
Exploring Cirrhosis: Insights into Advances in Therapeutic Strategies

Abstract

1. Introduction
2. Cellular Players in Cirrhosis Progression
3. Etiology and Pathophysiology of Liver Cirrhosis
4. Molecular Mechanisms Underlying Metabolic Dysfunction
5. Liver Fibrosis–Cirrhosis Correlation
6. Therapeutic Insights from Molecular Pathways

{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Key Interventions/Agents | Mechanism of Action | Key References |
|---|---|---|---|
| AMPK Activators | Metformin, AICAR | Activate AMPK → inhibit ACC → promote FAO, enhance mitochondrial biogenesis. | [84,85,86,87,88,89] |
| FGF21 Agonists | Pegbelfermin (FGF21 analogs) | Enhance FAO, mitochondrial respiration, and insulin sensitivity. | [64,91,92,93,94] |
| Mitochondrial Therapies | Urolithin A, MitoQ, SS-31, NAD+ precursors, mARC1 knockdown, NO-primed EVs | Restore mitophagy, antioxidant activity, mitochondrial biogenesis, and protein homeostasis. | [73,117,118,119,120,121,122,123,169] |
| Inflammatory Pathway Modulation | Mesenchymal stem cells, NLRP3 inhibitors, NF-kB pathway inhibitors | Reduce pro-inflammatory cytokines (e.g., TNF-α, IL-1β), inhibit fibrogenesis, and promote regeneration. | [124,125,126,127,129,130,131,170] |
| Hormonal and Endocrine Modulation | IGF-1 supplementation, leptin/adiponectin modulation, MSCs affecting the HPG axis | IGF-1 and adipocytokine imbalance, modulate the hormonal axes. | [132,133,134,138,139] |
| Glycogen Metabolism and Insulin Resistance Modulation | Metformin, Branched-Chain Amino Acids (BCAAs), leptin/adiponectin | Improve insulin signaling (IRS2/PI3K/Akt), promote glycogen synthesis, and muscle function. | [140,141,142,165] |
| Cardiovascular-Metabolic Interventions | SGLT2 inhibitors, thiazolidinediones (TZDs) | Improve insulin sensitivity, modulate cardiovascular risk factors. | [166,167] |
| Nutritional Interventions | Omega-3 fatty acids, vitamin E, and coenzyme Q10 | Antioxidant and anti-inflammatory effects enhance FAO and mitochondrial health. | [83,168] |
7. Summary
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gao, B.; Zhang, X.; Huang, Y.; Yang, Z.; Zhang, Y.; Zhang, W.; Gao, Z.H.; Xue, D. Coding and Non-Coding Gene Regulatory Networks Underlie the Immune Response in Liver Cirrhosis. PLoS ONE 2017, 12, e0174142. [Google Scholar] [CrossRef] [PubMed]
- Gribilas, G.; Ζάρρος, A.; Zira, A.; Giaginis, C.; Tsourouflis, G.; Liapi, C.; Spiliopoulou, C.; Theocharis, S. Involvement of Hepatic Stimulator Substance in Experimentally Induced Fibrosis and Cirrhosis in the Rat. Dig. Dis. Sci. 2008, 54, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Huai, Q.; Zhu, C.; Zhang, X.; Dai, H.; Li, X.; Wang, H. Mesenchymal stromal/stem cells and their extracellular vesicles in liver diseases: Insights on their immunomodulatory roles and clinical applications. Cell Biosci. 2023, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Abu Helal, R.; Muturi, H.T.; Lee, A.D.; Li, W.; Ghadieh, H.E.; Najjar, S.M. Aortic Fibrosis in Insulin-Sensitive Mice with Endothelial Cell-Specific Deletion of Ceacam1 Gene. Int. J. Mol. Sci. 2022, 23, 4335. [Google Scholar] [CrossRef]
- Yarde, S.S.; Cheng, X. Primary and Secondary Bile Acids Activate Hepatic Stellate Cells. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef]
- Qiu, Z.; Milichko, V.A.; Zhou, Y.; Jing, H.; Lu, Q.; Wang, H.; Kang, B.; Wang, X.; Li, N. “Kupffer Cell Teleportation” Strategy for Liver Fibrosis Alleviation Based on Hybrid Polymer- Bimetallic Sequential Delivery System. Adv. Funct. Mater. 2024, 34, 2309690. [Google Scholar] [CrossRef]
- Natarajan, V.; Moeller, M.; Casey, C.A.; Harris, E.N.; Kidambi, S. Matrix Stiffness Regulates Liver Sinusoidal Endothelial Cell Function Mimicking Responses in Fatty Liver Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liao, Y.; Zhou, C.; Duan, Y.; Liu, X.; Yue, J.; Li, X.; Wu, J.; Wan, C.; Zhang, L. Liver sinusoidal endothelial S1pr2 regulates experimental liver fibrosis through YAP/TGF-β signaling pathway. FASEB J. 2023, 37, e22905. [Google Scholar] [CrossRef]
- Micu, S.; Manea, M.; Popoiag, R.; Nikolić, D.; Andrada, D.; Patti, A.M.; Musat, M.; Bălălău, C.; Rogoveanu, A.; Rizzo, M.; et al. Alcoholic Liver Cirrhosis, More Than a Simple Hepatic Disease—A Brief Review of the Risk Factors Associated with Alcohol Abuse. J. Mind Med. Sci. 2019, 6, 232–236. [Google Scholar] [CrossRef]
- Mishra, D.; Dash, K.R.; Khatua, C.; Panigrahi, S.; Parida, P.K.; Behera, S.K.; Barik, R.; Pradhan, S.; Sahu, S.K.; Thakur, B.; et al. A Study on the Temporal Trends in the Etiology of Cirrhosis of Liver in Coastal Eastern Odisha. Euroasian J. Hepato Gastroenterol. 2020, 10, 1–6. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, Y.; Jin, W. Liver cirrhosis: Molecular mechanisms and therapeutic interventions. MedComm 2024, 5, e721. [Google Scholar] [CrossRef]
- Babuta, M.; Nagesh, P.T.; Datta, A.A.; Remotti, V.; Zhuang, Y.; Mehta, J.; Lami, F.; Wang, Y.; Szabo, G. Combined Insults of a MASH Diet and Alcohol Binges Activate Intercellular Communication and Neutrophil Recruitment via the NLRP3-IL-1β Axis in the Liver. Cells 2024, 13, 960. [Google Scholar] [CrossRef]
- Rezaei, N.; Asadi-Lari, M.; Sheidaei, A.; Khademi, S.; Gohari, K.; Delavari, F.; Delavari, A.; Abdolhamidi, E.; Chegini, M.; Rezaei, N.; et al. Liver Cirrhosis Mortality at National and Provincial Levels in Iran Between 1990 and 2015: A Meta Regression Analysis. PLoS ONE 2019, 14, e0198449. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Zhang, X.; Zhao, W.; Li, D.; Xu, C.; Wu, Z.; Xie, F.; Li, X. Lack of Mof Reduces Acute Liver Injury by Enhancing Transcriptional Activation of Igf1. J. Cell. Physiol. 2021, 236, 6559–6570. [Google Scholar] [CrossRef] [PubMed]
- Beudeker, B.J.B.; Groothuismink, Z.M.A.; Eijk, A.A.v.d.; Debes, J.D.; Boonstra, A. Circulating Cytokines Reflect the Etiology-Specific Immune Environment in Cirrhosis and HCC. Cancers 2022, 14, 4900. [Google Scholar] [CrossRef] [PubMed]
- Rowell, R.; Anstee, Q.M. An Overview of the Genetics, Mechanisms and Management of NAFLD and ALD. Clin. Med. 2015, 15, s77–s82. [Google Scholar] [CrossRef] [PubMed]
- Malnick, S.; Maor, Y. The Interplay Between Alcoholic Liver Disease, Obesity, and the Metabolic Syndrome. Visc. Med. 2020, 36, 198–205. [Google Scholar] [CrossRef]
