
Development of a Broad-Spectrum Pan-Mpox Vaccine via Immunoinformatic Approaches

 , , and
, , and
Abstract
1. Introduction
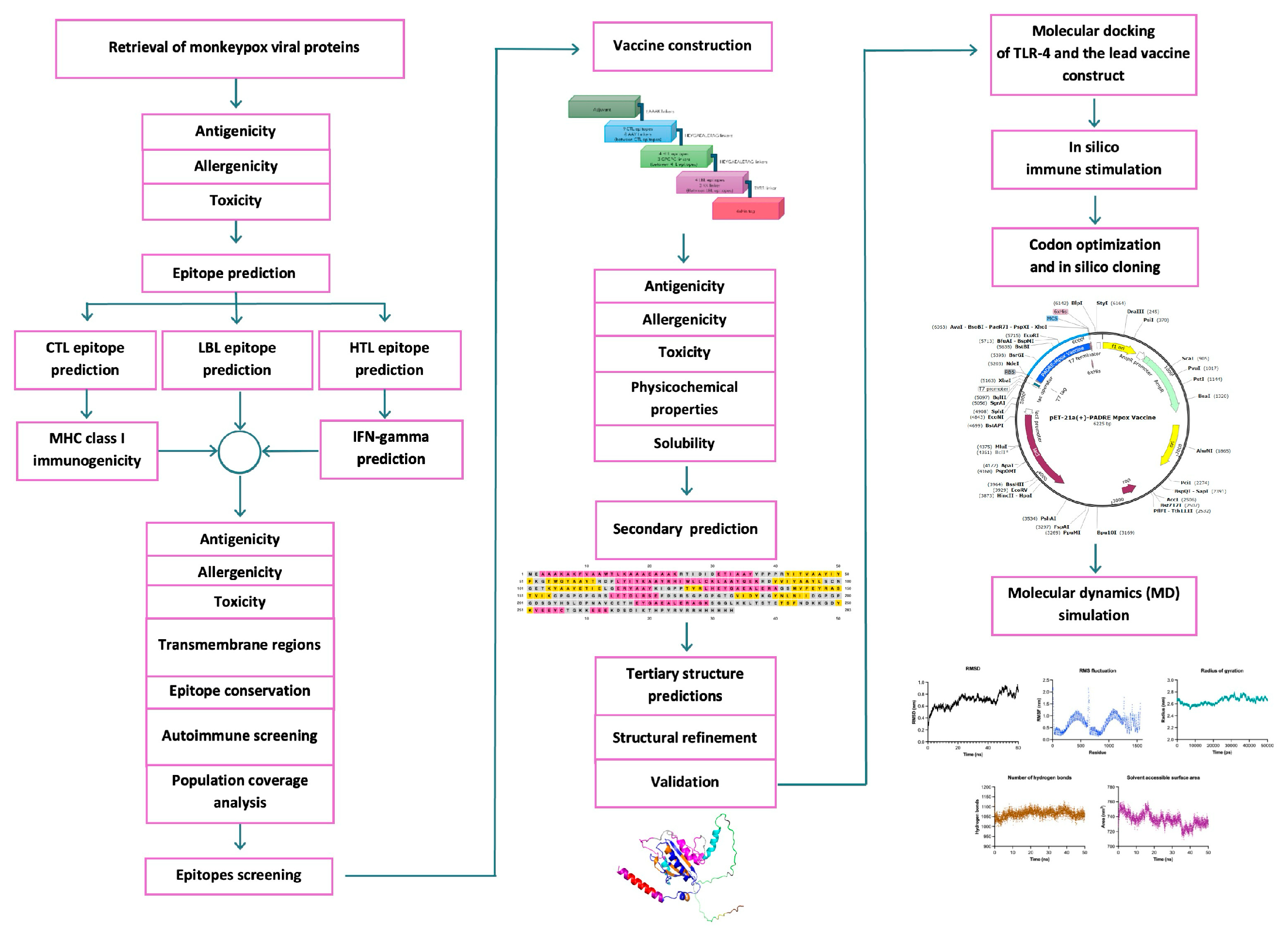
2. Results
2.1. Monkeypox Viral Protein Retrieval and Screening
2.2. Prediction of Epitope Candidates and Assessment of Allergenicity, Antigenicity, and Toxicity Parameters
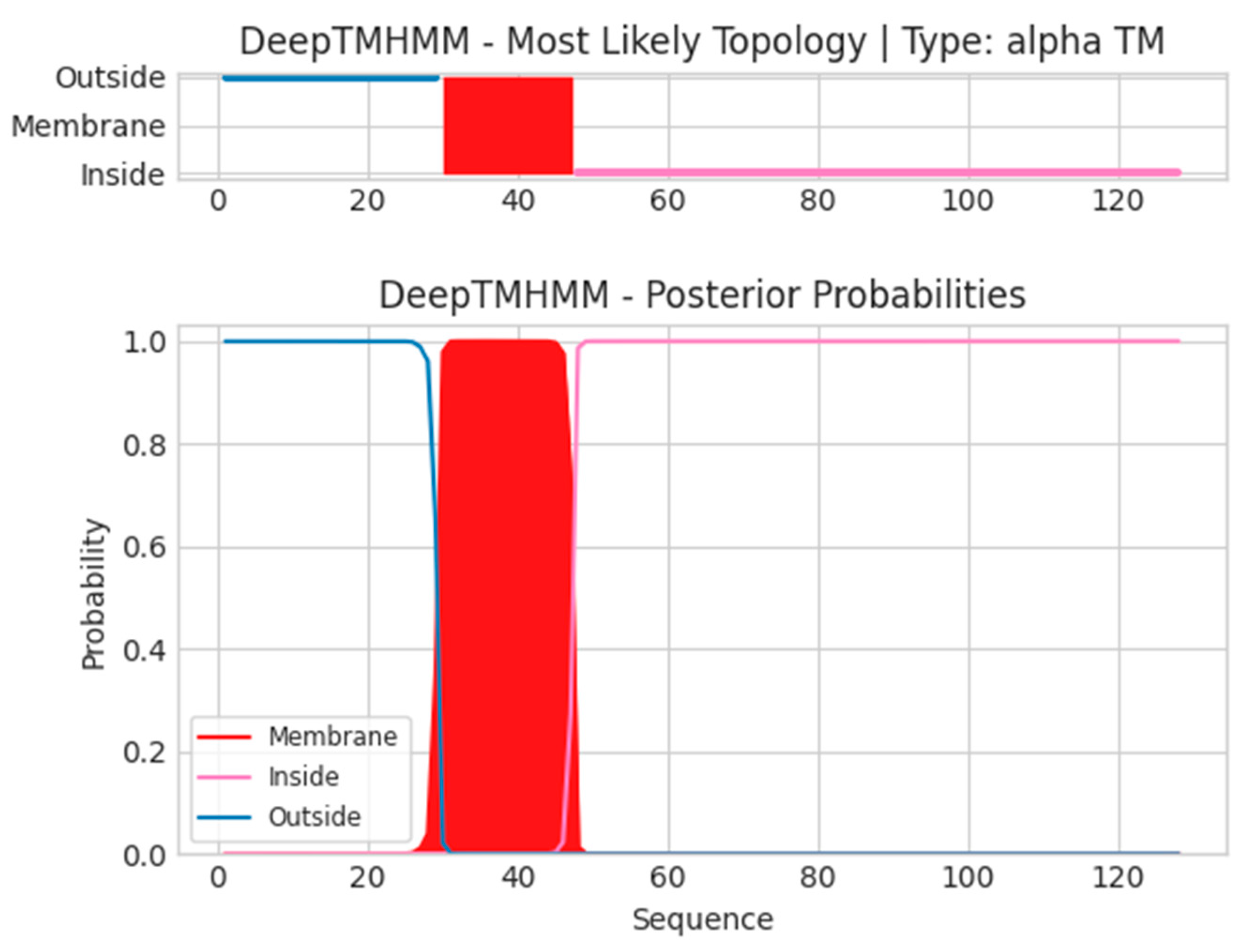
2.3. Transmembrane and Signal Peptide Screening
2.4. Epitope Conservancy for the Broad-Spectrum Coverage of Vaccines Across Different Clades
2.5. Epitope Homology Analysis with the Human Proteome
2.6. Final Epitope Selection and Worldwide Population Coverage
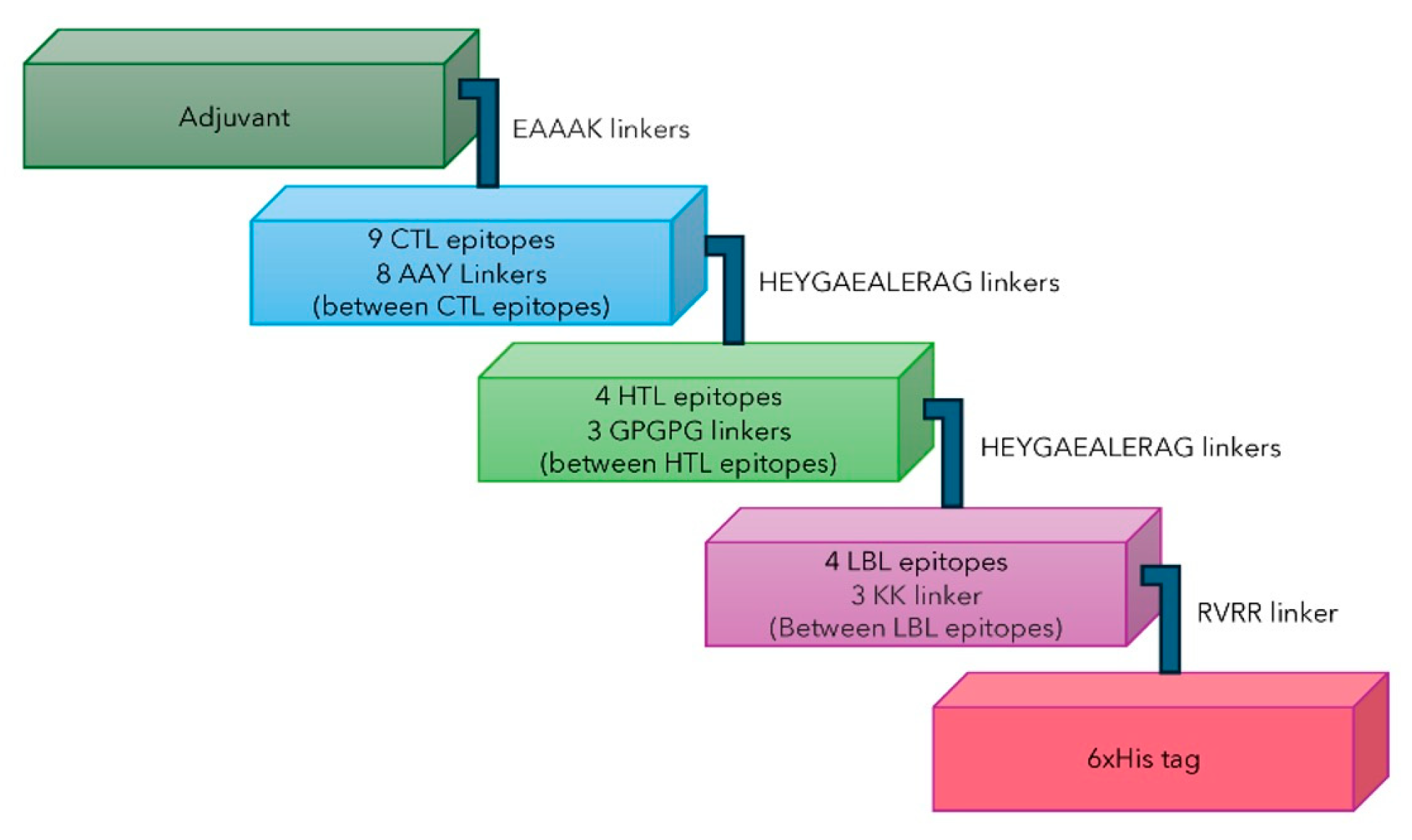
2.7. Multi-Epitope Subunit Vaccine Construct
2.8. Antigenicity, Allergenicity, Toxicity, and Physicochemical Properties
2.9. Secondary Structure Prediction, Three-Dimensional Structure Predictions, Refinement, and Validation
2.10. Molecular Docking of TLRs and Selected Vaccine Constructs
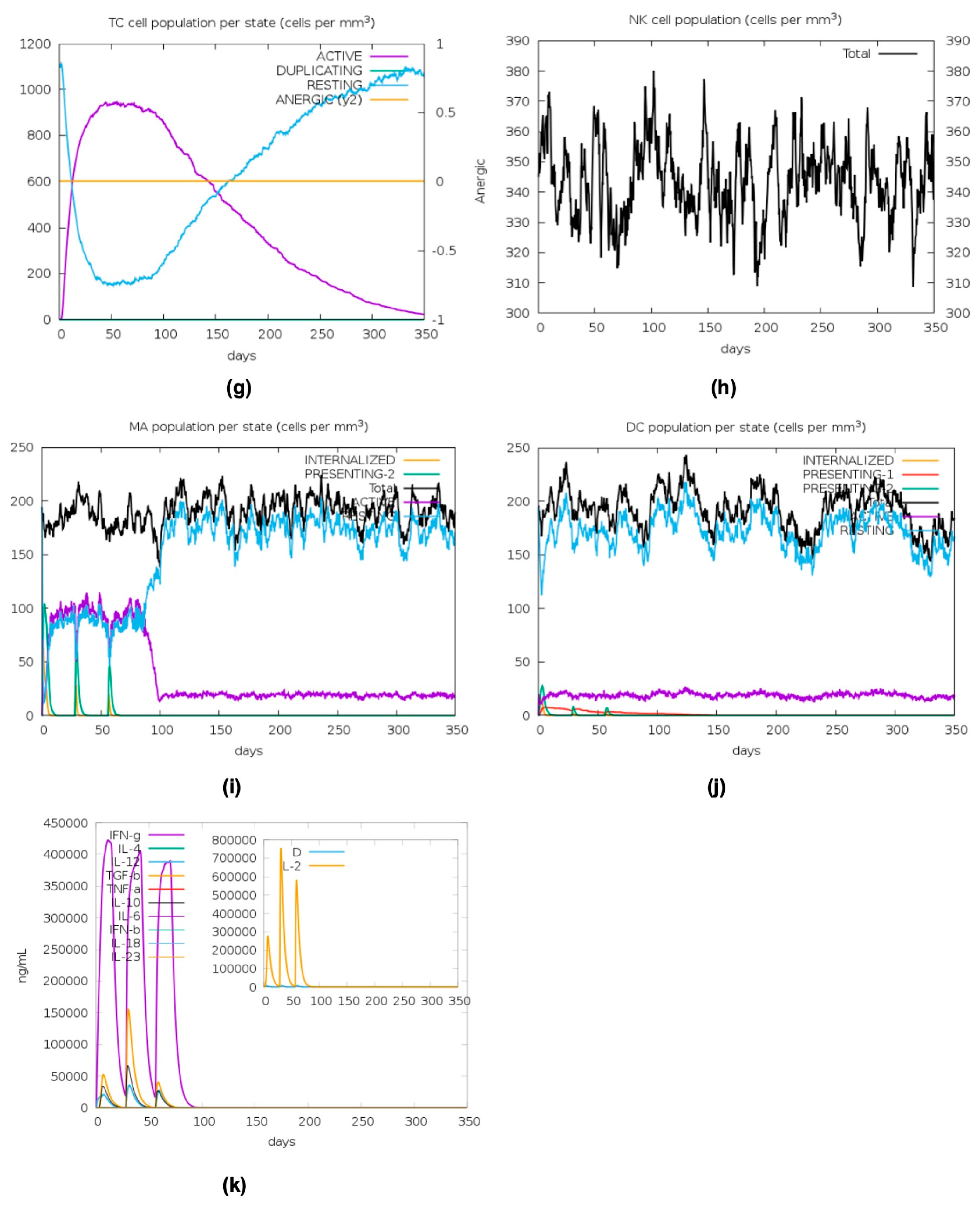
2.11. In Silico Immune Stimulation
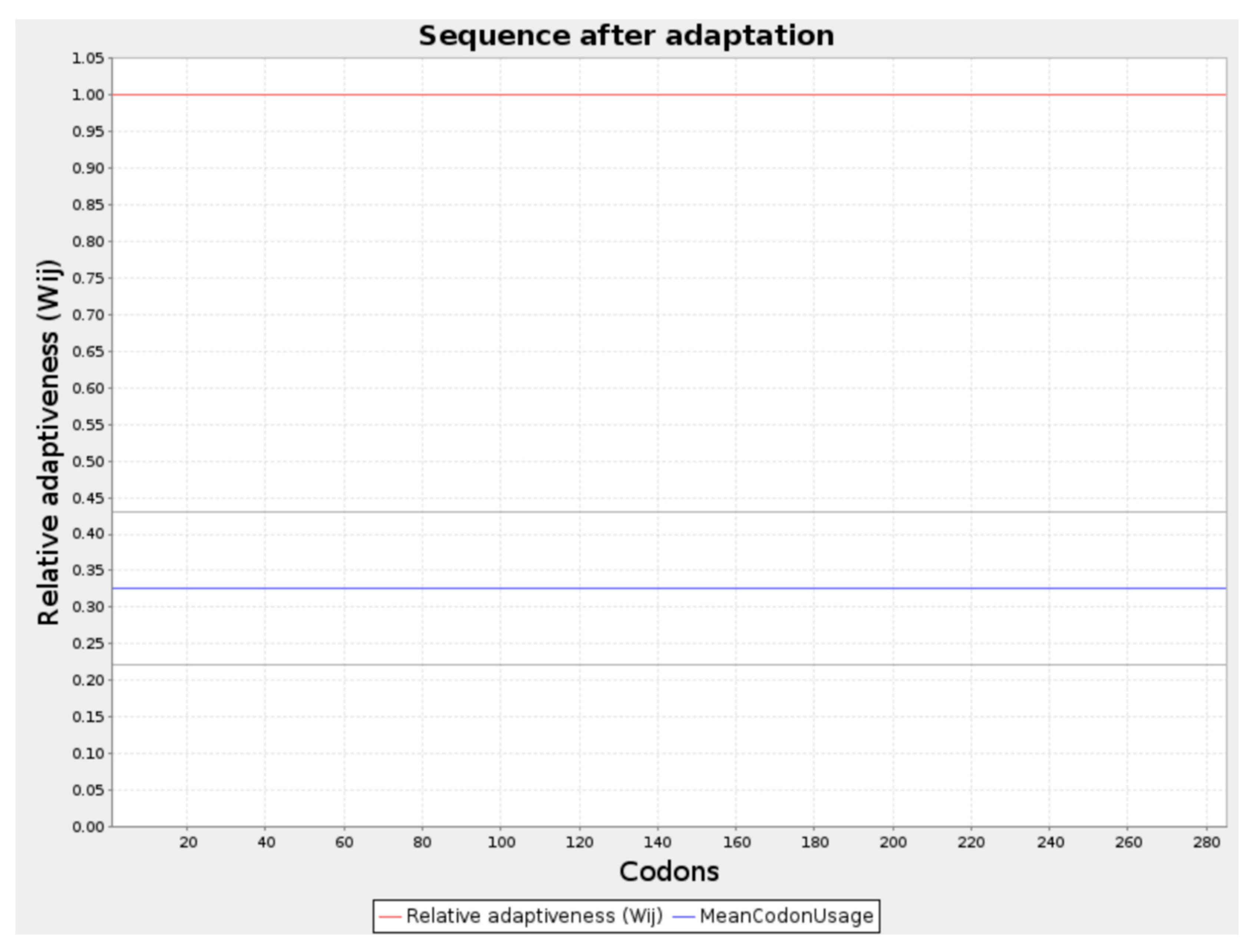
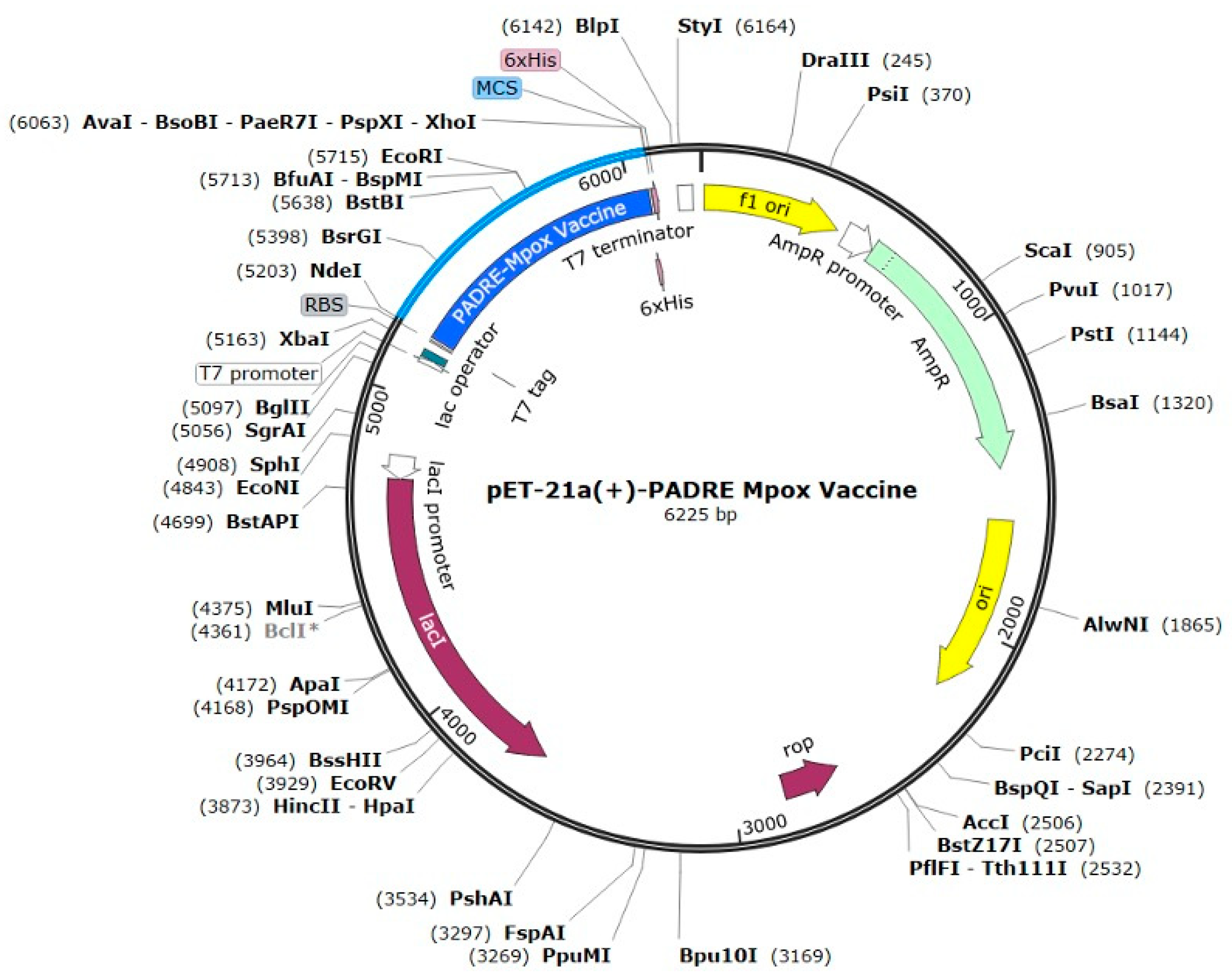
2.12. Codon Optimization and In Silico Cloning
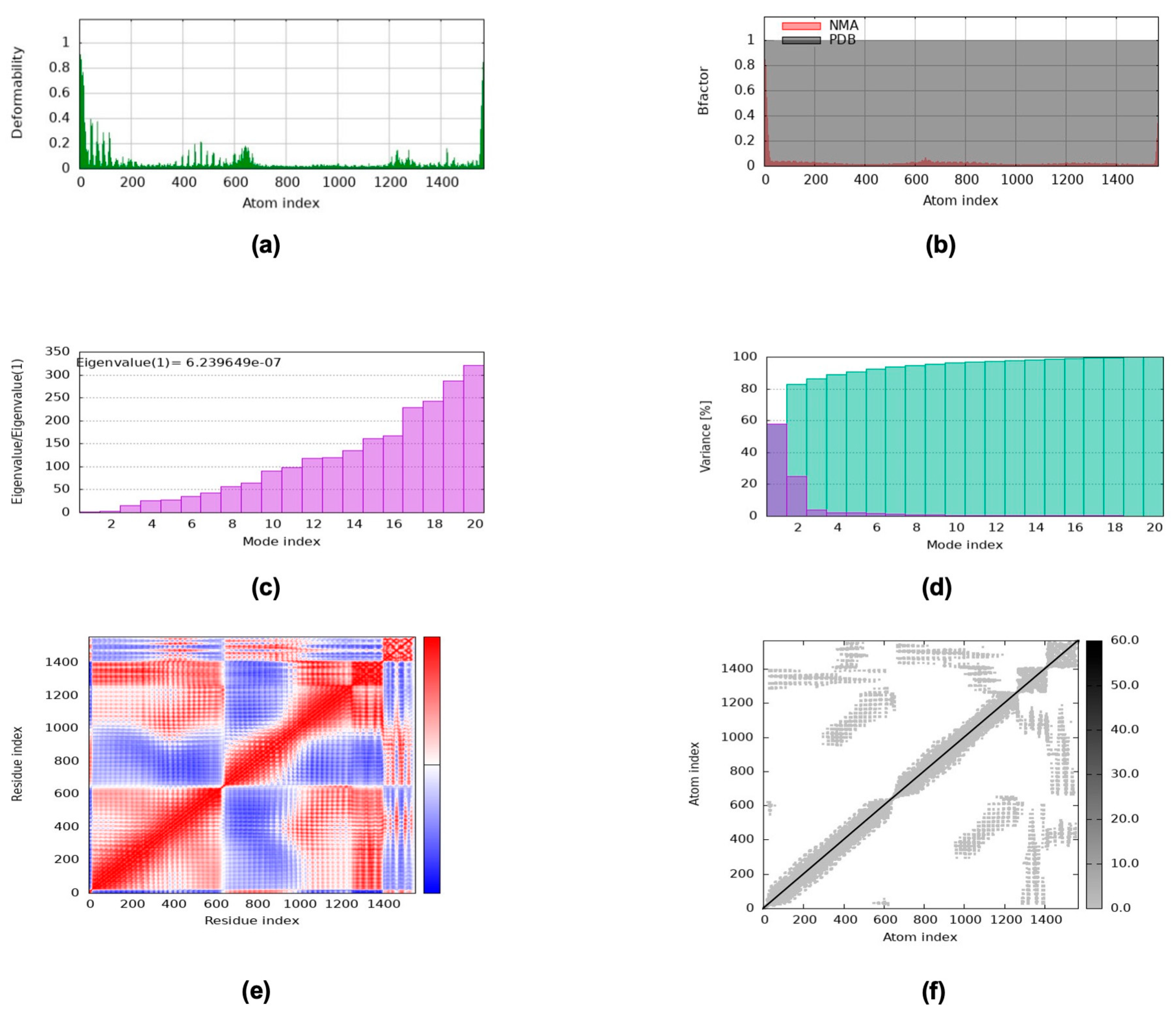
2.13. NMA via the iMODS Server
2.14. MD Analysis via GROMACS
3. Discussion
4. Materials and Methods
4.1. Retrieval of Monkeypox Viral Proteins
4.2. Prediction of Cytotoxic T Lymphocytes (CTLs), Helper T Lymphocytes (HTLs), and Linear B Lymphocytes (LBLs)
4.3. Effects of CTL, HTL, and B-Cell Epitopes on Allergenicity, Antigenicity, and Toxicity Parameters
4.4. Screening of Transmembrane Regions
4.5. Epitope Conservation for the Broad-Spectrum Coverage of Vaccines Across Different Clades
4.6. Autoimmune Screening by Homology Analysis of Epitopes with Human Proteomes
4.7. Population Coverage
4.8. Construction of the Multi-Epitope Subunit Vaccine
4.9. Physicochemical, Antigenicity, Allergenicity, Toxicity, and Solubility of the Vaccine
4.10. Secondary Prediction, Tertiary Structure Predictions, Refinement, and Validation
4.11. Molecular Docking of Toll-Like Receptor 4 (TLR4) and the Lead Vaccine Construct
4.12. In Silico Immune Stimulation
4.13. Codon Optimization and In Silico Cloning
4.14. Molecular Dynamics (MD) Simulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| MPXV | Monkeypox Virus |
| WHO | World Health Organization |
| CTB | Cholera Toxin B |
| dsDNA | Double-stranded DNA |
| MPX | Monkeypox |
| M1R | Myristylprotein |
| CTLs | Cytotoxic T Lymphocytes |
| HTLs | Helper T Lymphocytes |
| LBLs | Linear B Lymphocytes |
| SVM | Swiss-Prot-based Prediction Method |
| GRAVY | Grand Average Hydropathicity |
| pI | Isoelectric Point |
| JCAT | Java Codon Adaptation Tool |
| PME | Particle Mesh Ewald |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuations |
| Rg | Radius of Gyration |
| SASA | Solvent Accessible Surface Area |
| ΔG | Gibbs Free Energy |
| KD | Dissociation Constant |
| NK | Natural Killer Cells |
| Mas | Macrophages |
| DCs | Dendritic Cells |
| IFN-γ | Interferon-γ |
| TGF-β | Transforming Growth Factor-β |
| IL10 | Interleukin 10 |
| IL12 | Interleukin 12 |
| CAI | Codon Adaptation Index |
| ACIP | Advisory Committee on Immunization Practices |
| OMVs | Outer Membrane Vesicles |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) CTL Epitope | ||||||
| Protein | Epitope Sequence | Strain | Epitope length | Positions | Identity | Clade |
