exosome-based biomarkers for preclinical and clinical Alzheimer’s disease: A meta-analysis and systematic review. Neural Regen. Res. 2022, 17, 2381–2390. [Google Scholar]
- Jia, L.; Zhu, M.; Kong, C.; Pang, Y.; Zhang, H.; Qiu, Q.; Wei, C.; Tang, Y.; Wang, Q.; Li, Y.; et al. Blood neuro-exosomal synaptic proteins predict Alzheimer’s disease at the asymptomatic stage. Alzheimers Dement. 2021, 17, 49–60. [Google Scholar] [CrossRef]
- Visconte, C.; Fenoglio, C.; Serpente, M.; Muti, P.; Sacconi, A.; Rigoni, M.; Arighi, A.; Borracci, V.; Arcaro, M.; Arosio, B.; et al. Altered Extracellular Vesicle miRNA Profile in Prodromal Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14749. [Google Scholar] [CrossRef]
- Guimarães, T.R.; Swanson, E.; Kofler, J.; Thathiah, A. G protein-coupled receptor kinases are associated with Alzheimer’s disease pathology. Neuropathol. Appl. Neurobiol. 2021, 47, 942–957. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, P.; Ding, L.; Zhu, J.; Qi, X.; Zhang, Y.; Li, Y.; Xia, X.; Zheng, J.C. Circulating extracellular vesicle-containing microRNAs reveal potential pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 2022, 16, 955511. [Google Scholar] [CrossRef]
- Kaštelan, S.; Braš, M.; Pjevač, N.; Bakija, I.; Tomić, Z.; Pjevač Keleminić, N.; Gverović Antunica, A. Tear Biomarkers and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 13429. [Google Scholar] [CrossRef]
- Freeman, S.H.; Raju, S.; Hyman, B.T.; Frosch, M.P.; Irizarry, M.C. Plasma Abeta levels do not reflect brain Abeta levels. J. Neuropathol. Exp. Neurol. 2007, 66, 264–271. [Google Scholar] [CrossRef]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Locascio, J.J.; Peskind, E.R.; Li, G.; Hyman, B.T. A Luminex assay detects amyloid β oligomers in Alzheimer’s disease cerebrospinal fluid. PLoS ONE 2013, 8, e67898. [Google Scholar] [CrossRef]
- Song, F.; Poljak, A.; Valenzuela, M.; Mayeux, R.; Smythe, G.A.; Sachdev, P.S. Meta-analysis of plasma amyloid-beta levels in Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 365–375. [Google Scholar] [CrossRef]
- Pais, M.V.; Forlenza, O.V.; Diniz, B.S. Plasma Biomarkers of Alzheimer’s Disease: A Review of Available Assays, Recent Developments, and Implications for Clinical Practice. J. Alzheimers Dis. Rep. 2023, 7, 355–380. [Google Scholar] [CrossRef]
- Janelidze, S.; Bali, D.; Ashton, N.J.; Barthélemy, N.R.; Vanbrabant, J.; Stoops, E.; Vanmechelen, E.; He, Y.; Dolado, A.O.; Triana-Baltzer, G.; et al. Head-to-head comparison of 10 plasma phospho-tau assays in prodromal Alzheimer’s disease. Brain 2023, 146, 1592–1601. [Google Scholar] [CrossRef]
- Verberk, I.M.W.; Hendriksen, H.M.A.; van Harten, A.C.; Wesselman, L.M.P.; Verfaillie, S.C.J.; van den Bosch, K.A.; Slot, R.E.R.; Prins, N.D.; Scheltens, P.; Teunissen, C.E.; et al. Plasma amyloid is associated with the rate of cognitive decline in cognitively normal elderly: The SCIENCe project. Neurobiol. Aging 2020, 89, 99–107. [Google Scholar] [CrossRef]
- Janelidze, S.; Teunissen, C.E.; Zetterberg, H.; Allué, J.A.; Sarasa, L.; Eichenlaub, U.; Bittner, T.; Ovod, V.; Verberk, I.M.W.; Toba, K.; et al. Head-to-Head Comparison of 8 Plasma Amyloid-β 42/40 Assays in Alzheimer Disease. JAMA Neurol. 2021, 78, 1375–1382. [Google Scholar] [CrossRef]