- Decraecker, M.; Dutartre, D.; Hiriart, J.B.; Irlès-Depé, M.; Grottes, H.M.d.; Chermak, F.; Foucher, J.; Delamarre, A.; Lédinghen, V.d. Long-term Prognosis of Patients with Alcohol-related Liver Disease or Non-alcoholic Fatty Liver Disease According to Metabolic Syndrome or Alcohol Use. Liver Int. 2021, 42, 350–362. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Green, P.; Lowy, E.; Mun, E.J.; Berry, K. Differences in Hepatocellular Carcinoma Risk, Predictors and Trends Over Time According to Etiology of Cirrhosis. PLoS ONE 2018, 13, e0204412. [Google Scholar] [CrossRef]
- Abogalala, S.A.; El-Ghafar, M.T.A.; El-Sawy, A.A.; Al-Sheikh, M.R. Frequency of Spontaneous Bacterial Peritonitis in Patients of Liver Cirrhosis with Ascites at Tanta University Hospitals. Med. J. Cairo Univ. 2019, 87, 147–152. [Google Scholar] [CrossRef]
- Sharma, B.; Marwah, R.; Raina, S.; Sharma, N.; Kaushik, M.; Kaushal, S.S. A Study on the Etiology of Cirrhosis of Liver in Adults Living in the Hills of Himachal Pradesh, India. Trop. Gastroenterol. 2016, 37, 37–41. [Google Scholar] [CrossRef]
- Gao, L.; Meng, F.; Cheng, J.; Li, H.; Han, J.; Zhang, W. Prediction of Oesophageal Varices in Patients with Primary Biliary Cirrhosis by Non-Invasive Markers. Arch. Med. Sci. 2017, 2, 370–376. [Google Scholar] [CrossRef]
- Ren, A.; He, W.; Rao, J.; Ye, D.; Cheng, P.; Jian, Q.; Fu, Z.; Zhang, X.; Deng, R.; Gao, Y.; et al. Dysregulation of Innate Cell Types in the Hepatic Immune Microenvironment of Alcoholic Liver Cirrhosis. Front. Immunol. 2023, 14, 1034356. [Google Scholar] [CrossRef]
- Kim, M.Y.; Cho, M.-Y.; Baik, S.K.; Park, H.J.; Jeon, H.K.; Im, C.K.; Won, C.S.; Kim, J.W.; Kim, H.S.; Kwon, S.O.; et al. Histological Subclassification of Cirrhosis Using the Laennec Fibrosis Scoring System Correlates with Clinical Stage and Grade of Portal Hypertension. J. Hepatol. 2011, 55, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cui, X.; Li, P.; Feng, C.; Wang, L.; Wang, H.; Zhou, X.; Yang, B.; Lv, F.; Li, T. Suppression of MicroRNA-219-5p Activates Keratinocyte Growth Factor to Mitigate Severity of Experimental Cirrhosis. Cell. Physiol. Biochem. 2016, 40, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mazza, G.; Telese, A.; Al-Akkad, W.; Frenguelli, L.; Levi, A.; Marrali, M.; Longato, L.; Thanapirom, K.; Vilia, M.G.; Lombardi, B.; et al. Cirrhotic Human Liver Extracellular Matrix 3D Scaffolds Promote Smad-Dependent TGF-β1 Epithelial Mesenchymal Transition. Cells 2019, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ross, R.A.; Zhao, S.; Tu, W.; Liangpunsakul, S.; Wang, L. LncRNA AK054921 and AK128652 Are Potential Serum Biomarkers and Predictors of Patient Survival with Alcoholic Cirrhosis. Hepatol. Commun. 2017, 1, 513–523. [Google Scholar] [CrossRef]
- Amir, I.; Iqbal, M.; Qasim, M.; Solangi, R. Cirrhosis of Liver: Etiological Factors, Complicatins and Prognosis. J. Liaquat Univ. Med. Health Sci. 2008, 7, 61–66. [Google Scholar] [CrossRef]
- Ye, F.; Zhai, M.; Long, J.; Shu, B.; Liu, C.; Gong, Y.; Li, L.; Zhang, D.; He, C.; Liu, S. The Burden of Liver Cirrhosis in Mortality: Results from the Global Burden of Disease Study 2017. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Drolz, A.; Horvatits, T.; Roedl, K.; Rutter, K.; Brunner, R.; Zauner, C.; Schellongowski, P.; Heinz, G.; Funk, G.C.; Trauner, M.; et al. Acid–base Status and Its Clinical Implications in Critically Ill Patients with Cirrhosis, Acute-on-Chronic Liver Failure and Without Liver Disease. Ann. Intensive Care 2018, 8, 48. [Google Scholar] [CrossRef]
- Wang, X.; Ni, Y.; Wang, Z.; Li, C.; Hui, X.; Xu, H. Metabolic factors for liver cirrhosis: A Mendelian randomization study. Medicine 2024, 103, e40507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zou, Y.; Zhang, M.; Bai, S.; Tao, K.; Wu, J.; Shi, Y.; Wu, Y.; Lu, Y.; He, K.; et al. Single-cell RNA Sequencing Reveals the Heterogeneity and Intercellular Communication of Hepatic Stellate Cells and Macrophages During Liver Fibrosis. Medcomm 2023, 4, e378. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Li, J.; Xu, Y.; Yang, L.; Liu, X.; Yang, J.; Tai, W. Transcriptomic and Metabolomic Analysis of Liver Cirrhosis. Comb. Chem. High Throughput Screen. 2024, 27, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.H.; Yuan, X.; Li, J.F.; Xie, Y.; Zhang, A.Z.; Wang, X.L.; Yang, L.; Liu, C.X.; Liang, W.; Pang, L.; et al. Bioinformatics-Based Screening of Key Genes for Transformation of Liver Cirrhosis to Hepatocellular Carcinoma. J. Transl. Med. 2020, 18, 40. [Google Scholar] [CrossRef]
- Ye, J.; Lv, L.; Wu, W.; Li, Y.; Shi, D.; Fang, D.; Guo, F.; Jiang, H.; Yan, R.; Ye, W.; et al. Butyrate Protects Mice Against Methionine–Choline-Deficient Diet-Induced Non-Alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front. Microbiol. 2018, 9, 1967. [Google Scholar] [CrossRef]
- Huang, Y.C.; Niu, M.; Jing, J.; Zhang, Z.-T.; Zhao, X.; Chen, S.; Li, S.; Shi, Z.; Huang, A.; Zou, Z.; et al. Metabolomic Analysis Uncovers Energy Supply Disturbance as an Underlying Mechanism of the Development of Alcohol-Associated Liver Cirrhosis. Hepatol. Commun. 2021, 5, 961–975. [Google Scholar] [CrossRef]
- Shoaib, N.; Khan, Z.; Ibrahim, M.; Hafeez, A.; Fatima, A.; Imran, H.; Saleem, F.; Askari, S.M.H.; Gull, S. Dyslipidemia and Impaired Liver Function Biomarkers in Patients with Hepatitis B Liver Cirrhosis. J. Taibah Univ. Med. Sci. 2023, 18, 748–754. [Google Scholar] [CrossRef]
- Meikle, P.J.; Mundra, P.A.; Wong, G.; Rahman, K.; Huynh, K.; Barlow, C.K.; Duly, A.; Haber, P.; Whitfield, J.B.; Seth, D. Circulating Lipids Are Associated with Alcoholic Liver Cirrhosis and Represent Potential Biomarkers for Risk Assessment. PLoS ONE 2015, 10, e0130346. [Google Scholar] [CrossRef]
- Axley, P.; Ahmed, Z.; Arora, S.; Haas, A.; Kuo, Y.F.; Kamath, P.S.; Singal, A.K. NASH Is the Most Rapidly Growing Etiology for Acute-on-Chronic Liver Failure-Related Hospitalization and Disease Burden in the United States: A Population-Based Study. Liver Transplant. 2019, 25, 695–705. [Google Scholar] [CrossRef]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The Global, Regional, and National Burden of Cirrhosis by Cause in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Lee, B.-W. Association between Impaired Ketogenesis and Metabolic-Associated Fatty Liver Disease. Biomolecules 2023, 13, 1506. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, L.; Csiki, I.E.; Iacob, S.; Gheorghe, C.; Trifan, A.; Grigorescu, M.; Moţoc, A.; Suceveanu, A.I.; Curescu, M.; Căruntu, F.A.; et al. Hepatitis Delta Virus Infection in Romania: Prevalence and Risk Factors. J. Gastrointest. Liver Dis. 2015, 24, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Chvatal-Medina, M.; Li, Y.; Trillos-Almanza, M.C.; Post, A.; Connelly, M.A.; Moshage, H.; Bakker, S.J.L.; Meijer, V.E.d.; Blokzijl, H.; Dullaart, R.P.F. Plasma Beta-Hydroxybutyrate and All-Cause Mortality in Patients with Liver Cirrhosis. Biomedicines 2025, 13, 1120. [Google Scholar] [CrossRef]