| A40L | WKLDNHDIL | Zaire AF380138.1 | 9 | 47–55 | 100.00% | 1 |
| WKLDNHDIL | USA 2003 DQ011157.1 | 9 | 47–55 | 100.00% | 2a | |
| WKLDNHDIL | UK_P2 MT903344.1 | 9 | 47–55 | 100.00% | 2B-A.1 | |
| WKLDNHDIL | USA 2021 ON676708.1 | 9 | 47–55 | 100.00% | 2B-A.1.1 | |
| WKLDNHDIL | Nigeria 2018 NC-063383-1 | 9 | 47–55 | 100.00% | 2B-A.2 | |
| WKLDNHDIL | USA 2022 VA1 ON675438.1 | 9 | 47–55 | 100.00% | 2B-A.2 | |
| WKLDNHDIL | France 2022 ON622722-2 | 9 | 47–55 | 100.00% | 2B-B.1 | |
| B6R | LSCNGETKY | Zaire AF380138.1 | 9 | 99–107 | 100.00% | 1 |
| LSCNGETKY | USA 2003 DQ011157.1 | 9 | 99–107 | 100.00% | 2a | |
| LSCNGETKY | UK_P2 MT903344.1 | 9 | 99–107 | 100.00% | 2B-A.1 | |
| LSCNGETKY | USA 2021 ON676708.1 | 9 | 99–107 | 100.00% | 2B-A.1.1 | |
| LSCNGETKY | Nigeria 2018 NC-063383-1 | 9 | 99–107 | 100.00% | 2B-A.2 | |
| LSCNGETKY | USA 2022 VA1 ON675438.1 | 9 | 99–107 | 100.00% | 2B-A.2 | |
| LSCNGETKY | France 2022 ON622722-2 | 9 | 99–107 | 100.00% | 2B-B.1 | |
| B9R | KIGPPTVRL | Zaire AF380138.1 | 9 | 101–109 | 100.00% | 1 |
| KIGPPTVRL | USA 2003 DQ011157.1 | 9 | 101–109 | 100.00% | 2a | |
| KIGPPTVRL | UK_P2 MT903344.1 | 9 | 101–109 | 100.00% | 2B-A.1 | |
| KIGPPTVRL | USA 2021 ON676708.1 | 9 | 101–109 | 100.00% | 2B-A.1.1 | |
| KIGPPTVRL | Nigeria 2018 NC-063383-1 | 9 | 101–109 | 100.00% | 2B-A.2 | |
| KIGPPTVRL | USA 2022 VA1 ON675438.1 | 9 | 101–109 | 100.00% | 2B-A.2 | |
| KIGPPTVRL | France 2022 ON622722-2 | 9 | 101–109 | 100.00% | 2B-B.1 | |
| C2L | IYFKGTWQY | Zaire AF380138.1 | 9 | 173–181 | 100.00% | 1 |
| IYFKGTWQY | USA 2003 DQ011157.1 | 9 | 173–181 | 100.00% | 2a | |
| IYFKGTWQY | UK_P2 MT903344.1 | 9 | 173–181 | 100.00% | 2B-A.1 | |
| IYFKGTWQY | USA 2021 ON676708.1 | 9 | 173–181 | 100.00% | 2B-A.1.1 | |
| IYFKGTWQY | Nigeria 2018 NC-063383-1 | 9 | 173–181 | 100.00% | 2B-A.2 | |
| IYFKGTWQY | USA 2022 VA1 ON675438.1 | 9 | 173–181 | 100.00% | 2B-A.2 | |
| IYFKGTWQY | France 2022 ON622722-2 | 9 | 173–181 | 100.00% | 2B-B.1 | |
| C2L | TRDPLYIYK | Zaire AF380138.1 | 9 | 307–315 | 100.00% | 1 |
| TRDPLYIYK | USA 2003 DQ011157.1 | 9 | 307–315 | 100.00% | 2a | |
| TRDPLYIYK | UK_P2 MT903344.1 | 9 | 307–315 | 100.00% | 2B-A.1 | |
| TRDPLYIYK | USA 2021 ON676708.1 | 9 | 307–315 | 100.00% | 2B-A.1.1 | |
| TRDPLYIYK | Nigeria 2018 NC-063383-1 | 9 | 307–315 | 100.00% | 2B-A.2 | |
| TRDPLYIYK | USA 2022 VA1 ON675438.1 | 9 | 307–315 | 100.00% | 2B-A.2 | |
| TRDPLYIYK | France 2022 ON622722-2 | 9 | 307–315 | 100.00% | 2B-B.1 | |
| F8L | YPPPRYITV | Zaire AF380138.1 | 9 | 591–599 | 100.00% | 1 |
| YPPPRYITV | USA 2003 DQ011157.1 | 9 | 591–599 | 100.00% | 2a | |
| YPPPRYITV | UK_P2 MT903344.1 | 9 | 591–599 | 100.00% | 2B-A.1 | |
| YPPPRYITV | USA 2021 ON676708.1 | 9 | 591–599 | 100.00% | 2B-A.1.1 | |
| YPPPRYITV | Nigeria 2018 NC-063383-1 | 9 | 591–599 | 100.00% | 2B-A.2 | |
| YPPPRYITV | USA 2022 VA1 ON675438.1 | 9 | 591–599 | 100.00% | 2B-A.2 | |
| YPPPRYITV | France 2022 ON622722-2 | 9 | 591–599 | 100.00% | 2B-B.1 | |
| F8L | RTIDIDETI | Zaire AF380138.1 | 9 | 64–72 | 100.00% | 1 |
| RTIDIDETI | USA 2003 DQ011157.1 | 9 | 64–72 | 100.00% | 2a | |
| RTIDIDETI | UK_P2 MT903344.1 | 9 | 64–72 | 100.00% | 2B-A.1 | |
| RTIDIDETI | USA 2021 ON676708.1 | 9 | 64–72 | 100.00% | 2B-A.1.1 | |
| RTIDIDETI | Nigeria 2018 NC-063383-1 | 9 | 64–72 | 100.00% | 2B-A.2 | |
| RTIDIDETI | USA 2022 VA1 ON675438.1 | 9 | 64–72 | 100.00% | 2B-A.2 | |
| RTIDIDETI | France 2022 ON622722-2 | 9 | 64–72 | 100.00% | 2B-B.1 | |