- Yamashita, K.; Miura, M.; Watanabe, S.; Ishiki, K.; Arimatsu, Y.; Kawahira, J.; Kubo, T.; Sasaki, K.; Arai, T.; Hagino, K.; et al. Fully automated and highly specific plasma β-amyloid immunoassays predict β-amyloid status defined by amyloid positron emission tomography with high accuracy. Alzheimers Res. Ther. 2022, 14, 86. [Google Scholar] [CrossRef]
- Hu, Y.; Kirmess, K.M.; Meyer, M.R.; Rabinovici, G.D.; Gatsonis, C.; Siegel, B.A.; Whitmer, R.A.; Apgar, C.; Hanna, L.; Kanekiyo, M.; et al. Assessment of a Plasma Amyloid Probability Score to Estimate Amyloid Positron Emission Tomography Findings Among Adults with Cognitive Impairment. JAMA Netw. Open. 2022, 5, e228392. [Google Scholar] [CrossRef]
- West, T.; Kirmess, K.M.; Meyer, M.R.; Holubasch, M.S.; Knapik, S.S.; Hu, Y.; Contois, J.H.; Jackson, E.N.; Harpstrite, S.E.; Bateman, R.J.; et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: Findings from a multi cohort validity analysis. Mol. Neurodegener. 2021, 16, 30. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Stomrud, E.; Palmqvist, S.; van Westen, D.; Dage, J.L.; Mattsson-Carlgren, N.; Hansson, O. Plasma markers predict changes in amyloid, tau, atrophy and cognition in non-demented subjects. Brain 2021, 144, 2826–2836. [Google Scholar] [CrossRef]
- Tanner, J.A.; Rabinovici, G.D. Relationship Between Tau and Cognition in the Evolution of Alzheimer’s Disease: New Insights from Tau PET. J. Nucl. Med. 2021, 62, 612–613. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 33–356. [Google Scholar] [CrossRef]
- Chen, Y.R.; Liang, C.S.; Chu, H.; Voss, J.; Kang, X.L.; O’Connell, G.; Jen, H.J.; Liu, D.; Shen Hsiao, S.T.; Chou, K.R. Diagnostic accuracy of blood biomarkers for Alzheimer’s disease and amnestic mild cognitive impairment: A meta-analysis. Ageing Res. Rev. 2021, 71, 101446. [Google Scholar] [CrossRef]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef]
- Wilson, E.N.; Young, C.B.; Ramos Benitez, J.; Swarovski, M.S.; Feinstein, I.; Vandijck, M.; Le Guen, Y.; Kasireddy, N.M.; Shahid, M.; Corso, N.K.; et al. Performance of a fully-automated Lumipulse plasma phospho-tau181 assay for Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 172. [Google Scholar] [CrossRef]
- Tissot, C.; Therriault, J.; Kunach, P.; Benedet, A.L.; Pascoal, T.A.; Ashton, N.J.; Karikari, T.K.; Servaes, S.; Lussier, F.Z.; Chamoun, M.; et al. Comparing tau status determined via plasma pTau181, pTau231 and [18F]MK6240 tau-PET. EBioMedicine 2022, 76, 103837. [Google Scholar] [CrossRef]
- Groot, C.; Smith, R.; Stomrud, E.; Binette, A.P.; Leuzy, A.; Wuestefeld, A.; Wisse, L.E.M.; Palmqvist, S.; Mattsson-Carlgren, N.; Janelidze, S.; et al. Phospho-tau with subthreshold tau-PET predicts increased tau accumulation rates in amyloid-positive individuals. Brain 2023, 146, 1580–1591. [Google Scholar] [CrossRef]
- Doré, V.; Doecke, J.D.; Saad, Z.S.; Triana-Baltzer, G.; Slemmon, R.; Krishnadas, N.; Bourgeat, P.; Huang, K.; Burnham, S.; Fowler, C.; et al. Plasma p217+tau versus NAV4694 amyloid and MK6240 tau PET across the Alzheimer’s continuum. Alzheimers Dement. 2022, 14, e12307. [Google Scholar] [CrossRef]