- Li, M.; Wei, Z.; Su, J.; Wu, X.; Xie, X.; You, H.; Jia, J.; Kong, Y. Changing Spectrum and Mortality Disparities of Etiology of Liver Cirrhosis in Beijing, China. J. Med. Virol. 2024, 96, e29405. [Google Scholar] [CrossRef]
- Mokdad, A.A.; López, A.D.; Shahraz, S.; Lozano, R.; Mokdad, A.H.; Stanaway, J.D.; Murray, C.J.L.; Naghavi, M. Liver Cirrhosis Mortality in 187 Countries Between 1980 and 2010: A Systematic Analysis. BMC Med. 2014, 12, 145. [Google Scholar] [CrossRef]
- Kumar, K.V.S.H.; Pawah, A.K.; Manrai, M. Occult Endocrine Dysfunction in Patients with Cirrhosis of Liver. J. Fam. Med. Prim. Care 2016, 5, 576. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Cheng, L.; Guo, M.; Li, D.; Li, X.; Tan, Y.J.; Ma, S.; Li, S.; Yang, Y.; et al. CD137-Mediated Pathogenesis from Chronic Hepatitis to Hepatocellular Carcinoma in Hepatitis B Virus-Transgenic Mice. J. Immunol. 2010, 185, 7654–7662. [Google Scholar] [CrossRef]
- Zaghla, H.; Omer, E. The Incidence of Myocardial Diastolic Dysfunction in Patients with Decompensated Liver Disease. World J. Cardiovasc. Dis. 2019, 9, 404–418. [Google Scholar] [CrossRef]
- Rani, R.; Gandhi, C.R. Stellate cell in hepatic inflammation and acute injury. J. Cell. Physiol. 2023, 238, 1226–1236. [Google Scholar] [CrossRef]
- Zeng, H.; Fang, L.; Yang, Z.; Zhao, X.; Chen, H.; Xing, P.; Niu, Z.; Li, Z.; Li, Z.; Zhao, J.; et al. Prognostic and Predictive Effects of New Steatotic Liver Disease Nomenclatures: A Large Population-based Study. Medcomm 2025, 6, e70087. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, Y.; Liu, Y.; Hu, H.; Cheng, Q.; Yang, F.; Pei, L.; Zhou, Y.; Li, Y.; Lin, S. High Albumin Level Is Associated with Regression of Glucose Metabolism Disorders Upon Resolution of Acute Liver Inflammation in Hepatitis B-Related Cirrhosis. Front. Cell. Infect. Microbiol. 2022, 12, 721138. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Xue, C.; Li, S.; Zao, X.; Li, X.; Liu, Q.; Cao, X.; Wang, W.; Qi, W.; Du, H.; et al. Liver cirrhosis: Current status and treatment options using western or traditional Chinese medicine. Front. Pharmacol. 2024, 15, 1381476. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; Terrault, N.A.; Tacke, F.; Gluud, L.L.; Arrese, M.; Bugianesi, E.; Loomba, R. Global epidemiology of cirrhosis—Aetiology, trends and predictions. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 388–398. [Google Scholar] [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of Liver Fibrosis and Its Role in Liver Cancer. Exp. Biol. Med. 2020, 245, 96–108. [Google Scholar] [CrossRef]
- Kim, S.M.; Choi, J.E.; Hur, W.; Kim, J.H.; Hong, S.W.; Lee, E.B.; Lee, J.H.; Li, T.Z.; Sung, P.S.; Yoon, S.K. RAR-Related Orphan Receptor Gamma (ROR-γ) Mediates Epithelial-Mesenchymal Transition of Hepatocytes During Hepatic Fibrosis. J. Cell. Biochem. 2017, 118, 2026–2036. [Google Scholar] [CrossRef]
- Wang, H.; Chang, Y.; Liu, X.; Liu, L.; Hua, M.; Li, A. Protective Effects of Baicalin on Diethyl nitrosamine-induced Liver Cirrhosis by Suppressing Oxidative Stress and Inflammation. Chem. Biol. Drug Des. 2023, 103, e14386. [Google Scholar] [CrossRef]
- Lin, L.; Zhou, F.; Shen, S.; Zhang, T. Fighting Liver Fibrosis with Naturally Occurring Antioxidants. Planta Medica 2018, 84, 1318–1333. [Google Scholar] [CrossRef]
- Kojima-Yuasa, A.; Goto, M.; Yoshikawa, E.; Morita, Y.; Sekiguchi, H.; Sutoh, K.; Usumi, K.; Matsui-Yuasa, I. Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt Against Ethanol-Induced Liver Injury in Rats. Mar. Drugs 2016, 14, 232. [Google Scholar] [CrossRef]
- Issa, D.; Patel, V.; Sanyal, A.J. Future Therapy for Non-alcoholic Fatty Liver Disease. Liver Int. 2018, 38, 56–63. [Google Scholar] [CrossRef]
- Fausther, M.; Lavoie, É.G.; Dranoff, J.A. Contribution of Myofibroblasts of Different Origins to Liver Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.; Weiss, E.; Paradis, V.; Wan, J.; Mabire, M.; Sukriti, S.; Rautou, P.E.; Albuquerque, M.; Picq, O.; Gupta, A.C.; et al. Mucosal-Associated Invariant T Cells Are a Profibrogenic Immune Cell Population in the Liver. Nat. Commun. 2018, 9, 2146. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Peng, Y.; Yang, S. MicroRNA-146a Regulates the Transformation from Liver Fibrosis to Cirrhosis in Patients with Hepatitis B via Interleukin-6. Exp. Ther. Med. 2019, 17, 4670–4676. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, Y.B.; Hu, H. Metabolic Role of Fibroblast Growth Factor 21 in Liver, Adipose and Nervous System Tissues. Biomed. Rep. 2017, 6, 495–502. [Google Scholar] [CrossRef]
- Razdan, A.; Singh, S.; Akhtar, S.; Yogi, J.P.; Fiza, B.; Sinha, M. Evaluation of Thyroid Hormones in Liver Cirrhosis Patients on the Basis of Child Pugh Score. Int. J. Health Sci. 2022, 6, 2462–2470. [Google Scholar] [CrossRef]
- Ge, L.; Shi, B.; Song, Y.E.; Li, Y.; Wang, S.; Wang, X. Clinical Value of Real-Time Elastography Quantitative Parameters in Evaluating the Stage of Liver Fibrosis and Cirrhosis. Exp. Ther. Med. 2015, 10, 983–990. [Google Scholar] [CrossRef]
- Pawlak, M.; Baugé, E.; Bourguet, W.; Bosscher, K.D.; Lalloyer, F.; Tailleux, A.; Lebherz, C.; Lefèbvre, P.; Staels, B. The Transrepressive Activity of Peroxisome Proliferator-Activated Receptor Alpha Is Necessary and Sufficient to Prevent Liver Fibrosis in Mice. Hepatology 2014, 60, 1593–1606. [Google Scholar] [CrossRef]
- Verloh, N.; Utpatel, K.; Haimerl, M.; Zeman, F.; Beyer, L.P.; Fellner, C.; Brennfleck, F.W.; Dahlke, M.H.; Stroszczynski, C.; Evert, M.; et al. Detecting Liver Fibrosis with Gd-Eob-Dtpa-Enhanced MRI: A Confirmatory Study. Sci. Rep. 2018, 8, 6207. [Google Scholar] [CrossRef]
- Hu, Z.; Kurihara, T.; Sun, Y.; Cetin, Z.; Florentino, R.M.; Faccioli, L.A.P.; Liu, Z.; Yang, B.R.; Ostrowska, A.; Soto–Gutiérrez, A.; et al. A Rat Model of Cirrhosis with Well-Differentiated Hepatocellular Carcinoma Induced by Thioacetamide. Front. Gastroenterol. 2024, 3, 1427820. [Google Scholar] [CrossRef]