| F8L | ETIELGERY | Zaire AF380138.1 | 9 | 920–928 | 100.00% | 1 |
| ETIELGERY | USA 2003 DQ011157.1 | 9 | 920–928 | 100.00% | 2a | |
| ETIELGERY | UK_P2 MT903344.1 | 9 | 920–928 | 100.00% | 2B-A.1 | |
| ETIELGERY | USA 2021 ON676708.1 | 9 | 920–928 | 100.00% | 2B-A.1.1 | |
| ETIELGERY | Nigeria 2018 NC-063383-1 | 9 | 920–928 | 100.00% | 2B-A.2 | |
| ETIELGERY | USA 2022 VA1 ON675438.1 | 9 | 920–928 | 100.00% | 2B-A.2 | |
| ETIELGERY | France 2022 ON622722-2 | 9 | 920–928 | 100.00% | 2B-B.1 | |
| H3L | QEKRDVVIV | Zaire AF380138.1 | 9 | 44–52 | 100.00% | 1 |
| QEKRDVVIV | USA 2003 DQ011157.1 | 9 | 44–52 | 100.00% | 2a | |
| QEKRDVVIV | UK_P2 MT903344.1 | 9 | 44–52 | 100.00% | 2B-A.1 | |
| QEKRDVVIV | USA 2021 ON676708.1 | 9 | 44–52 | 100.00% | 2B-A.1.1 | |
| QEKRDVVIV | Nigeria 2018 NC-063383-1 | 9 | 44–52 | 100.00% | 2B-A.2 | |
| QEKRDVVIV | USA 2022 VA1 ON675438.1 | 9 | 44–52 | 100.00% | 2B-A.2 | |
| QEKRDVVIV | France 2022 ON622722-2 | 9 | 44–52 | 100.00% | 2B-B.1 | |
| (b) HTL Epitope | ||||||
| Protein | Epitope sequence | Strain | Epitope length | Positions | Identity | Clade |
| B6R | DSGYHSLDPNAVCET | Zaire AF380138.1 | 15 | 49–63 | 100.00% | 1 |
| DSGYHSLDPNAVCET | USA 2003 DQ011157.1 | 15 | 49–63 | 100.00% | 2a | |
| DSGYHSLDPNAVCET | UK_P2 MT903344.1 | 15 | 49–63 | 100.00% | 2B-A.1 | |
| DSGYHSLDPNAVCET | USA 2021 ON676708.1 | 15 | 49–63 | 100.00% | 2B-A.1.1 | |
| DSGYHSLDPNAVCET | Nigeria 2018 NC-063383-1 | 15 | 49–63 | 100.00% | 2B-A.2 | |
| DSGYHSLDPNAVCET | USA 2022 VA1 ON675438.1 | 15 | 49–63 | 100.00% | 2B-A.2 | |
| DSGYHSLDPNAVCET | France 2022 ON622722-2 | 15 | 49–63 | 100.00% | 2B-B.1 | |
| E4R | TGVIDYKGYNLNIID | Zaire AF380138.1 | 15 | 100–114 | 100.00% | 1 |
| TGVIDYKGYNLNIID | USA 2003 DQ011157.1 | 15 | 100–114 | 100.00% | 2a | |
| TGVIDYKGYNLNIID | UK_P2 MT903344.1 | 15 | 100–114 | 100.00% | 2B-A.1 | |
| TGVIDYKGYNLNIID | USA 2021 ON676708.1 | 15 | 100–114 | 100.00% | 2B-A.1.1 | |
| TGVIDYKGYNLNIID | Nigeria 2018 NC-063383-1 | 15 | 100–114 | 100.00% | 2B-A.2 | |
| TGVIDYKGYNLNIID | USA 2022 VA1 ON675438.1 | 15 | 100–114 | 100.00% | 2B-A.2 | |
| TGVIDYKGYNLNIID | France 2022 ON622722-2 | 15 | 100–114 | 100.00% | 2B-B.1 | |
| F8L | RSLETDLRSEFDSRS | Zaire AF380138.1 | 15 | 870–884 | 100.00% | 1 |
| RSLETDLRSEFDSRS | USA 2003 DQ011157.1 | 15 | 870–884 | 100.00% | 2a | |
| RSLETDLRSEFDSRS | UK_P2 MT903344.1 | 15 | 870–884 | 100.00% | 2B-A.1 | |
| RSLETDLRSEFDSRS | USA 2021 ON676708.1 | 15 | 870–884 | 100.00% | 2B-A.1.1 | |
| RSLETDLRSEFDSRS | Nigeria 2018 NC-063383-1 | 15 | 870–884 | 100.00% | 2B-A.2 | |
| RSLETDLRSEFDSRS | USA 2022 VA1 ON675438.1 | 15 | 870–884 | 100.00% | 2B-A.2 | |
| RSLETDLRSEFDSRS | France 2022 ON622722-2 | 15 | 870–884 | 100.00% | 2B-B.1 | |
| F8L | SMVFEYRASTVIKGP | Zaire AF380138.1 | 15 | 491–505 | 100.00% | 1 |
| SMVFEYRASTIIKGP | USA 2003 DQ011157.1 | 15 | 491–505 | 93.33% | 2a | |
| SMVFEYRASTIIKGP | UK_P2 MT903344.1 | 15 | 491–505 | 93.33% | 2B-A.1 | |
| SMVFEYRASTIIKGP | USA 2021 ON676708.1 | 15 | 491–505 | 93.33% | 2B-A.1.1 | |
| SMVFEYRASTIIKGP | Nigeria 2018 NC-063383-1 | 15 | 491–505 | 93.33% | 2B-A.2 | |
| SMVFEYRASTIIKGP | USA 2022 VA1 ON675438.1 | 15 | 491–505 | 93.33% | 2B-A.2 | |
| SMVFEYRASTIIKGP | France 2022 ON622722-2 | 15 | 491–505 | 93.33% | 2B-B.1 | |
| (c) LBL Epitope | ||||||
| Protein | Epitope sequence | Strain | Epitope length | Positions | Identity | Clade |