- Shen, X.N.; Huang, Y.Y.; Chen, S.D.; Guo, Y.; Tan, L.; Dong, Q.; Yu, J.T.; Alzheimer’s Disease Neuroimaging Initiative. Plasma phosphorylated-tau181 as a predictive biomarker for Alzheimer’s amyloid, tau and FDG PET status. Transl. Psychiatry 2021, 11, 585. [Google Scholar] [CrossRef]
- Gerards, M.; Schild, A.K.; Meiberth, D.; Rostamzadeh, A.; Vehreschild, J.J.; Wingen-Heimann, S.; Johannis, W.; Martino Adami, P.; Onur, O.A.; Ramirez, A.; et al. Alzheimer’s Disease Plasma Biomarkers Distinguish Clinical Diagnostic Groups in Memory Clinic Patients. Dement. Geriatr. Cogn. Disord. 2022, 51, 182–192. [Google Scholar] [CrossRef]
- Roses, A.; Saunders, A.; Corder, E.H.; Risch, N.; Haines, J.; Pericak-Vance, M.; Han, S.H.; Einstein, G.; Hulette, C.; Schmechel, D.; et al. Apolipoprotein E4 and E2 are susceptibility genes that affect the rate of Alzheimer disease expressivity. J. Cell. Biochem. 1995, 21, 86. [Google Scholar]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Ashton, N.J.; Tegg, M.; Goozee, K.; Singh, A.K.; Karikari, T.K.; Simrén, J.; Vanmechelen, E.; Armstrong, N.J.; et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer’s disease. Alzheimers Dement. 2022, 18, 1141–1154. [Google Scholar] [CrossRef]
- Asken, B.M.; VandeVrede, L.; Rojas, J.C.; Fonseca, C.; Staffaroni, A.M.; Elahi, F.M.; Lindbergh, C.A.; Apple, A.C.; You, M.; Weiner-Light, S.; et al. Lower White Matter Volume and Worse Executive Functioning Reflected in Higher Levels of Plasma GFAP among Older Adults with and Without Cognitive Impairment. J. Int. Neuropsychol. Soc. 2022, 28, 588–599. [Google Scholar] [CrossRef]
- Oeckl, P.; Anderl-Straub, S.; Von Armin, C.A.F.; Baldeiras, I.; Diehl-Schmid, J.; Grimmer, T.; Halbgebauer, S.; Kort, A.M.; Lima, M.; Marques, T.M.; et al. Serum GFAP differentiates Alzheimer’s disease from frontotemporal dementia and predicts MCI-to-dementia conversion. J. Neurol. Neurosurg. Psychiatry 2022, 93, 659–667. [Google Scholar] [CrossRef]
- Hansson, O.; Edelmayer, R.M.; Boxer, A.L.; Carillo, M.C.; Mielke, M.M.; Rabinovici, G.D.; Salloway, S.; Sperling, R.; Zetterberg, K.; Teunissen, C.E. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 2022, 18, 2669–2686. [Google Scholar] [CrossRef]
- Wu, J.; Xiao, Z.; Wang, M.; Wu, W.; Ma, X.; Liang, X.; Zheng, L.; Ding, S.; Luo, J.; Cao, Y.; et al. The impact of kidney function on plasma neurofilament light and phospho-tau181 in a community-based cohort: The Shanghai Aging Study. Alzheimers Res. Ther. 2024, 16, 32. [Google Scholar] [CrossRef]
- Ashton, N.J.; Keshavan, A.; Brum, W.S.; Andreasson, U.; Arslan, B.; Droescher, M.; Barghorn, S.; Vanbrabant, J.; Lambrechts, C.; Van Loo, M.; et al. The Alzheimer’s Association Global Biomarker Standardization Consortium (GBSC) plasma phospho-tau Round Robin study. Alzheimers Dement. 2025, 21, e14508. [Google Scholar] [CrossRef]
- Ibanez, L.; Liu, M.; Beric, A.; Timsina, J.; Kohlfeld, P.; Bergmann, K.; Lowery, J.; Sykora, N.; Sanchez-Montejo, B.; Brock, W.; et al. Benchmarking of a multi-biomarker low-volume panel for Alzheimer’s disease and related dementia research. Alzheimers Dement. 2025, 21, e14413. [Google Scholar] [CrossRef]
- Mielke, M.M.; Fowler, N.R. Alzheimer disease blood biomarkers: Considerations for population-level use. Nat. Rev. Neurol. 2024, 20, 495–504. [Google Scholar] [CrossRef]