- Iwasa, M.; Kobayashi, Y.; Mifuji-Moroka, R.; Hara, N.; Miyachi, H.; Sugimoto, R.; Tanaka, H.; Fujita, N.; Gabazza, E.C.; Takei, Y. Branched-Chain Amino Acid Supplementation Reduces Oxidative Stress and Prolongs Survival in Rats with Advanced Liver Cirrhosis. PLoS ONE 2013, 8, e70309. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, L.; Li, X.; Song, J.; Wu, X.; Zhou, S. Inositol-Requiring Enzyme 1-Mediated Endoplasmic Reticulum Stress Triggers Apoptosis and Fibrosis Formation in Liver Cirrhosis Rat Models. Mol. Med. Rep. 2014, 11, 2941–2946. [Google Scholar] [CrossRef]
- Zhou, L.; Shen, H.; Li, X.; Wang, H. Endoplasmic Reticulum Stress in Innate Immune Cells—A Significant Contribution to Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 951406. [Google Scholar] [CrossRef] [PubMed]
- Türkseven, S.; Bolognesi, M.; Brocca, A.; Pesce, P.; Angeli, P.; Pascoli, M.D. Mitochondria-Targeted Antioxidant Mitoquinone Attenuates Liver Inflammation and Fibrosis in Cirrhotic Rats. AJP Gastrointest. Liver Physiol. 2020, 318, G298–G304. [Google Scholar] [CrossRef] [PubMed]
- Stefano, J.T.; Pereira, I.V.A.; Torres, M.M.; Bida, P.M.; Coelho, A.M.M.; Xerfan, M.P.; Cogliati, B.; Barbeiro, D.F.; Mazo, D.F.d.C.; Kubrusly, M.S.; et al. Sorafenib Prevents Liver Fibrosis in a Non-Alcoholic Steatohepatitis (NASH) Rodent Model. Braz. J. Med. Biol. Res. 2015, 48, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lin, D.; Zhang, Y.; Zhang, Y.; Li, N. Plasma microRNAs as Predictive Biomarkers of Liver Fibrosis and Cirrhosis in Patients with HBV-associated Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Luerken, L.; Dollinger, M.; Goetz, A.; Utpatel, K.; Doppler, M.; Weiss, J.; Uller, W.; Ignee, A.; Verloh, N.; Haimerl, M. Diagnostic Accuracy of Indocyanine Green Clearance Test for Different Stages of Liver Fibrosis and Cirrhosis. Diagnostics 2023, 13, 2663. [Google Scholar] [CrossRef]
- Thabet, K.; Asimakopoulos, A.; Shojaei, M.; Romero-Gómez, M.; Mangia, A.; Irving, W.L.; Berg, T.; Dore, G.J.; Grønbæk, H.; Sheridan, D.; et al. MBOAT7 Rs641738 Increases Risk of Liver Inflammation and Transition to Fibrosis in Chronic Hepatitis C. Nat. Commun. 2016, 7, 12757. [Google Scholar] [CrossRef]
- Cross, T.J.; Calvaruso, V.; Maimone, S.; Carey, I.; Chang, T.S.; Pleguezuelo, M.; Manousou, P.; Quaglia, A.; Grillo, F.; Dhillon, A.P.; et al. Prospective Comparison of Fibroscan, King’s Score and Liver Biopsy for the Assessment of Cirrhosis in Chronic Hepatitis C Infection. J. Viral Hepat. 2010, 17, 546–554. [Google Scholar] [CrossRef]
- Lédinghen, V.d.; Vergniol, J. Transient Elastography for the Diagnosis of Liver Fibrosis. Expert Rev. Med. Devices 2010, 7, 811–823. [Google Scholar] [CrossRef]
- Hu, W.C. TGFβ Producing Regulatory T Cells Related Tolerable Immunities and Their Relations to Scleroderma and End Organ Failures Including Heart Failure, Liver Cirrhosis, Uremia, and Pulmonary Fibrosis. Preprints 2024. [Google Scholar] [CrossRef]
- Borssén, Å.D.; Palmqvist, R.; Kechagias, S.; Marschall, H.U.; Bergquist, A.; Rorsman, F.; Weiland, O.; Verbaan, H.; Nyhlin, N.; Nilsson, E.; et al. Histological Improvement of Liver Fibrosis in Well-Treated Patients with Autoimmune Hepatitis. Medicine 2017, 96, e7708. [Google Scholar] [CrossRef]
- Alboraie, M.; Khairy, M.; Elsharkawy, A.; Asem, N.; El-Kassas, M.; Elgendy, A.-E.A.-E.; Nagdy, H.; Shady, Z.; Eliwa, A.; El-Ansary, A.R.; et al. Role of Liver Biopsy Versus Non-Invasive Biomarkers for Diagnosis of Significant Fibrosis and Cirrhosis: A Web-Based Survey. Egypt. Liver J. 2021, 11, 94. [Google Scholar] [CrossRef]
- Hoge, A.; Tabar, V.; Donneau, A.-F.; Dardenne, N.; Degée, S.; Timmermans, M.; Nisolle, M.; Guillaume, M.; Castronovo, V. Imbalance Between Omega-6 and Omega-3 Polyunsaturated Fatty Acids in Early Pregnancy Is Predictive of Postpartum Depression in a Belgian Cohort. Nutrients 2019, 11, 876. [Google Scholar] [CrossRef]
- Henriksen, B.S.; Curtis, M.E.; Fillmore, N.; Cardon, B.; Thomson, D.M.; Hancock, C.R. The Effects of Chronic AMPK Activation on Hepatic Triglyceride Accumulation and Glycerol 3-Phosphate Acyltransferase Activity with High Fat Feeding. Diabetol. Metab. Syndr. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Wu, F.; Chen, G.; Dong, H.; Li, J.; Zhao, Y.; Xu, L.; Zou, X.; Lu, F. Diosgenin Ameliorates Palmitic Acid-Induced Lipid Accumulation via AMPK/ACC/CPT-1A and SREBP-1c/FAS Signaling Pathways in LO2 Cells. BMC Complement. Altern. Med. 2019, 19, 255. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vančura, A. Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Efstathiou, N.E.; Konstantinou, E.K.; Maidana, D.E.; Miller, J.W.; Young, L.H.; Vavvas, D.G. AICAR Suppresses TNF-α-induced Complement Factor B in RPE Cells. Sci. Rep. 2017, 7, 17651. [Google Scholar] [CrossRef]
- Zheng, T.; Yang, X.; Wu, D.; Xing, S.; Bian, F.; Li, W.; Chi, J.; Bai, X.; Wu, G.; Chen, X.; et al. Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3K/Akt/GSK3β pathway. Br. J. Pharmacol. 2015, 172, 3284–3301. [Google Scholar] [CrossRef]
- Logie, L.; Lees, Z.; Allwood, J.W.; McDougall, G.J.; Beall, C.; Rena, G. Regulation of Hepatic Glucose Production and AMPK by AICAR but Not by Metformin Depends on Drug Uptake Through the Equilibrative Nucleoside Transporter 1 (ENT1). Diabetes Obes. Metab. 2018, 20, 2748–2758. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, L.; Zhu, X.; Bian, H.; Gao, X.; Xia, M. Advances in Management of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Mechanisms to Therapeutics. Lipids Health Dis. 2024, 23, 95. [Google Scholar] [CrossRef]
- Boparai, R.K.; Arum, O.; Miquet, J.G.; Masternak, M.M.; Bartke, A.; Khardori, R. Resistance to the Beneficial Metabolic Effects and Hepatic Antioxidant Defense Actions of Fibroblast Growth Factor 21 Treatment in Growth Hormone-Overexpressing Transgenic Mice. Int. J. Endocrinol. 2015, 2015, 282375. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast Growth Factor 21 Regulates Energy Metabolism by Activating the AMPK–SIRT1–PGC-1α Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jeleník, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.A.; Minnich, A.; Sanyal, A.J.; Loomba, R.; Du, S.; Schwarz, J.H.; Ehman, R.L.; Karsdal, M.A.; Leeming, D.J.; Cizza, G.; et al. Effect of Pegbelfermin on NASH and Fibrosis-Related Biomarkers and Correlation with Histological Response in the FALCON 1 Trial. JHEP Rep. 2023, 5, 100661. [Google Scholar] [CrossRef]
- Anjeza, E.; Régis, M. The regulation of FGF21 gene expression by metabolic factors and nutrients. Horm. Mol. Biol. Clin. Investig. 2017, 30, 20160016. [Google Scholar]
- Hyunbae, K.; Roberto, M.; Ze, Z.; Lin, C.; Juan, C.; Ren, Z.; Kezhong, Z. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 2014, 155, 769–782. [Google Scholar]