| B6R | LTSTETSFND | Zaire AF380138.1 | 10 | 31–40 | 100.00% | 1 |
| LTSTETSFND | USA 2003 DQ011157.1 | 10 | 31–40 | 100.00% | 2a | |
| LTSTETSFND | UK_P2 MT903344.1 | 10 | 31–40 | 100.00% | 2B-A.1 | |
| LTSTETSFND | USA 2021 ON676708.1 | 10 | 31–40 | 100.00% | 2B-A.1.1 | |
| LTSTETSFND | Nigeria 2018 NC-063383-1 | 10 | 31–40 | 100.00% | 2B-A.2 | |
| LTSTETSFND | USA 2022 VA1 ON675438.1 | 10 | 31–40 | 100.00% | 2B-A.2 | |
| LTSTETSFND | France 2022 ON622722-2 | 10 | 31–40 | 100.00% | 2B-B.1 | |
| B9R | GDYKVEEYCTG | Zaire AF380138.1 | 11 | 85–95 | 100.00% | 1 |
| GDYKVEEYCTG | USA 2003 DQ011157.1 | 11 | 85–95 | 100.00% | 2a | |
| GDYKVEEYCTG | UK_P2 MT903344.1 | 11 | 85–95 | 100.00% | 2B-A.1 | |
| GDYKVEEYCTG | USA 2021 ON676708.1 | 11 | 85–95 | 100.00% | 2B-A.1.1 | |
| GDYKVEEYCTG | Nigeria 2018 NC-063383-1 | 11 | 85–95 | 100.00% | 2B-A.2 | |
| GDYKVEEYCTG | USA 2022 VA1 ON675438.1 | 11 | 85–95 | 100.00% | 2B-A.2 | |
| GDYKVEEYCTG | France 2022 ON622722-2 | 11 | 85–95 | 100.00% | 2B-B.1 | |
| H3L | KSGGL | Zaire AF380138.1 | 5 | 204–209 | 100.00% | 1 |
| KSGGL | USA 2003 DQ011157.1 | 5 | 204–209 | 100.00% | 2a | |
| KSGGL | UK_P2 MT903344.1 | 5 | 204–209 | 100.00% | 2B-A.1 | |
| KSGGL | USA 2021 ON676708.1 | 5 | 204–209 | 100.00% | 2B-A.1.1 | |
| KSGGL | Nigeria 2018 NC-063383-1 | 5 | 204–209 | 100.00% | 2B-A.2 | |
| KSGGL | USA 2022 VA1 ON675438.1 | 5 | 204–209 | 100.00% | 2B-A.2 | |
| KSGGL | France 2022 ON622722-2 | 5 | 204–209 | 100.00% | 2B-B.1 | |
| J3R/J1L | EEEKDSDIKTHPV | Zaire AF380138.1 | 13 | 162–174 | 100.00% | 1 |
| EEEKDSDIKTHPV | USA 2003 DQ011157.1 | 13 | 162–174 | 100.00% | 2a | |
| EEEKDSDIKTHPV | UK_P2 MT903344.1 | 13 | 162–174 | 100.00% | 2B-A.1 | |
| EEEKDSDIKTHPV | USA 2021 ON676708.1 | 13 | 162–174 | 100.00% | 2B-A.1.1 | |
| EEEKDSDIKTHPV | Nigeria 2018 NC-063383-1 | 13 | 162–174 | 100.00% | 2B-A.2 | |
| EEEKDSDIKTHPV | USA 2022 VA1 ON675438.1 | 13 | 162–174 | 100.00% | 2B-A.2 | |
| EEEKDSDIKTHPV | France 2022 ON622722-2 | 13 | 162–174 | 100.00% | 2B-B.1 | |
| Parameter | PADRE Vaccine-TLR4 Complex Cluster 1 | PADRE Vaccine-TLR4 Complex Cluster 2 | PADRE Vaccine-TLR4 Complex Cluster 3 | PADRE Vaccine-TLR4 Complex Cluster 4 |
|---|---|---|---|---|
| KD (M) at 25 °C | 7.0 × 10−14 | 2.3 × 10−14 | 8.7 × 10−14 | 5.3 × 10−14 |
| ΔG (kcal mol−1) | −18.7 | −19.3 | −18.5 | −18.8 |
| ICs charged-charged | 19 | 16 | 17 | 15 |
| ICs charged-polar | 24 | 25 | 21 | 20 |
| ICs charged-apolar | 46 | 39 | 40 | 42 |
| ICs polar-polar | 12 | 10 | 10 | 10 |
| ICs polar-apolar | 40 | 46 | 41 | 43 |
| ICs apolar-apolar | 31 | 24 | 28 | 31 |
| NIS charged | 20.33 | 20.88 | 20.32 | 20.23 |
| NIS apolar | 40.66 | 40.72 | 40.48 | 41.41 |
- A14L
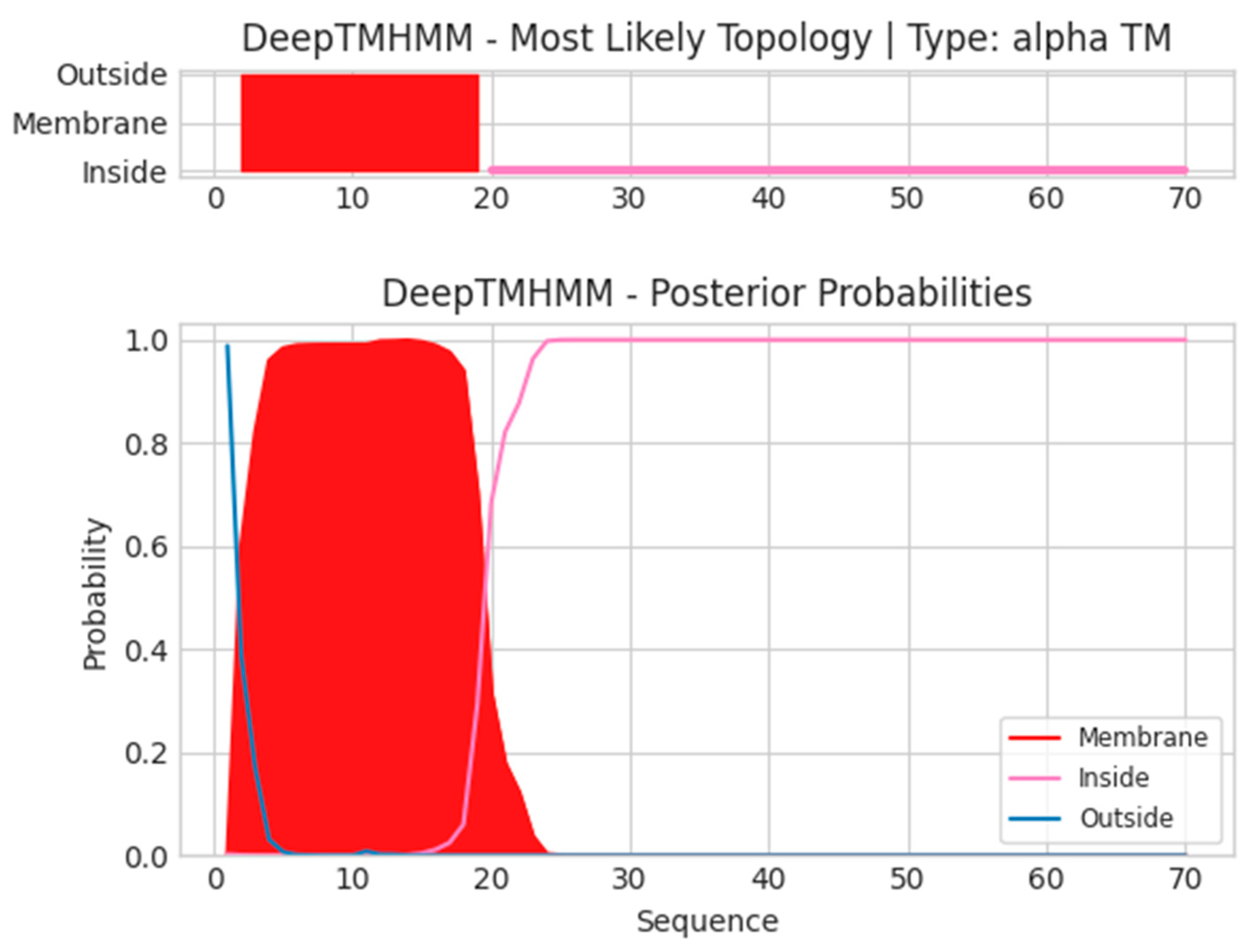
- Unnamed Tmhelix 2 19
- MIGILLLIGICVAVTVAILYTLYNKIKNPQNPNPSPNLNSPPPETRNTKFVNNLEKDH
- ISSLYNLVKSSA
- A15L
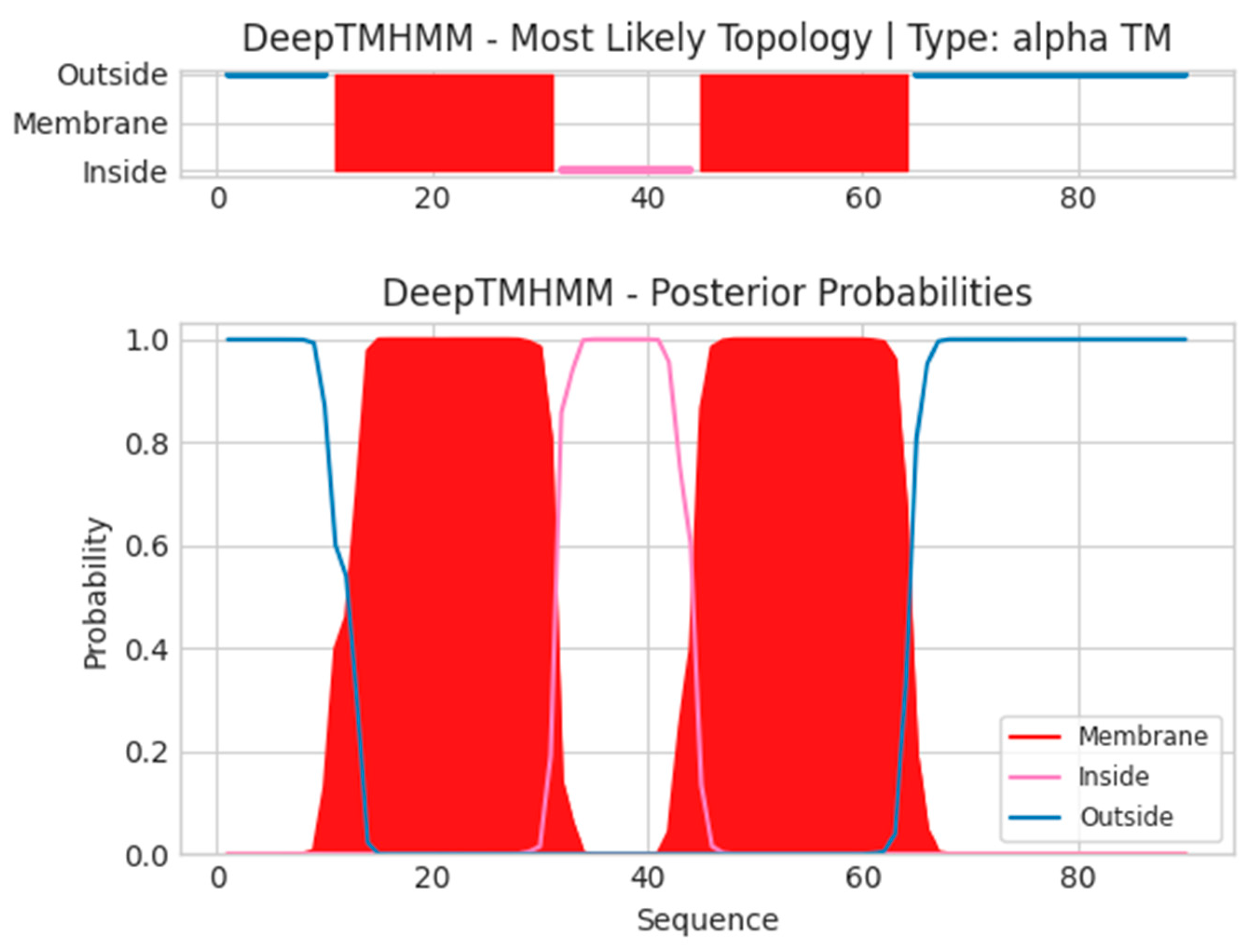
- Unnamed Tmhelix 11 31
- Unnamed Tmhelix 45 64
- MDMMLMIGNYFSGVLIAGIILLILSCIFAFIDFSKSTSPTRTWKVLSIMAFILGIIITV
- GMLIYSMWGKHCAPHRVSGVIHTNHSDISMN
- A15.5L
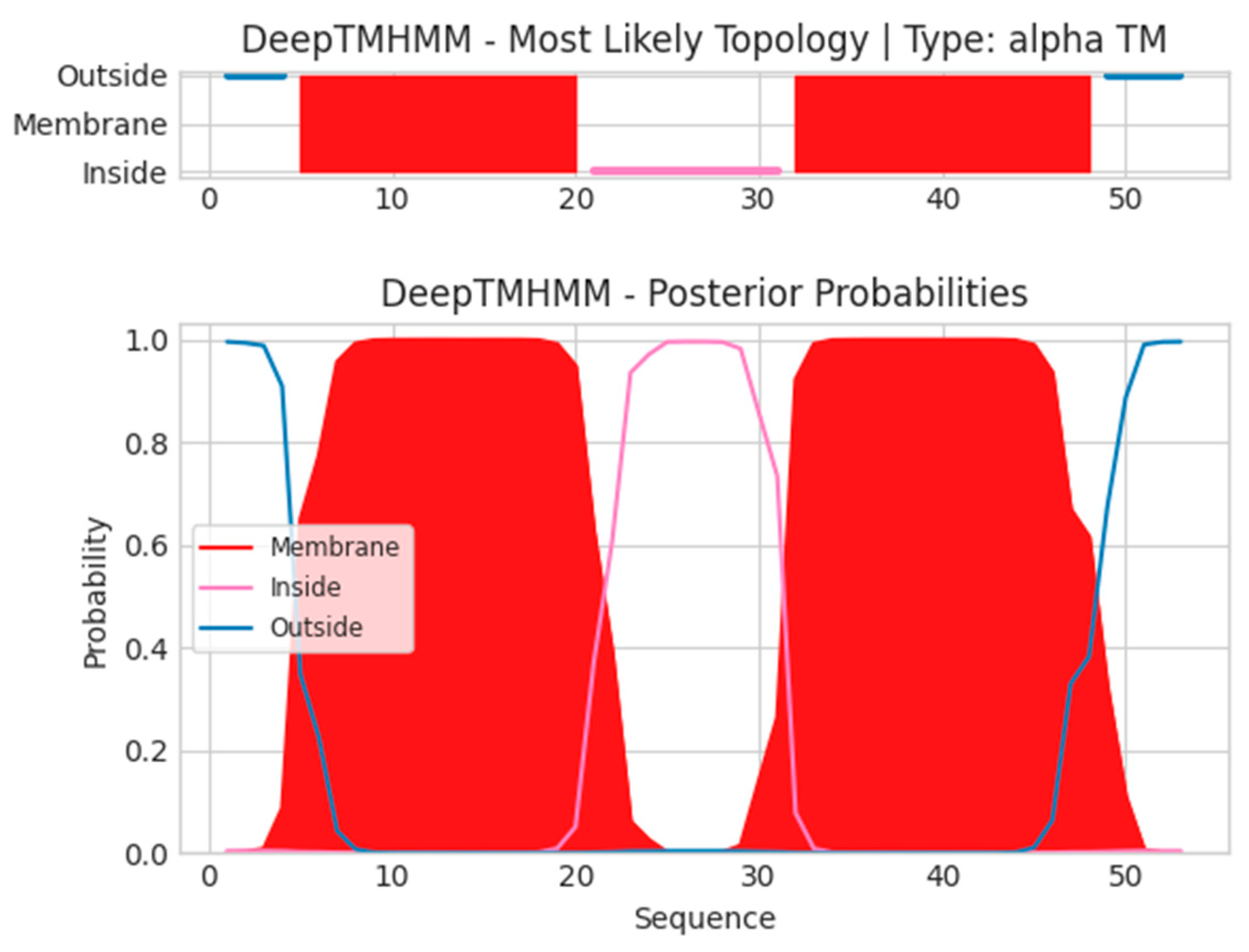
- Unnamed Tmhelix 5 20
- Unnamed Tmhelix 32 48
- MISNYEPLLLLVITCCVLLFNFTISSKTKIDIIFAVQTIVFIWFIFHFVYSAI
- MISNNFTISSKTKIDVYSAI
- A40L
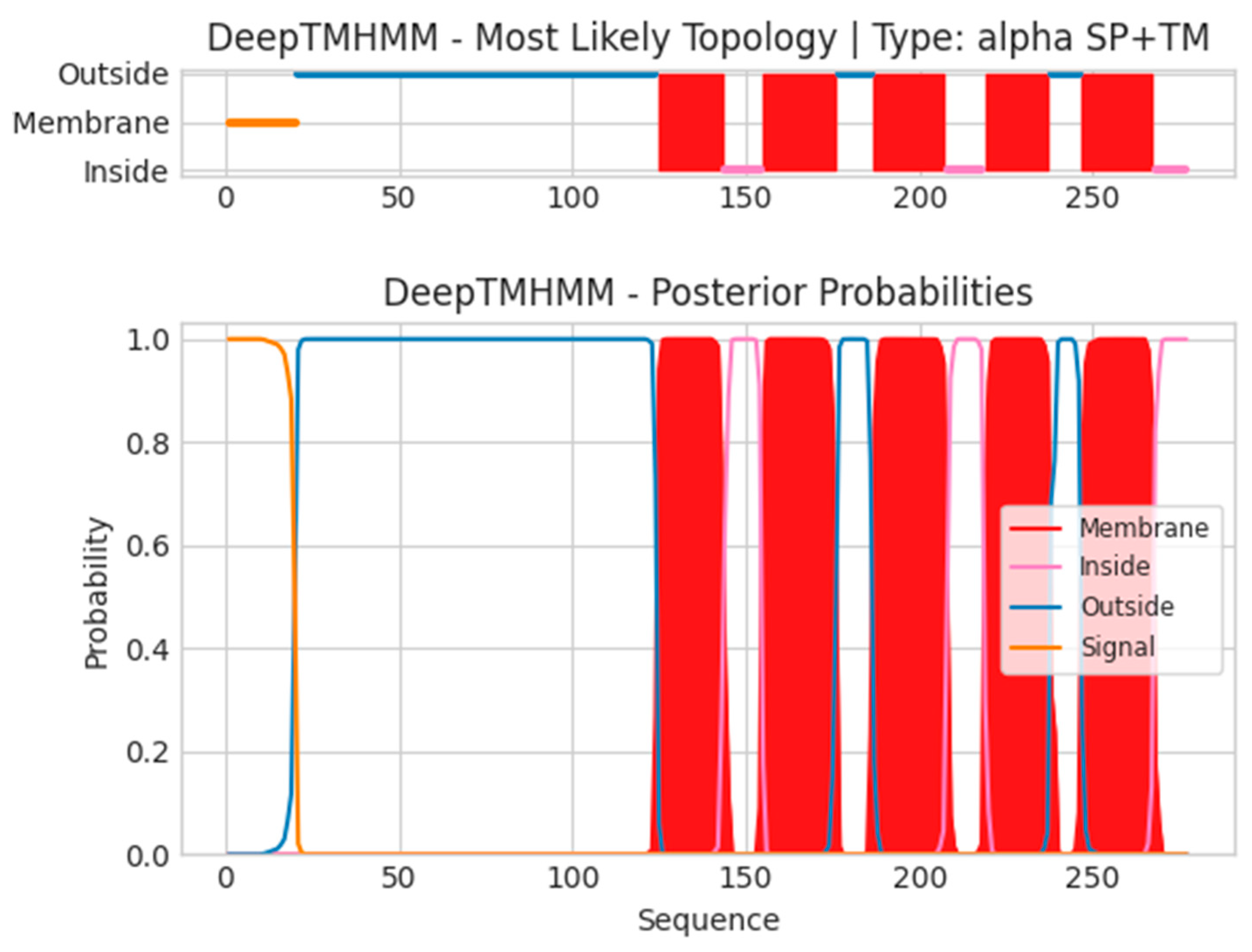
- Unnamed Tmhelix 125 143
- Unnamed Tmhelix 155 176
- Unnamed Tmhelix 187 207
- Unnamed Tmhelix 219 237
- Unnamed Tmhelix 247 267
- MLRVRILLIYLCTFVVITSTKTIEYTACNDTIIIPCIIDNPTKYIRWKLDNHDILTYNK