- Nunkoo, V.S.; Cristian, A.; Jurcau, A.; Diaconu, R.G.; Jurcau, M.C. The Quest for Eternal Youth: Hallmarks of Aging and Rejuvenating Therapeutic Strategies. Biomedicines 2024, 12, 2540. [Google Scholar] [CrossRef]
- Justice, J.N.; Leng, X.I.; LeBrasseur, N.K.; Tchkonia, T.; Kirkland, J.L.; Mitin, N.; Liu, Y.; Kritchevsky, S.B.; Nicklas, B.J.; Ding, J. Caloric Restriction Intervention Alters Specific Circulating Biomarkers of the Senescence-Associated Secretome in Middle-Aged and Older Adults with Obesity and Prediabetes in an 18-Week Randomized Controlled Trial. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glad214. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Gatsonis, C.; Apgar, C.; Chaudhary, K.; Gareen, I.; Hanna, L.; Hendrix, J.; Hillner, B.E.; Olson, C.; Lesman-Segev, O.H.; et al. Association of Amyloid Positron Emission Tomography with Subsequent Change in Clinical Management Among Medicare Beneficiaries with Mild Cognitive Impairment or Dementia. JAMA 2019, 321, 1286–1294. [Google Scholar] [CrossRef]
- Suemoto, C.K.; Ferretti-Rebustini, R.E.; Rodriguez, R.D.; Leite, R.E.; Soterio, L.; Brucki, S.M.; Spera, R.R.; Cippiciani, T.M.; Farfel, J.M.; Chiavegatto Filho, A.; et al. Neuropathological diagnoses and clinical correlates in older adults in Brazil: A cross-sectional study. PLoS Med. 2017, 14, e1002267. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 2022, 28, 1797–1801. [Google Scholar] [CrossRef]
- Palmqvist, S.; Stomrud, E.; Cullen, N.; Janelidze, S.; Manuilova, E.; Jethwa, A.; Bittner, T.; Eichenlaub, U.; Suridjan, I.; Kollmorgen, G.; et al. An accurate fully automated panel of plasma biomarkers for Alzheimer’s disease. Alzheimers Dement. 2023, 19, 1204–1215. [Google Scholar] [CrossRef]
- Tsiknia, A.A.; Edland, S.D.; Sundermann, E.E.; Reas, E.T.; Brewer, J.B.; Galasko, D.; Banks, S.J.; Alzheimer’s Disease Neuroimaging Initiative. Sex differences in plasma p-tau181 associations with Alzheimer’s disease biomarkers, cognitive decline, and clinical progression. Mol. Psychiatry 2022, 27, 4314–4322. [Google Scholar] [CrossRef]
- Angioni, D.; Delrieu, J.; Hansson, O.; Fillit, H.; Aisen, P.; Cummings, J.; Sims, J.R.; Braunstein, J.B.; Sabbagh, M.; Bittner, T.; et al. Blood Biomarkers from Research Use to Clinical Practice: What Must Be Done? A Report from the EU/US CTAD Task Force. J. Prev. Alzheimers Dis. 2022, 9, 569–579. [Google Scholar] [CrossRef]
- Mattke, S.; Cho, S.K.; Bittner, T.; Hlavka, J.; Hansson, M. Blood-based biomarkers for Alzheimer’s pathology and the diagnostic process for a disease-modifying treatment: Projecting the impact on the cost and wait times. Alzheimers Dement. 2020, 12, e12081. [Google Scholar] [CrossRef]
- Angioni, D.; Hansson, O.; Bateman, R.J.; Rabe, C.; Toloue, M.; Braunstein, J.B.; Agus, S.; Sims, J.R.; Bittner, T.; Carrillo, M.C.; et al. Can We Use Blood Biomarkers as Entry Criteria and for Monitoring Drug Treatment Effects in Clinical Trials? A Report from the EU/US CTAD Task Force. J. Prev. Alzheimers Dis. 2023, 10, 418–425. [Google Scholar] [CrossRef]