- Alison, I. Nutritional Regulation of the Hepatokine FGF21 in the Liver: Interdependence of the Transcription Factors ChREBP and PPARα. Ph.D. Dissertation, Université Sorbonne-Paris-Cité, Paris, France, 2017. [Google Scholar]
- Xiao-shan, W.; Xuan, W.; Jian, X.; Xiaokun, L.; Xiaokun, L.; Huiping, Z. Corrigendum to “ATF4- and CHOP-Dependent Induction of FGF21 through Endoplasmic Reticulum Stress”. BioMed Res. Int. 2018, 2018, 3218606. [Google Scholar] [CrossRef]
- Yasaman, B.; Weijuan, S.; Lili, T.; Tianru, J. 370-OR: Hepatic FGF21 Expression Is Under the Regulation of the Canonical Wnt Signaling Pathway. Diabetes 2020, 69, 370-OR. [Google Scholar] [CrossRef]
- Shuqin, C.; Huating, L.; Jing, Z.; Shan, J.; Mingliang, Z.; Yilan, X.; Kun, D.; Ying, Y.; Qichen, F.; Weiping, J. Identification of Sp1 as a Transcription Activator to Regulate Fibroblast Growth Factor 21 Gene Expression. BioMed Res. Int. 2017, 2017, 8402035. [Google Scholar] [CrossRef]
- Mark, F.M. Practical prospects for boosting hepatic production of the pro-longevity hormone FGF21. Horm. Mol. Biol. Clin. Investig. 2017, 30, 20150057. [Google Scholar]
- Masahiro, M.; Masahiro, M.; Nadeem, S.; Sakie, K.; Christopher, R.; Ola, L.; Takeshi, N.; Bahareh, H.; Bahareh, H.; Akinori, T.; et al. Hepatic posttranscriptional network comprised of CCR4–NOT deadenylase and FGF21 maintains systemic metabolic homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 7973–7981. [Google Scholar]
- Kaori, N.; Chisaki, I.; Seiko, I.; Ryohei, Y.; Makoto, N.; Izumi, N.; Toshiki, M.; Keiko, Y.-T. Serum FGF21 levels are altered by various factors including lifestyle behaviors in male subjects. Sci. Rep. 2021, 11, 22632. [Google Scholar] [CrossRef]
- Allison Ross, E.; Heather, Y.H.; Nancy, L.H.; Mary Ann, O.R.; Danielle, L.; Julia, C.K.; Sarah, E.S.; Grace, A.M. Fibroblast Growth Factor 21 Is Elevated in HIV and Associated with Interleukin-6. J. Acquir. Immune Defic. Syndr. 2020, 83, e30–e33. [Google Scholar]
- Takeshi, I. Research Perspectives on the Regulation and Physiological Functions of FGF21 and its Association with NAFLD. Front. Endocrinol. 2015, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Ryuto, M.; Makoto, S.; Juan, L.; Jun, I.; Ryuichiro, S. Fibroblast growth factor 21 induction by activating transcription factor 4 is regulated through three amino acid response elements in its promoter region. Biosci. Biotechnol. Biochem. 2016, 80, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, Y.; Yuan, J.; Yang, W.; Mo, Z. Compound C Protects Mice from HFD-Induced Obesity and Nonalcoholic Fatty Liver Disease. Int. J. Endocrinol. 2019, 2019, 3206587. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Hong, K.; Luo, F.; Gu, Q.; Lu, N.; Bai, A. AMPK inhibition blocks ROS-NFκB signaling and attenuates endotoxemia-induced liver injury. PLoS ONE 2014, 9, e86881. [Google Scholar] [CrossRef]
- Petereit, J.; Duncan, O.; Murcha, M.W.; Fenske, R.; Cincu, E.; Cahn, J.; Pružinská, A.; Ivanova, A.; Kollipara, L.; Wortelkamp, S.; et al. Mitochondrial CLPP2 Assists Coordination and Homeostasis of Respiratory Complexes. Plant Physiol. 2020, 184, 148–164. [Google Scholar] [CrossRef]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-Sensitive Mitochondrial Accumulation of Cystathionine Β-Synthase Mediated by Lon Protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef]
- Lee, H.J.; Chung, K.S.; Lee, H.J.; Lee, K.; Lim, J.H.; Song, J. Downregulation of Mitochondrial Lon Protease Impairs Mitochondrial Function and Causes Hepatic Insulin Resistance in Human Liver SK-HEP-1 Cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y.L. Overexpression of Lon Contributes to Survival and Aggressive Phenotype of Cancer Cells Through Mitochondrial Complex I-Mediated Generation of Reactive Oxygen Species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Guillon, B.; Bulteau, A.L.; Wattenhofer-Donzé, M.; Schmucker, S.; Friguet, B.; Puccio, H.; Drapier, J.C.; Bouton, C. Frataxin Deficiency Causes Upregulation of Mitochondrial Lon and ClpP Proteases and Severe Loss of Mitochondrial Fe–S Proteins. FEBS J. 2009, 276, 1036–1047. [Google Scholar] [CrossRef]
- Raut, M.P.; Couto, N.; Pham, T.K.; Evans, C.A.; Noirel, J.; Wright, P.C. Quantitative Proteomic Analysis of the Influence of Lignin on Biofuel Production by Clostridium Acetobutylicum ATCC 824. Biotechnol. Biofuels 2016, 9, 113. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.R.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of Mitochondrial Protease ClpP Protects Mice from Diet-induced Obesity and Insulin Resistance. Embo Rep. 2018, 19, e45009. [Google Scholar] [CrossRef] [PubMed]
- Milić, N.Š.; Dolinar, K.; Miš, K.; Matkovič, U.; Bizjak, M.; Pavlin, M.; Podbregar, M.; Pirkmajer, S. Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1α. Int. J. Mol. Sci. 2021, 22, 8610. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Parekh, M.; Deshpande, N.; Cheung, Q.; Shatz, N.; Kumar, V.; Jurkunas, U.V. Mitochondria-Targeted Antioxidant (MitoQ) and Nontargeted Antioxidant (Idebenone) Mitigate Mitochondrial Dysfunction in Corneal Endothelial Cells. Cornea 2025, 44, 492–503. [Google Scholar] [CrossRef]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef]
- Coyne, E.S.; Nie, Y.; Lee, D.; Pandovski, S.; Yang, T.; Zhou, H.; Rosahl, T.W.; Carballo-Jane, E.; Abdurrachim, D.; Zhou, Y.; et al. Loss of mitochondrial amidoxime-reducing component 1 (mARC1) prevents disease progression by reducing fibrosis in multiple mouse models of chronic liver disease. Hepatol. Commun. 2025, 9, e0637. [Google Scholar] [CrossRef]
- Lewis, L.C.; Chen, L.; Hameed, L.S.; Kitchen, R.R.; Maroteau, C.; Nagarajan, S.R.; Norlin, J.; Daly, C.E.; Szczerbinska, I.; Hjuler, S.T.; et al. Hepatocyte mARC1 promotes fatty liver disease. JHEP Rep. 2023, 5, 100693. [Google Scholar] [CrossRef]
- Peng, F.; Chen, X.; Wu, L.; He, J.; Li, Z.; Hong, Q.; Zhao, Q.; Qian, M.; Wang, X.; Shen, W.; et al. Nitric Oxide-Primed Engineered Extracellular Vesicles Restore Bioenergetics in Acute Kidney Injury via Mitochondrial Transfer. Theranostics 2025, 15, 5499–5517. [Google Scholar] [CrossRef]