- TSKTTILSKWHTSARLHSLSDSDVSLIMEYKDILPGTYTCGDNTGIKSTVKLVQRH
- TNWFNDYQTMLMFIFTGITLFLLFLEIAYTSISVVFSTNLGILQVFGCVIAMIELCG′
- AFLFYPSMFTLRHIIGLLMMTLPSIFLIITKVFSFWLLCKLSCAVHLIIYYQLAGYIL
- TVLGLGLSLKECVDGTLLLSGLGTIMVSEHFGLLFLVCFPSTQRDYY
- B6R
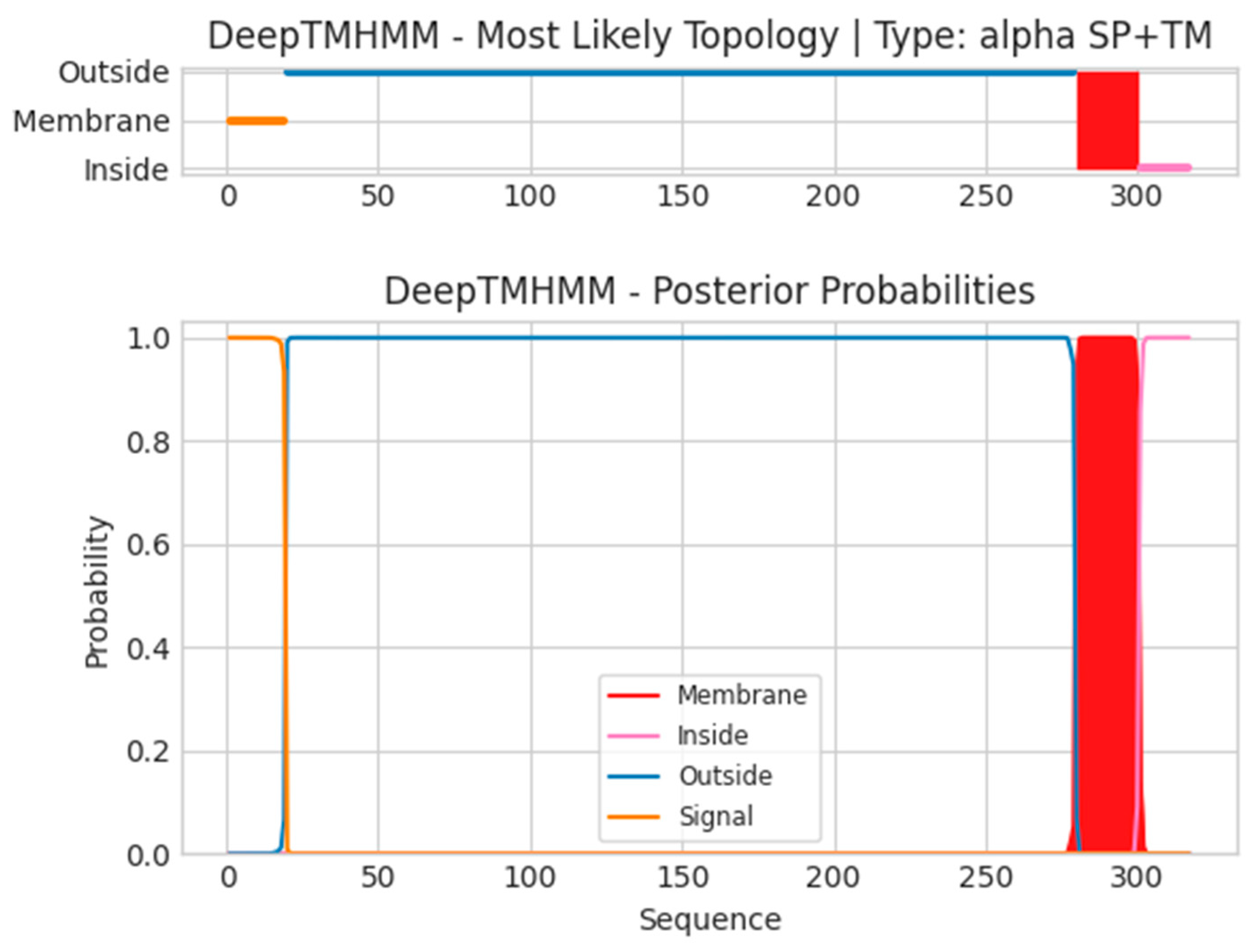
- Unnamed Tmhelix 280 300
- MKTISVVTLLCVLPAVVYSTCTVPTMNNAKLTSTETSFNDKQKVTFTCDSGYHSL
- DPNAVCETDKWKYENPCKKMCTVSDYVSELYDKPLYEVNSTMTLSCNGETKYF
- RCEEKNGNTSWNDTVTCPNAECQPLQLEHGSCQPVKEKYSFGEYMTINCDVGY
- EVIGVSYISCTANSWNVIPSCQQKCDIPSLSNGLISGSTFSIGGVIHLSCKSGFTLTG
- SPSSTCIDGKWNPILPTCVRSNEEFDPVDDGPDDETDLSKLSKDVVQYEQEIESLE
- ATYHIIIMALTIMGVIFLISIIVLVCSCDKNNDQYKFHKLLP
- B21R
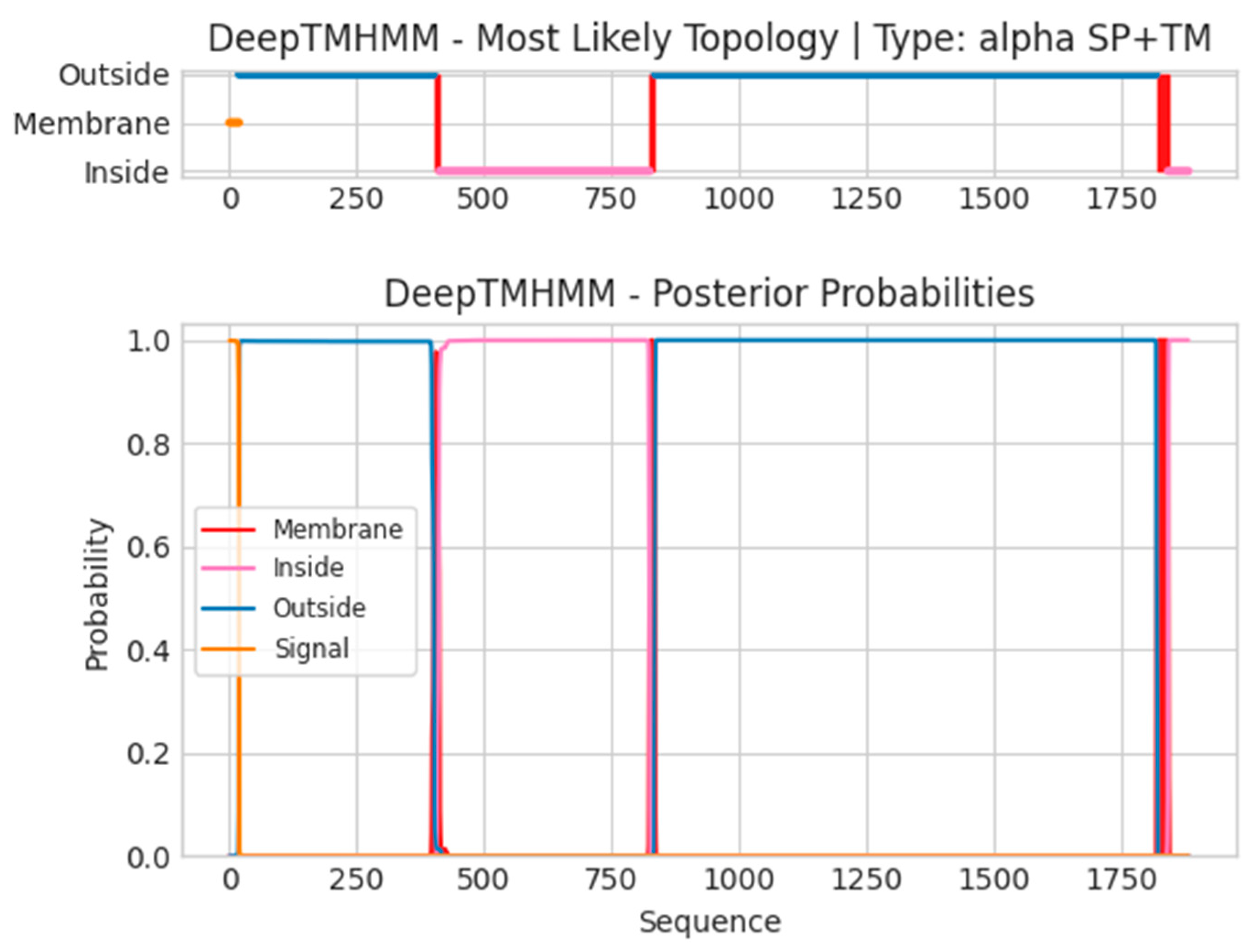
- Unnamed Tmhelix 402 411
- Unnamed Tmhelix 824 833
- Unnamed Tmhelix 1817 1839
- MNLQKLSLAIYLTVTCSWCYETCMRKTALYHDIQLEHVEDNKDSVASLPYKYLQ
- VVKQRERSRLLATFNWTDIAEGVRNEFIKICDINGTYLYNYTIDVSIIIDSTEELPTV
- TPITTYEPSIYNYTIDYSTVITTEELQVTPTYAPVTTPLPTSAVPYDQRSNNNVSTISI
- QILSKILGVNETELTNYLIMHKNDTVDNNTMVDDETSDNNTLHGNIGFLEINNCY
- NVSVSDASFRITLVNDTSEEILLMLTGTSSSDTFISSTNITECLKTLINNVSINDVLIT
- QNMNVTSNCDKCSMNLMASVIPAVNEFNNTLMKIGVKDDENNTVYNYYICKLT
- TNSTCDELINLDEVINNITLTNIIRNSVSTTNSRKRRDLNGEFEFSTSKELDCLYESY
- GVNDDISHCFASPRRRRSDDKKEYMDMKLFDHAKKDLGIDSVIPRGTTHFQVGA
- SGASGGVVGDSFPFQNVKSRASLLAEKIMPRVPITATEADLYATVNRQPKLPAGVK
- STPFTEALASTINQKLSNVREVTYASLNLPGSSGYVHRPSDSVIYSSIRRSRLPSDS
- DSDYEDIQTVVKEYNERYGRSVSRTQSSSSESDFEDIDTVVREYRQKYGNAMAK
- GRSSSPKPDPLYSTVKKTTKSLSTGVDIVTKQSDYSLLPDVNTGSSIVSPLTRKGAT
- RRRPRRPTNDGLQSPNPPLRNPLPQHDDYSPPQVHRPPPLPPKPVQNPPQLPPRPV
- GQLPPPIDQPDKGFSKFVSPRRCRRASSGVICGMIQSKPNDDTYSLLQRPKIEPEYA
- EVGNGIPKNNVPVIGNKHSKKYTSTMSKISTKFDKSTAFGAAMLLTGQQAISQQT
- RSTTLSRKDQMSKEEKIFEAVTMSLSTIGSTLTSAGMTGGPKLMIAGMAITAITGII
- DTIKDIYYMFSGQERPVDPVIKLFNKYAGLMSDNNKMGVRKCLTPGDDTLIYIAY
- RNDTSFKQNTDAMALYFLDVIDSEILYLNTSNLVLEYQLKVACPIGTLRSVDVDIT
- AYTILYDTADNIKKYKFIRMATLLSKHPVIRLTCGLAATLVIKPYEVPISDMQLLKM
- ATPGEPESTKSIPSDVCDRYPLKKFYLLAGGCPYDTSQTFIVHTTCSILLRTATRDQ
- FRNRWVLQNPFRQEGTYKQLFTFSKYDFNDTIIDPNGVVGHASFCTNRSSNQCF
- WSEPMILEDVSSCSSRTRKIYVKLGIFNAEGFNSFVLNCPTGSTPTYIKHKNADSN
- NVIIELPVGDYGTAKLYSATKPSRIAVFCTHNYDKRFKSDIIVLMFNKNSGIPFWS
- MYTGSVTSKNRMFTTLARGMPFRSTYCDNRRRSGCYYAGIPFHEDSVETDIHY
- GPEIMLKETYDINSIDPRVITKSKTHFPAPLSVKFMVDNLGNGYDNPNSFWEDAK
- TKKRTYSAMTIKVLPCTVRNKNIDFGYNYGDIISNMVYLQSTSQDYGDGTKYTF
- KSVTRSDHECESSLDLTSKEVTVTCPAFSIPRNISTYEGLCFSVTTSKDHCATGIGW
- LKSSGYGKEDADKPRACFHHWNYYTLSLDYYCSYEDIWRSTWPDYDPCKSYIHI
- EYRDTWIESNVLQQPPYTFEFIHDNSNEYVDKEISNKLNDLYNEYKKIMEYSDGS
- LPASINRLAKALTSEGREIASVNIDGNLLDIAYQADKEKMADIQTRINDIIRDLFIHT
- LSDKDIKDIIESEEGKRCCIIDVKNNRVKKYYSIDNYLCGTLDDYIYTSVEYNKSY
- VLVNDTYMSYDYLESSGVVVLSCYEMTIISLDTKDAKDAIEDVIVASAVAEALND
- MFKEFDKNVSAIIIKEEDNYLNSSPDIYHIIYIIGGTILLLLVIILILAIYIARNKYRTR
- KYEIMKYDNMSIKSDHHDSLETVSMEIIDNRY
- H3L
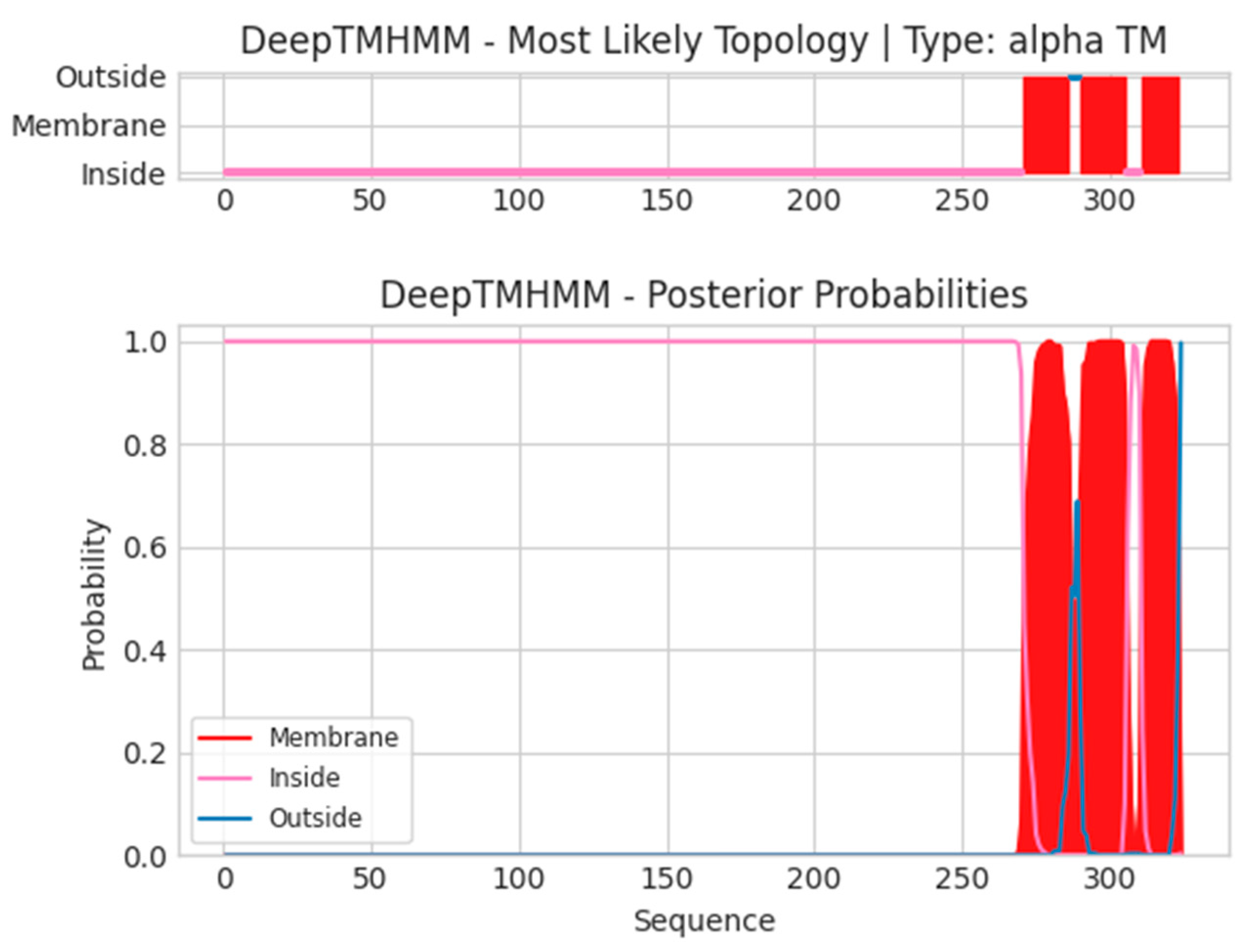
- Unnamed Tmhelix 271 286
- Unnamed Tmhelix 290 305
- Unnamed Tmhelix 311 323
- MAAAKTPVIVVPVIDRPPSETFPNVHEHINDQKFDDVKDNEVMQEKRDVVIVND
- DPDHYKDYVFIQWTGGNIRDDDKYTHFFSGFCNTMCTEETKRNIARHLALWDSK
- FFIELENKNVEYVVIIENDNVIEDITFLRPVLKAIHDKKIDILQMREIITGNKVKTEL
- VIDKDHAIFTYTGGYDVSLSAYIIRVTTALNIVDEIIKSGGLSSGFYFEIARIENEMKI
- NRQIMDNSAKYVEHDPRLVAEHRFETMKPNFWSRIGTVAAKRYPGVMYTFTTPLI
- SFFGLFDINVIGLIVILFIMFMLIFNVKSKLLWFLTGTFVTAFI
- L5L
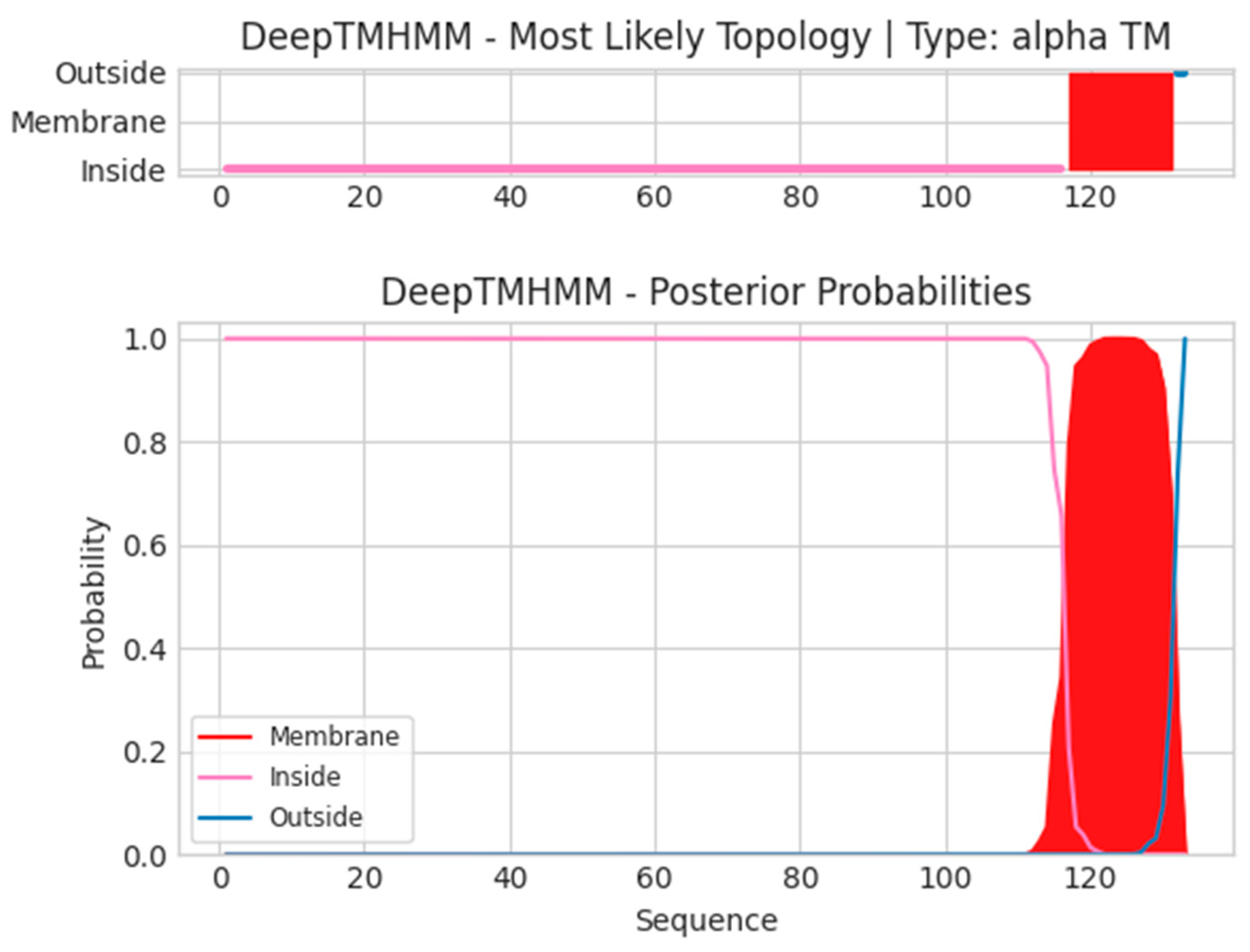
- Unnamed Tmhelix 117 131
- MTDEQIYAFCDANKDDIRCKCIYPDKSIVRIGIDTRLPYYCWYEPCKRSDALLPAS
- LKKNISRCNVSDCTISLGNVSITDSKLDVNNVCDSKRVATENIAVRYLNQEIRYPII
- DIKWLPIGLLALAILILAFF
- M1R

- Unnamed TMhelix 184 204