- Kubota, M.; Bun, S.; Takahata, K.; Kurose, S.; Momota, Y.; Iwabuchi, Y.; Tezuka, T.; Tabuchi, H.; Seki, M.; Yamamoto, Y.; et al. Plasma biomarkers for early detection of Alzheimer’s disease: A cross-sectional study in a Japanese cohort. Alzheimers Res. Ther. 2025, 17, 131. [Google Scholar] [CrossRef]
- Burton, J.; Brothers, H.M.; Hutchison, R.M.; Murphy, J.; Sun, T.; Dent, G.; Curiale, G.; Kowalski, K. Lower baseline amyloid beta burden is associated with greater percent of amyloid beta positron emission tomography reduction and better clinical outcomes in the aducanumab Phase 3 trials ENGAGE and EMERGE in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2025, 100202, Epub ahead of print. [Google Scholar] [CrossRef]
- Ornago, A.M.; Pinardi, E.; Grande, G.; Valletta, M.; Calderón-Larrañaga, A.; Andersson, S.; Calvani, R.; Picca, A.; Marzetti, E.; Winblad, B.; et al. Blood biomarkers of Alzheimer’s disease and 12-year muscle strength trajectories in community-dwelling older adults: A cohort study. Lancet Healthy Longev. 2025, 6, 100715. [Google Scholar] [CrossRef]
- Stevenson-Hoare, J.; Heslegrave, A.; Leonenko, G.; Fathalla, D.; Bellou, E.; Luckcuck, L.; Marshall, R.; Sims, R.; Morgan, B.P.; Hardy, J.; et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer’s disease. Brain 2023, 146, 690–699. [Google Scholar] [CrossRef]
- Tanne, J.H. FDA approves blood test to diagnose Alzheimer’s. BMJ 2025, 389, r1082. [Google Scholar] [CrossRef]
- Bagger, F.O.; Borgwardt, L.; Jespersen, A.S.; Hansen, A.R.; Bertelsen, B.; Kodama, M.; Nielsen, F.C. Whole genome sequencing in clinical practice. BMC Med. Genom. 2024, 17, 39. [Google Scholar] [CrossRef]
- Rankin, S.M.; Marskell, L.; Hamad, L.; Machin, L. The UK National screening committee, the newborn genomes programme, and the ethical conundrum for UK newborn screening. J. Community Genet 2025. Epub ahead of print. [Google Scholar] [CrossRef]
| Stage | Amyloid-PET | Tau-PET Medial Temporal | Tau-PET Moderate Neocortical Uptake | Tau-PET High Neocortical Uptake | AT2 Notation |
|---|---|---|---|---|---|
| A | + | − | − | − | A + T2− |
| B | + | + | − | − | A + T2MTL+ |
| C | + | + | + | − | A + T2MOD+ |
| D | + | + | + | + | A + T2HIGH+ |
| Pathology | miRNA | Modification | Significance |
|---|---|---|---|
| Amyloid burden | hsa-miR-145-5p | increased | p < 0.001 |
| hsa-miR-483-3p | increased | p < 0.001 | |
| hsa-miR-374a-3p | increased | p < 0.001 | |
| hsa-miR-339-5p | decreased | p < 0.001 | |
| hsa-miR-652-3p | decreased | p < 0.001 | |
| hsa-miR-95-30 | decreased | p < 0.001 | |
| hsa-miR-628-5p | decreased | p < 0.001 | |
| hsa-miR190a-5p | decreased | p < 0.001 | |
| hsa-miR-3679-5p | decreased | p < 0.01 | |
| Tau pathology | hsa-miR-1224-5p | increased | p < 0.001 |
| hsa-miR-337-5p | increased | p < 0.001 | |
| hsa-miR-1180-3p | increased | p < 0.001 | |
| hsa-miR-190a-5p | increased | p < 0.001 | |
| hsa-miR-1255b-5p | decreased | p < 0.001 | |
| hsa-miR-369-5p | decreased | p < 0.01 | |
| Neurodegeneration | hsa-miR-1224-5p | increased | p < 0.001 |
| hsa-miR-337-5p | increased | p < 0.001 | |
| hsa-miR-1180-3p | increased | p < 0.001 | |
| hsa-miR-1255b-5p | decreased | p < 0.001 | |
| hsa-miR-941 | decreased | p < 0.001 | |
| hsa-miR-369-5p | decreased | p < 0.001 | |
| hsa-miR-215-5p | decreased | p < 0.001 |
| Biomarker | Advantages | Limitations |
|---|---|---|