- Sen, B.; Rastogi, A.; Nath, R.; Shasthry, S.M.; Pamecha, V.; Pandey, S.; Gupta, K.J.; Sarin, S.K.; Trehanpati, N.; Ramakrishna, G. Senescent Hepatocytes in Decompensated Liver Show Reduced UPRMT and Its Key Player, CLPP, Attenuates Senescence In Vitro. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-C.; Chiou, T.-W.; Lin, Z.-S.; Huang, K.-C.; Lin, Y.-C.; Huang, P.-C.; Syu, W.-S.; Harn, H.-J.; Lin, S.Z. A Proposed Novel Stem Cell Therapy Protocol for Liver Cirrhosis. Cell Transplant. 2015, 24, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhou, Y.; Han, C.-S.; Chen, L.-J.; Wang, Y.; Zhuang, Z.-M.; Lin, S.; Zhou, Y.; Jiang, J.; Yang, R. Stem Cells from Human Exfoliated Deciduous Teeth Alleviate Liver Cirrhosis via Inhibition of Gasdermin D-Executed Hepatocyte Pyroptosis. Front. Immunol. 2022, 13, 860225. [Google Scholar] [CrossRef] [PubMed]
- Abo-Aziza, F.A.M.; Zaki, A.K.A.; El-Maaty, A.M.A. Bone Marrow-Derived Mesenchymal Stem Cell (BM-MSC): A Tool of Cell Therapy in Hydatid Experimentally Infected Rats. Cell Regen. 2019, 8, 58–71. [Google Scholar] [CrossRef]
- Jeng, K.S.; Lu, S.-J.; Wang, C.-H.; Chang, C.F. Liver Fibrosis and Inflammation Under the Control of ERK2. Int. J. Mol. Sci. 2020, 21, 3796. [Google Scholar] [CrossRef]
- Ye, Z.; Lu, W.; Liang, L.; Tang, M.; Wang, Y.; Li, Z.; Zeng, H.; Wang, A.; Lin, M.; Huang, L.; et al. Mesenchymal Stem Cells Overexpressing Hepatocyte Nuclear Factor-4 Alpha Alleviate Liver Injury by Modulating Anti-Inflammatory Functions in Mice. Stem Cell Res. Ther. 2019, 10, 149. [Google Scholar] [CrossRef]
- Lei, J.; Xie, Q.; Li, Q.; Qin, S.; Wu, J.; Jiang, H.; Yu, B.; Luo, W. Resveratrol Alleviates Liver Fibrosis by Targeting Cross-Talk Between TLR2/MyD88/ERK and NF-κB/NLRP3 Inflammasome Pathways in Macrophages. J. Biochem. Mol. Toxicol. 2025, 39, e70208. [Google Scholar] [CrossRef]
- Kyung, E.; Kim, H.B.; Hwang, E.S.; Lee, S.; Choi, B.K.; Kim, J.W.; Kim, H.J.; Lim, S.M.; Kwon, O.I.; Woo, E.J. Evaluation of Hepatoprotective Effect of Curcumin on Liver Cirrhosis Using a Combination of Biochemical Analysis and Magnetic Resonance-Based Electrical Conductivity Imaging. Mediat. Inflamm. 2018, 2018, 5491797. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, H.; Xu, J.; Liu, L.; Tan, J.; Li, M.; Xu, N.; Luo, S.; Wang, J.; Yang, F.; et al. Liver-Targeted siRNA Lipid Nanoparticles Treat Hepatic Cirrhosis by Dual Antifibrotic and Anti-Inflammatory Activities. ACS Nano 2020, 14, 6305–6322. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Y.; Xu, M.; Meng, H.; Dai, C.; Yao, Z.; Lin, N. Bone Marrow Mesenchymal Stem Cells Inhibit Hepatic Fibrosis via the AABR07028795.2/rno-miR-667-5p Axis. Stem Cell Res. Ther. 2022, 13, 375. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and Hepatic Fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef]
- Kalra, S.; Bhattacharya, S.; Rawal, P. Hepatocrinology. Med. Sci. 2021, 9, 39. [Google Scholar] [CrossRef]
- Fu, X.; Jiang, B.; Zheng, B.; Yan, Y.; Wang, J.; Duan, Y.; Li, S.; Yan, L.; Wang, H.; Chen, B.; et al. Heterogenic Transplantation of Bone Marrow-Derived Rhesus Macaque Mesenchymal Stem Cells Ameliorates Liver Fibrosis Induced by Carbon Tetrachloride in Mouse. PeerJ 2018, 6, e4336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Cheng, H.; Jin, N.; Jin, S.; Liu, Z.; Xu, J.; Xie, J. Human Umbilical Cord Mesenchymal Stem Cells Enhance Liver Regeneration and Decrease Collagen Content in Fibrosis Mice After Partial Hepatectomy by Activating WNT/β-Catenin Signaling. Acta Biochim. Biophys. Sin. 2024, 57, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, M.-J.; Han, J.; Yoon, M.H.; Jung, Y. Mesenchymal Stem Cells Influence Activation of Hepatic Stellate Cells, and Constitute a Promising Therapy for Liver Fibrosis. Biomedicines 2021, 9, 1598. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Wang, H. Progress of Interference of Traditional Chinese Medicine on Cirrhosis Treated with Bone Marrow Mesenchymal Stem Cells. Evid. Based Complement. Altern. Med. 2021, 2021, 5569274. [Google Scholar] [CrossRef]
- Kwak, K.-A.; Cho, H.J.; Yang, J.-Y.; Park, Y.S. Current Perspectives Regarding Stem Cell-Based Therapy for Liver Cirrhosis. Can. J. Gastroenterol. Hepatol. 2018, 2018, 4197857. [Google Scholar] [CrossRef]
- Xu, H.X.; Zhou, Y.; Liu, Y.; Ping, J.; Shou, Q.; Chen, F.M.; Ruo, R. Metformin Improves Hepatic IRS2/PI3K/Akt Signaling in Insulin-Resistant Rats of NASH and Cirrhosis. J. Endocrinol. 2016, 229, 133–144. [Google Scholar] [CrossRef]
- Naseer, M.; Turse, E.P.; Syed, A.; Dailey, F.; Zatreh, M.; Tahan, V. Interventions to Improve Sarcopenia in Cirrhosis: A Systematic Review. World J. Clin. Cases 2019, 7, 156–170. [Google Scholar] [CrossRef]
- Ebadi, M.; Bhanji, R.A.; Mazurak, V.C.; Montaño-Loza, A.J. Sarcopenia in Cirrhosis: From Pathogenesis to Interventions. J. Gastroenterol. 2019, 54, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.W.-T.; Poon, R.T.P. Role of Branched-chain Amino Acids in Management of Cirrhosis and Hepatocellular Carcinoma. Hepatol. Res. 2008, 38, S107–S115. [Google Scholar] [CrossRef] [PubMed]
- Swansson, W.D.; Anderson, B.; Yeoh, S.W.; Lewis, D.J. Management of Minimal and Overt Hepatic Encephalopathy with Branched-Chain Amino Acids: A Review of the Evidence. Eur. J. Gastroenterol. Hepatol. 2023, 35, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, K.C.; Lin, C.J.; Hsu, S.W.; Fang, W.C.; Hsu, T.F.; Chiu, C.C.; Chang, H.W.; Hsu, C.H.; Lee, A.Y.L. Obtusilactone a and (−)-sesamin Induce Apoptosis in Human Lung Cancer Cells by Inhibiting Mitochondrial Lon Protease and Activating DNA Damage Checkpoints. Cancer Sci. 2010, 101, 2612–2620. [Google Scholar] [CrossRef]
- Ohno, T.; Tanaka, Y.; Sugauchi, F.; Orito, E.; Hasegawa, I.; Nukaya, H.; Kato, A.; Matunaga, S.; Endo, M.; Tanaka, Y.; et al. Suppressive Effect of Oral Administration of Branched-chain Amino Acid Granules on Oxidative Stress and Inflammation in HCV-positive Patients with Liver Cirrhosis. Hepatol. Res. 2008, 38, 683–688. [Google Scholar] [CrossRef]
- Francisco, B.; Javier, U.; Nixa, O.; Paula, H.; Daniel, C.; Romero-Gómez, M. The Janus of a disease: Diabetes and metabolic dysfunction-associated fatty liver disease. Ann. Hepatol. 2024, 29, 101501. [Google Scholar] [CrossRef]
- Weijing, Z.; Wenyan, S.; Weiyu, C.; Zoucheng, P.; Jiawei, Z.; Li, F.; Jie, L. Metabolic dysfunction-associated steatotic liver disease-related hepatic fibrosis increases risk of insulin resistance, type 2 diabetes, and chronic kidney disease. Eur. J. Gastroenterol. Hepatol. 2024, 36, 802–810. [Google Scholar]
- Michael, P.C.; Luisa, V.; Sven, M.F. MASLD/MASH and type 2 diabetes: Two sides of the same coin? From single PPAR to pan-PPAR agonists. Diabetes Res. Clin. Pract. 2024, 212, 111688. [Google Scholar]