- MGAAASIQTTVNTLSERISSKLEQEANASAQTKCDIEIGNFYIRQNHGCNITVKNM
- CSADADAQLDAVLSAATETYSGLTPEQKAYVPAMFTAALNIQTSVNTVVRDFEN
- YVKQTCNSSAVVDNKLKIQNVIIDECYGAPGSPTNLEFINTGSSKGNCAIKALMQ
- LTTKATTQIAPRQVAGTGVQFYMIVIGVIILAALFMYYAKRMLFTSTNDKIKLILA
- NKENVHWTTYMDTFFRTSPMIIATTDIQN
- M5R
- Unnamed Tmhelix 30 47
- MENVPNVYFNPVFIEPTFKHSLLSVYKHRLIVLFEVFVVFILIYVFFRSELNMFFMP
- KRKIPDPIDRLRRANLACEDDKLMIYGLPWITTQTSALSINSKPIVYKDCAKLLRSI
- NGSQPVSLNDVL
- Codon optimization (PADRE adjuvant)
- Codon Usage adapted to Escherichia coli (strain K12)
- Codon-optimized cDNA sequence
- ATGGAAGCTGCTGCTAAAGCTAAATTCGTTGCTGCTTGGACCCTGAAAGCTGCTGCTGAAGCTGCTGCTAAACGTACCATCGACATCGACGAAACCATCGCTGCTTACTACCCGCCGCCGCGTTACATCACCGTTGCTGCTTACATCTACTTCAAAGGTACCTGGCAGTACGCTGCTTACACCCGTGACCCGCTGTACATCTACAAAGCTGCTTACCGTCACATCTGGCTGCTGTGCAAACTGGCTGCTTACCAGGAAAAACGTGACGTTGTTATCGTTGCTGCTTACCTGTCTTGCAACGGTGAAACCAAATACGCTGCTTACGAAACCATCGAACTGGGTGAACGTTACGCTGCTTACAAAATCGGTCCGCCGACCGTTCGTCTGCACGAATACGGTGCTGAAGCTCTGGAACGTGCTGGTTCTATGGTTTTCGAATACCGTGCTTCTACCGTTATCAAAGGTCCGGGTCCGGGTCCGGGTCGTTCTCTGGAAACCGACCTGCGTTCTGAATTCGACTCTCGTTCTGGTCCGGGTCCGGGTACCGGTGTTATCGACTACAAAGGTTACAACCTGAACATCATCGACGGTCCGGGTCCGGGTGACTCTGGTTACCACTCTCTGGACCCGAACGCTGTTTGCGAAACCCACGAATACGGTGCTGAAGCTCTGGAACGTGCTGGTAAATCTGGTGGTCTGTCTAAAAAACTGACCTCTACCGAAACCTCTTTCAACGACAAAAAAAAAGGTGACTACAAAGTTGAAGAATACTGCACCGGTAAAAAAGAAGAAGAAAAAGACTCTGACATCAAAACCCACCCGGTTCGTGTTCGTCGTCACCACCACCACCACCAC
- CAI-Value of the improved sequence: 1.0
- GC-Content of the improved sequence: 51.11
- GC-Content of Escherichia coli (strain K12): 50.73
- Dataset 1. Multi-epitope vaccine models
- (1)
- CTL, HTL, and LBL
- (2)
- Adjuvant
- (3)
- 2 EAAAK linkers (vaccine with adjuvant)
- (4)
- 8 AAY Linkers (between CTL epitopes)
- (5)
- 1 KK linker (Between LBL epitopes)
- (6)
- 2 HEYGAEALERAG linkers (Between the last CTL epitope and the first HTL epitope, and the last HTL epitope and the first LBL epitope)
- (7)
- 1 RVRR linker (between the last LBL epitopes and Histag or HHHHHH)
- (8)
- 3 GPGPG linkers (between HTL epitopes)
- PADRE adjuvant
- MEAAAKAKFVAAWTLKAAAEAAAKRTIDIDETIAAYYPPPRYITVAAYIYFKGTWQYAAYTRDPLYIYKAAYRHIWLLCKLAAYQEKRDVVIVAAYLSCNGETKYAAYETIELGERYAAYKIGPPTVRLHEYGAEALERAGSMVFEYRASTVIKGPGPGPGRSLETDLRSEFDSRSGPGPGTGVIDYKGYNLNIIDGPGPGDSGYHSLDPNAVCETHEYGAEALERAGKSGGLKKLTSTETSFNDKKGDYKVEEYCTGKKEEEKDSDIKTHPVRVRRHHHHHH
- RS09 adjuvant
- MEAAAKAPPHALSEAAAKRTIDIDETIAAYYPPPRYITVAAYIYFKGTWQYAAYTRDPLYIYKAAYRHIWLLCKLAAYQEKRDVVIVAAYLSCNGETKYAAYETIELGERYAAYKIGPPTVRLHEYGAEALERAGSMVFEYRASTVIKGPGPGPGRSLETDLRSEFDSRSGPGPGTGVIDYKGYNLNIIDGPGPGDSGYHSLDPNAVCETHEYGAEALERAGKSGGLKKLTSTETSFNDKKGDYKVEEYCTGKKEEEKDSDIKTHPVRVRRHHHHHH
- 50S Ribosomal L7/L12 adjuvant
- MEAAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVAVAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVSGLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTVKEAAAKRTIDIDETIAAYYPPPRYITVAAYIYFKGTWQYAAYTRDPLYIYKAAYRHIWLLCKLAAYQEKRDVVIVAAYLSCNGETKYAAYETIELGERYAAYKIGPPTVRLHEYGAEALERAGSMVFEYRASTVIKGPGPGPGRSLETDLRSEFDSRSGPGPGTGVIDYKGYNLNIIDGPGPGDSGYHSLDPNAVCETHEYGAEALERAGKSGGLKKLTSTETSFNDKKGDYKVEEYCTGKKEEEKDSDIKTHPVRVRRHHHHHH
- Beta-defensin adjuvant
- MEAAAKGIINTLCKYYCRVRGGRCCVCSCCPKEEQIGKCSTRGRKCCRRKKQEKIPFHLQISKQVIEAAAKRTIDIDETIAAYYPPPRYITVAAYIYFKGTWQYAAYTRDPLYIYKAAYRHIWLLCKLAAYQEKRDVVIVAAYLSCNGETKYAAYETIELGERYAAYKIGPPTVRLHEYGAEALERAGSMVFEYRASTVIKGPGPGPGRSLETDLRSEFDSRSGPGPGTGVIDYKGYNLNIIDGPGPGDSGYHSLDPNAVCETHEYGAEALERAGKSGGLKKLTSTETSFNDKKGDYKVEEYCTGKKEEEKDSDIKTHPVRVRRHHHHHH