| Genetic tests | Risk assessment. Insight into an individual’s genetic Predisposition. | Cost and accessibility. Incomplete penetrance and gene expression. Interactions with other risk factors. Limited predictive accuracy. |
| Brain MRI | Detailed information on microstructural and functional alterations of the brain. Quantification of brain atrophy Monitoring of disease progression. | Expensive and has limited availability. Expertise required for analysis of images. Requires patient cooperation, time-consuming. Side effects of contrast material. |
| PET scans | Objective assessment of extent and distribution of Aβ and tau pathology in the brain. Monitoring disease progression. Differentiation of AD from other dementias. | Expensive and with limited availability. Exposure to ionizing radiations. Expertise required for the analysis of images. Time-consuming. Contrast side effects. |
| CSF bio- markers | High concentration of biomarkers due to proximity to the brain. Standardized procedures. Can test numerous biomarkers. Allows for longitudinal monitoring of disease progression. Can be used as outcome measure to assess efficacy of therapies. | Invasive procedure, requiring hospitalization. Risk of complications. Biomarkers may vary with age, gender, underlying health conditions, making the interpretation of the results rather complex. Relatively expensive. |
| Blood biomarkers | Simple sampling procedure. Widespread use possible. Can test large numbers of biomarkers. Reproducible. Can serve as initial diagnostic examination in a more complex diagnostic procedure. | Relatively low concentrations of potential biomarkers due to the presence of the BBB. Unreliable findings because biomarkers may have other sources aside from the brain. Still low sensitivity and specificity of blood biomarkers. |
| Population, Sample Size | Biomarker | Analytical Platform | Cut-Off/Threshold | Reference |
|---|---|---|---|---|
| 397 participants (MissionAD1 and MissionAD2 trials. | Aβ1-40 Aβ1-42 | ECL | Plasma Aβ1-42/Aβ1-40 ratio of 0.102 (AUC 0.94, sensitivity 96%, specificity 83.5%). | [159] |
| 249 patients from the Plasma Test for Amyloidosis Risk Screening (PARIS) cohort + 437 patients form the MissionAD trial. | Plasma Aβ1-40 Plasma Aβ1-42 | LC-MS | PARIS cohort: Plasma Aβ1-42/Aβ1-40 ratio ≥ 0.089 (AUC 0.79, sensitivity 85%, specificity 63%). Mission AD cohort: Plasma Aβ1-42/Aβ1-40 ratio ≥ 0.092 (AUC 0.86, sensitivity 90%, specificity 71%). | [160] |
| 414 plasma samples from 6 independent US cohorts. | Plasma Aβ1-40 Plasma Aβ1-42 | LC-MS | Plasma Aβ1-42/Aβ1-40 ratio ≥ 0.0975 (AUC 0.81). | [161] |
| 317 participants from the Swedish BioFINDER-2 cohort. | Plasma Aβ1-40 Aβ1-42 | ECL immunoassay | Plasma Aβ1-42/Aβ1-40 cut-off to differentiate between healthy and AD patients = 0.16. No data on accuracy, sensitivity, or specificity provided. | [162] |
| Population, Sample Size | Biomarker | Analytical Platform | Cut-Off/Threshold | Reference |
|---|---|---|---|---|
| 648 individuals (107 with MCI and 78 with AD) from the Stanford Aging and Memory study (SAMS) and the Stanford University Alzheimer’s Disease Research Center (ADRC) cohorts. | Plasma p-tau181. | Automated chemiluminescent enzyme immunoassay (Lumipulse). | p-tau181: 2.35 pg/mL (AUC 0.96, sensitivity 70.6%, specificity 93.3%). | [167] |