- Hanai, T.; Shiraki, M.; Watanabe, S.; Kochi, T.; Imai, K.; Suetsugu, A.; Takai, K.; Moriwaki, H.; Shimizu, M. Sarcopenia Predicts Minimal Hepatic Encephalopathy in Patients with Liver Cirrhosis. Hepatol. Res. 2017, 47, 1359–1367. [Google Scholar] [CrossRef]
- Bonvini, A.; Coqueiro, A.Y.; Tirapegui, J.; Calder, P.C.; Rogero, M.M. Immunomodulatory Role of Branched-Chain Amino Acids. Nutr. Rev. 2018, 76, 840–856. [Google Scholar] [CrossRef]
- Moriwaki, H.; Shiraki, M.; Fukushima, H.; Shimizu, M.; Iwasa, J.; Naiki, T.; Nagaki, M. Long-term Outcome of Branched-chain Amino Acid Treatment in Patients with Liver Cirrhosis. Hepatol. Res. 2008, 38, S102–S106. [Google Scholar] [CrossRef]
- Zhang, X.; Ojanen, X.; Zhuang, H.; Wu, N.; Cheng, S.; Wiklund, P. Branched-Chain and Aromatic Amino Acids Are Associated with Insulin Resistance During Pubertal Development in Girls. J. Adolesc. Health 2019, 65, 337–343. [Google Scholar] [CrossRef]
- Shi, X.; Wei, X.; Yin, X.; Wang, Y.; Zhang, M.; Zhao, C.; Zhao, H.; McClain, C.J.; Feng, W.; Zhang, X. Hepatic and Fecal Metabolomic Analysis of the Effects of Lactobacillus rhamnosus GG on Alcoholic Fatty Liver Disease in Mice. J. Proteome Res. 2015, 14, 1174–1182. [Google Scholar] [CrossRef]
- Lü, Z.; Sun, G.; Pan, X.-A.; Qu, X.-H.; Yang, P.; Chen, Z.; Han, X.J.; Wang, T. BCATc Inhibitor 2 Ameliorated Mitochondrial Dysfunction and Apoptosis in Oleic Acid-Induced Non-Alcoholic Fatty Liver Disease Model. Front. Pharmacol. 2022, 13, 1025551. [Google Scholar] [CrossRef]
- Kumar, A.; Harrelson, T.F.; Lewis, N.E.; Gallagher, E.J.; LeRoith, D.; Shiloach, J.; Betenbaugh, M.J. Multi-Tissue Computational Modeling Analyzes Pathophysiology of Type 2 Diabetes in MKR Mice. PLoS ONE 2014, 9, e102319. [Google Scholar] [CrossRef]
- Nong, X.-z.; Zhang, C.; Wang, J.; Ding, P.; Ji, G.; Wu, T. The Mechanism of Branched-Chain Amino Acid Transferases in Different Diseases: Research Progress and Future Prospects. Front. Oncol. 2022, 12, 988290. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Taniguchi, E.; Itou, M.; Sumie, S.; Oriishi, T.; Matsuoka, H.; Nagao, Y.; Sata, M. Branched-chain Amino Acids Improve Insulin Resistance in Patients with Hepatitis C Virus-related Liver Disease: Report of Two Cases. Liver Int. 2007, 27, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Barceló, A.; Bauçà, J.M.; Peña-Zarza, J.A.; Morell-García, D.; Yáñez, A.M.; Pérez, G.; Piérola, J.; Toledo, N.; Peña, M.d.l. Circulating Branched-Chain Amino Acids in Children with Obstructive Sleep Apnea. Pediatr. Pulmonol. 2017, 52, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ye, D.; Xie, W.; Hua, F.; Yang, Y.; Wu, J.; Gu, A.; Ren, Y.; Mao, K. Defect of Branched-Chain Amino Acid Metabolism Promotes the Development of Alzheimer’s Disease by Targeting the mTOR Signaling. Biosci. Rep. 2018, 38, BSR20180127. [Google Scholar] [CrossRef]
- Alkhayal, F.A.; Haddad, S.; Bakraa, R.; Alqahtani, A. Acrodermatitis Dysmetabolica Secondary to Isoleucine Deficiency in Infant with Maple Syrup Urine Disease. Dermatol. Rep. 2023, 15, 9750. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhan, L.; Zhang, L.; Shi, Q.; Li, L. Branched-Chain Amino Acids in Liver Diseases: Complexity and Controversy. Nutrients 2024, 16, 1875. [Google Scholar] [CrossRef]
- Burrage, L.C.; Nagamani, S.C.S.; Campeau, P.M.; Lee, B.H. Branched-Chain Amino Acid Metabolism: From Rare Mendelian Diseases to More Common Disorders. Human. Mol. Genet. 2014, 23, R1–R8. [Google Scholar] [CrossRef]
- Avery, C.L.; Howard, A.G.; Lee, H.; Downie, C.G.; Lee, M.P.; Koenigsberg, S.H.; Ballou, A.F.; Preuss, M.; Raffield, L.M.; Yarosh, R.; et al. Branched Chain Amino Acids Harbor Distinct and Often Opposing Effects on Health and Disease. Commun. Med. 2023, 3, 172. [Google Scholar] [CrossRef]
- Košuta, I.; Mrzljak, A.; Kolarić, B.; Lovrenčić, M.V. Leptin as a Key Player in Insulin Resistance of Liver Cirrhosis? A Cross-Sectional Study in Liver Transplant Candidates. J. Clin. Med. 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.S.; Wei, J.C.C.; Chiu, L.T.; Hsu, C.C.; Hou, M.C.; Hwu, C.M. Thiazolidinediones Were Associated with Higher Risk of Cardiovascular Events in Patients with Type 2 Diabetes and Cirrhosis. Liver Int. 2020, 41, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Dhoop, S.; Ghazaleh, S.; Roberts, L.; Shehada, M.; Patel, M.; Lee-Smith, W.; Rabeeah, S.; Sawaf, B.; Vadehra, P.; Hart, B.; et al. Sodium-Glucose Cotransporter-2 Inhibitors in Liver Cirrhosis: A Systematic Review of Their Role in Ascites Management, Slowing Disease Progression, and Safety. Int. J. Mol. Sci. 2025, 26, 4781. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Nguyen, H.H.; Hwang, S.Y.; Lee, S.S. Oxidative Mechanisms and Cardiovascular Abnormalities of Cirrhosis and Portal Hypertension. Int. J. Mol. Sci. 2023, 24, 16805. [Google Scholar] [CrossRef]
- Srivastava, S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef]
- Ye, F.; Zhai, M.; Long, J.; Gong, Y.; Ren, C.; Zhang, D.; Lin, X.; Liu, S. The burden of liver cirrhosis in mortality: Results from the global burden of disease study. Front. Public Health 2022, 10, 909455. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Lan, Y.; Wang, H.; Weng, H.; Xu, X.; Yu, X.; Tu, H.; Gong, K.; Yao, J.; Ye, S.; Shi, Y.; et al. The burden of liver cirrhosis and underlying etiologies: Results from the Global Burden of Disease Study 2019. Hepatol. Commun. 2023, 7, e0026. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, V. Pathophysiology, diagnosis and treatment of ascites in cirrhosis. Ann. Hepatol. 2002, 1, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Trebicka, J. Portal hypertension in cirrhosis: Pathophysiological mechanisms and therapy. JHEP Rep. 2021, 3, 100316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wei, S.; Niu, M.; Wang, J.; Wang, J.; Su, H.; Luo, S.-q.; Zhang, X.; Guo, Y.; Liu, L.; et al. A Network Pharmacology Approach to Discover Active Compounds and Action Mechanisms of San-Cao Granule for Treatment of Liver Fibrosis. Drug Des. Dev. Ther. 2016, 10, 733. [Google Scholar] [CrossRef]
- Chan, Y.-T.; Wang, N.; Tan, H.Y.; Li, S.; Feng, Y. Targeting Hepatic Stellate Cells for the Treatment of Liver Fibrosis by Natural Products: Is It the Dawning of a New Era? Front. Pharmacol. 2020, 11, 548. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D.A. Hepatic Stellate Cells and the Reversal of Fibrosis. J. Gastroenterol. Hepatol. 2006, 21, S84–S87. [Google Scholar] [CrossRef]