- Cholera Toxin B subunit (CTB)
- MEAAAKTPQNITDLCAEYHNTQIYTLNDKIFSYTESLAGKREMAIITFKNGAIFQVEVPGSQHIDSQKKAIERMKDTLRIAYLTEAKVEKLCVWNNKTPHAIAAISMANEAAAKRTIDIDETIAAYYPPPRYITVAAYIYFKGTWQYAAYTRDPLYIYKAAYRHIWLLCKLAAYQEKRDVVIVAAYLSCNGETKYAAYETIELGERYAAYKIGPPTVRLHEYGAEALERAGSMVFEYRASTVIKGPGPGPGRSLETDLRSEFDSRSGPGPGTGVIDYKGYNLNIIDGPGPGDSGYHSLDPNAVCETHEYGAEALERAGKSGGLKKLTSTETSFNDKKGDYKVEEYCTGKKEEEKDSDIKTHPVRVRRHHHHHH
References
- Das, T.; Nandy, S.; Ghosh, A.; Chandran, D.; Sharma, A.K.; Dhama, K.; Dey, A. Efficacy of Smallpox Approved Tecovirimat (Tpoxx) Drug against Monkeypox: Current Update and Futuristic Prospects. Int. J. Surg. 2023, 109, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-S.; Lin, C.-Y.; Urbina, A.N.; Wang, W.-H.; Assavalapsakul, W.; Tseng, S.-P.; Lu, P.-L.; Chen, Y.-H.; Yu, M.-L.; Wang, S.-F. The First Case of Monkeypox Virus Infection Detected in Taiwan: Awareness and Preparation. Int. J. Infect. Dis. 2022, 122, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Damon, I.K. Status of Human Monkeypox: Clinical Disease, Epidemiology and Research. Vaccine 2011, 29, D54–D59. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mu, L.; Wang, W. Monkeypox: Epidemiology, Pathogenesis, Treatment and Prevention. Signal Transduct. Target. Ther. 2022, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human Monkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and Prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Mpox Trends. Available online: https://worldhealthorg.shinyapps.io/mpx_global/#overview (accessed on 12 July 2025).
- Shamim, M.A.; Padhi, B.K.; Satapathy, P.; Veeramachaneni, S.D.; Chatterjee, C.; Tripathy, S.; Akhtar, N.; Pradhan, A.; Dwivedi, P.; Mohanty, A.; et al. The Use of Antivirals in the Treatment of Human Monkeypox Outbreaks: A Systematic Review. Int. J. Infect. Dis. 2023, 127, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Paul, D.; Mohanty, A.; Shah, A.; Mohanasundaram, A.S.; Padhi, B.K. Monkeypox (Mpox) Vaccines and Their Side Effects: The Other Side of the Coin. Int. J. Surg. 2023, 109, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Humayun, M.; Baig, E.; Farooq, M.; Hussain, H.G.; Shahid, M.U.; Cheema, H.A.; Chandran, D.; Yatoo, M.I.; Sharma, A.K.; et al. FDA’s Authorized “JYNNEOS” Vaccine for Counteracting Monkeypox Global Public Health Emergency; an Update—Correspondence. Int. J. Surg. 2022, 107, 106971. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.W.; Kabamba, J.; McCollum, A.M.; Lushima, R.S.; Wemakoy, E.O.; Muyembe Tamfum, J.-J.; Nguete, B.; Hughes, C.M.; Monroe, B.P.; Reynolds, M.G. Vaccinating against Monkeypox in the Democratic Republic of the Congo. Antivir. Res. 2019, 162, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Titanji, B.K.; Tegomoh, B.; Nematollahi, S.; Konomos, M.; Kulkarni, P.A. Monkeypox: A Contemporary Review for Healthcare Professionals. Open Forum Infect. Dis. 2022, 9, ofac310. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Zhu, F.; Pan, P.; Wu, A.; Li, C. Development of Multi-Epitope Vaccines against the Monkeypox Virus Based on Envelope Proteins Using Immunoinformatics Approaches. Front. Immunol. 2023, 14, 1112816. [Google Scholar] [CrossRef] [PubMed]
- Kenner, J.; Cameron, F.; Empig, C.; Jobes, D.V.; Gurwith, M. LC16m8: An Attenuated Smallpox Vaccine. Vaccine 2006, 24, 7009–7022. [Google Scholar] [CrossRef] [PubMed]