| 234 individuals from the Translational Biomarkers in Aging and Dementia (TRIAD) cohort (60 MCI and 32 AD patients). | Plasma p-tau181 and p-tau231. | Simoa. | p-tau181: 15.085 pg/mL (AUC 0.96, sensitivity 87.5%, specificity 93.3%). p-tau231: 17.652 pg/mL (AUC 0.94, sensitivity 81.2%, specificity 93.3%). | [168] |
| A community-based cohort including 1329 participants (153 with MCI and 15 with dementia). | Plasma p-tau181 and p-tau217. | ECL-based assay. | p-tau181 ≥ 1.57 pg/mL (AUC 0.80). p-tau217 ≥ 0.25 pg/mL (AUC 0.85). | [63] |
| 231 participants from the BioFINDER 2 cohort, of which 135 were cognitively unimpaired, and 96 were amyloid-PET positive, 18 being also tau-PET positive. | Plasma p-tau217. | Simoa and ECL. | Correlation with Aβ positivity: p-tau217 ≥ 0.07 pg/mL with Simoa (AUC 0.85, sensitivity 75%, specificity 85%). p-tau ≥ 0.26 pg/mL with ECL (AUC 0.88, sensitivity 84%, specificity 81%). Progression to AD dementia: p-tau ≥ 0.08 pg/mL with Simoa (AUC 0.88, sensitivity 81%). p-tau217 ≥ 0.31 pg/mL with ECL AUC 0.89, sensitivity 85%, specificity 84%). | [169] |
| 397 participants, 22% with MCI and 21% with dementia. | Plasma p-tau217. | Simoa. | Correlation with positive Aβ-PET: p-tau217 ≥ 126.7 fg/mL (sensitivity 79%, specificity 89%). Identifying cognitively impaired participants: p-tau217 ≥ 126.7 fg/mL (sensitivity 82%, specificity 83%). | [170] |
| 1189 participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. | Plasma p-tau181. | Simoa. | p-tau181 ≥ 18.85 pg/mL (AUC 0.84, sensitivity 81.1%, specificity 81.6%). | [171] |
| 144 patients recruited by the Centre for Memory Disorders at the University Hospital of Cologne, 54 with AD. | Plasma p-tau181. | Simoa. | p-tau181 ≥ 12.2 pg/mL (AUC 0.85, sensitivity 80%, specificity 79%). | [172] |
| 317 participants from the Swedish BioFINDER-2 cohort. | Plasma p-tau181 and p-tau217. | ECL. | Thresholds: p-tau181: 7.48 pg/mL; p-tau217: 3.04 pg/mL. | [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunkoo, V.S.; Jurcau, A.; Les, M.; Cristian, A.; Militaru, M.; Marge, C.; Iovanovici, D.C.; Jurcau, M.C. Circulating Biomarkers for the Early Diagnosis of Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 7268. https://doi.org/10.3390/ijms26157268
Nunkoo VS, Jurcau A, Les M, Cristian A, Militaru M, Marge C, Iovanovici DC, Jurcau MC. Circulating Biomarkers for the Early Diagnosis of Alzheimer’s Disease. International Journal of Molecular Sciences. 2025; 26(15):7268. https://doi.org/10.3390/ijms26157268
Chicago/Turabian StyleNunkoo, Vharoon Sharma, Anamaria Jurcau, Mihaela Les, Alexander Cristian, Marius Militaru, Cristian Marge, Diana Carina Iovanovici, and Maria Carolina Jurcau. 2025. "Circulating Biomarkers for the Early Diagnosis of Alzheimer’s Disease" International Journal of Molecular Sciences 26, no. 15: 7268. https://doi.org/10.3390/ijms26157268
APA StyleNunkoo, V. S., Jurcau, A., Les, M., Cristian, A., Militaru, M., Marge, C., Iovanovici, D. C., & Jurcau, M. C. (2025). Circulating Biomarkers for the Early Diagnosis of Alzheimer’s Disease. International Journal of Molecular Sciences, 26(15), 7268. https://doi.org/10.3390/ijms26157268