- Zheng, K.; Yuan, S.; Dong, M.; Zhang, H.; Jiang, X.; Yan, C.; Ye, R.; Zhou, H.; Chen, L.; Jiang, R.; et al. Dihydroergotamine Ameliorates Liver Fibrosis by Targeting Transforming Growth Factor Β Type II Receptor. World J. Gastroenterol. 2023, 29, 3103–3118. [Google Scholar] [CrossRef]
- Wang, X.; Zai, Q.; He, Y.; Xie, Q. MicroRNAs as Critical Regulators in Liver Fibrosis. Portal Hypertens. Cirrhosis 2023, 2, 144–148. [Google Scholar] [CrossRef]
- Massey, V.; Cabezas, J.; Bataller, R. Epigenetics in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 219–230. [Google Scholar] [CrossRef]
- Ji, D.; Chen, G.F.; Wang, J.; Cao, L.; Lu, F.; Mu, X.; Zhang, X.; Lu, X. Identification of TAF1, HNF4A, and CALM2 as Potential Therapeutic Target Genes for Liver Fibrosis. J. Cell. Physiol. 2018, 234, 9045–9051. [Google Scholar] [CrossRef]
- Semenovich, D.S.; Andrianova, N.V.; Zorova, L.D.; Pevzner, I.B.; Abramicheva, P.A.; Elchaninov, A.; Mapкoвa, O.B.; Petrukhina, A.S.; Zorov, D.B.; Plotnikov, E.Y. Fibrosis Development Linked to Alterations in Glucose and Energy Metabolism and Prooxidant–Antioxidant Balance in Experimental Models of Liver Injury. Antioxidants 2023, 12, 1604. [Google Scholar] [CrossRef]
- Li, R.; Tai, Y.; Zhang, X.; Liu, Z.; Si, H.; Kong, D.; Zhao, L.; Li, J.; Midgley, A.C. Tissue-Microenvironment-Responsive Self-Assembling Peptide Nanoshells Boost Pirfenidone Efficacy in the Treatment of Liver Fibrosis. Adv. Healthc. Mater. 2025, 14, 2500101. [Google Scholar] [CrossRef]
- Xi, Y.; Li, Y.; Xu, P.; Li, S.; Liu, Z.; Tung, H.C.; Cai, X.; Wang, J.; Huang, H.; Wang, M.; et al. The Anti-Fibrotic Drug Pirfenidone Inhibits Liver Fibrosis by Targeting the Small Oxidoreductase Glutaredoxin-1. Sci. Adv. 2021, 7, eabg9241. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ericson, M.; Fanjul, A.; Erion, D.M.; Paraskevopoulou, M.D.; Smith, E.N.; Cole, B.K.; Feaver, R.E.; Holub, C.; Gavva, N.; et al. Genome-Wide CRISPR Screening to Identify Drivers of TGF-β-Induced Liver Fibrosis in Human Hepatic Stellate Cells. ACS Chem. Biol. 2022, 17, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, O.S.; Attia, H.G.; Mohamed, B.M.S.A.; Elbaset, M.A.; Fayed, H.M. Current Investigations for Liver Fibrosis Treatment: Between Repurposing the FDA-approved Drugs and the Other Emerging Approaches. J. Pharm. Pharm. Sci. 2023, 26, 11808. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhao, G.; Lin, Q.; Zhuang, G.; Zhu, J.; Jin, J. A Network Pharmacology Approach to Explore the Molecular Mechanism of Active Peptide Ingredients of Carapax trionycis on Liver Fibrosis. Pept. Sci. 2023, 116, e24335. [Google Scholar] [CrossRef]
- Huang, L.; Yu, Q.; Peng, H.; Zhen, Z. The Mechanism of Peach Kernel and Safflower Herb-Pair for the Treatment of Liver Fibrosis Based on Network Pharmacology and Molecular Docking Technology: A Review. Medicine 2023, 102, e33593. [Google Scholar] [CrossRef]
- Liu, S.; Han, D.; Xu, C.; Yang, F.; Li, Y.; Zhang, K.; Zhao, X.; Zhang, J.; Lü, T.; Lu, S.; et al. Antibody-Drug Conjugates Targeting CD248 Inhibits Liver Fibrosis Through Specific Killing on Myofibroblasts. Mol. Med. 2022, 28, 37. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; Yu, Q.; Zhan, S.; Mao, M. Exploring the Mechanism of Action of Bidens pilosa L. in Combating Hepatic Fibrosis Through Network Pharmacology and Molecular Docking: An. Observational Study. Medicine 2024, 103, e39725. [Google Scholar] [CrossRef]
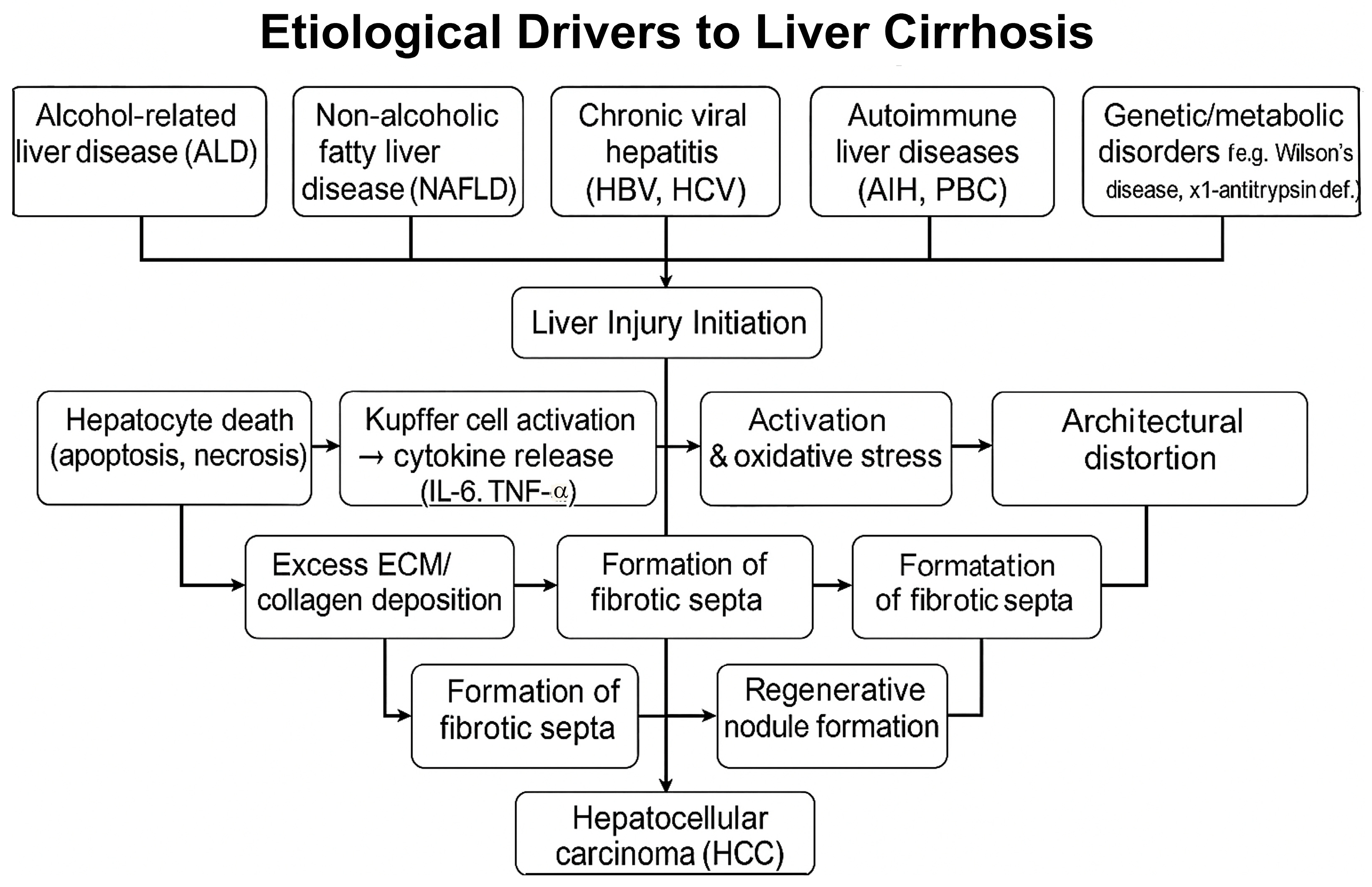

| Etiological/Pathophysiological Factor | Description | Key References |
|---|---|---|
| Hepatitis C virus (HCV) | Hepatitis C virus (HCV) is a major etiological factor of cirrhosis, leading to chronic inflammation, hepatocyte injury, and progressive fibrosis that can culminate in cirrhotic transformation. | [29,30] |
| Alcohol-Related Liver Disease (ALD) | Chronic alcohol consumption leads to liver injury and fibrosis; alcohol is the primary etiological factor in 62.9% of cirrhosis cases and exacerbates other liver diseases. | [10,11,12,31] |
| Non-Alcoholic Fatty Liver Disease (NAFLD) | Closely associated with obesity and metabolic syndrome, NAFLD progresses from steatosis to NASH, fibrosis, cirrhosis, and eventually HCC. | [14,15,32] |
| Autoimmune and Genetic Disorders | Autoimmune hepatitis and primary biliary cholangitis drive chronic inflammation; genetic disorders such as Wilson’s disease and α1-antitrypsin deficiency also contribute. | [21,22] |
| Hepatic Stellate Cell Activation | Injury triggers the transformation of hepatic stellate cells into collagen-producing myofibroblast-like cells, driving fibrogenesis. | [23] |
| Chronic Hepatitis and Regeneration | Hepatocyte death and compensatory regeneration in chronic hepatitis lead to fibrotic scarring and regenerative nodule formation, resulting in portal hypertension. | [23,24] |
| Histological Features | Liver histology reveals fibrotic septa, regenerative nodules, and architectural distortion; the Laennec score is used to assess the severity of fibrosis. | [21,25] |
| Cellular Heterogeneity | Single-cell RNA sequencing reveals immune cell heterogeneity, particularly in macrophages and lymphocytes, which are involved in fibrotic and inflammatory signaling pathways. | [33,34] |
| Progression to Hepatocellular Carcinoma (HCC) | Cirrhosis predisposes individuals to hepatocellular carcinoma (HCC) through chronic oxidative stress, immune dysregulation, insulin resistance, and hepatocyte senescence; biomarkers, such as microRNAs, show diagnostic potential. | [23,26,28,35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiacek, M.; Adam, A.; Studnicki, R.; Zubrzycki, I.Z. Exploring Cirrhosis: Insights into Advances in Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 7226. https://doi.org/10.3390/ijms26157226
Wiacek M, Adam A, Studnicki R, Zubrzycki IZ. Exploring Cirrhosis: Insights into Advances in Therapeutic Strategies. International Journal of Molecular Sciences. 2025; 26(15):7226. https://doi.org/10.3390/ijms26157226
Chicago/Turabian StyleWiacek, Magdalena, Anna Adam, Rafał Studnicki, and Igor Z. Zubrzycki. 2025. "Exploring Cirrhosis: Insights into Advances in Therapeutic Strategies" International Journal of Molecular Sciences 26, no. 15: 7226. https://doi.org/10.3390/ijms26157226
APA StyleWiacek, M., Adam, A., Studnicki, R., & Zubrzycki, I. Z. (2025). Exploring Cirrhosis: Insights into Advances in Therapeutic Strategies. International Journal of Molecular Sciences, 26(15), 7226. https://doi.org/10.3390/ijms26157226