- Bamouh, Z.; Hamdi, J.; Fellahi, S.; Khayi, S.; Jazouli, M.; Tadlaoui, K.O.; Fihri, O.F.; Tuppurainen, E.; Elharrak, M. Investigation of Post Vaccination Reactions of Two Live Attenuated Vaccines against Lumpy Skin Disease of Cattle. Vaccines 2021, 9, 621. [Google Scholar] [CrossRef] [PubMed]
- Yokote, H.; Shinmura, Y.; Kanehara, T.; Maruno, S.; Kuranaga, M.; Matsui, H.; Hashizume, S. Safety of Attenuated Smallpox Vaccine LC16m8 in Immunodeficient Mice. Clin. Vaccine Immunol. 2014, 21, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.F. Current Status of Monkeypox Vaccines. npj Vaccines 2022, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic Characterization and Signs of Microevolution in the 2022 Multi-Country Outbreak of Monkeypox Virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Sanami, S.; Nazarian, S.; Ahmad, S.; Raeisi, E.; ul Qamar, M.T.; Tahmasebian, S.; Pazoki-Toroudi, H.; Fazeli, M.; Samani, M.G. In Silico Design and Immunoinformatics Analysis of a Universal Multi-Epitope Vaccine against Monkeypox Virus. PLoS ONE 2023, 18, e0286224. [Google Scholar] [CrossRef] [PubMed]
- Bidmos, F.A.; Siris, S.; Gladstone, C.A.; Langford, P.R. Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 2018, 9, 2315. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Rappuoli, R.; Seib, K.L. Vaccines, Reverse Vaccinology, and Bacterial Pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a012476. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.; Almajhdi, F.N.; Waqas, M.; Ullah, I.; Salim, M.A.; Khan, N.A.; Ali, A. Contriving Multi-Epitope Vaccine Ensemble for Monkeypox Disease Using an Immunoinformatics Approach. Front. Immunol. 2022, 13, 1004804. [Google Scholar] [CrossRef] [PubMed]
- Amanna, I.J.; Slifka, M.K. Contributions of Humoral and Cellular Immunity to Vaccine-Induced Protection in Humans. Virology 2011, 411, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Fantin, R.F.; Coelho, C.H. Human Antibody Responses to Circulating Monkeypox Virus Emphasise the Need for the First Mpox-Specific Vaccine. Lancet Microbe 2024, 5, e204–e205. [Google Scholar] [CrossRef] [PubMed]
- Abrahim, M.; Guterres, A.; Neves, P.C.d.C.; Bom, A.P.D.A. The Emergence of New Lineages of the Mpox Virus Could Affect the 2022 Outbreak. bioRxiv 2023. [Google Scholar] [CrossRef]
- Song, H.; Sidney, J.; Wiseman, R.W.; Josleyn, N.; Cohen, M.; Blaney, J.E.; Jahrling, P.B.; Sette, A. Characterizing Monkeypox Virus Specific CD8+ T Cell Epitopes in Rhesus Macaques. Virology 2013, 447, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Liu, X.; Lu, J.; Fan, H.; Xu, X.; Sun, K.; Wang, R.; Li, C.; Dan, D.; Du, H.; et al. Recombinant Proteins A29L, M1R, A35R, and B6R Vaccination Protects Mice from Mpox Virus Challenge. Front. Immunol. 2023, 14, 1203410. [Google Scholar] [CrossRef] [PubMed]
- Keasey, S.; Pugh, C.; Tikhonov, A.; Chen, G.; Schweitzer, B.; Nalca, A.; Ulrich, R.G. Proteomic Basis of the Antibody Response to Monkeypox Virus Infection Examined in Cynomolgus Macaques and a Comparison to Human Smallpox Vaccination. PLoS ONE 2010, 5, e15547. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ren, Z.; Chen, J.; Li, M.; Jiang, Y.; Liu, Y.; Zhang, B.; Lu, H.; Zhao, W.; Shen, C.; et al. Analysis of Binding and Authentic Virus-Neutralizing Activities of Immune Sera Induced by Various Monkeypox Virus Antigens. Immunol. Res. 2024, 72, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Wang, X.; He, J.; Wu, W.; Jiang, W.; Xue, S.; Zhang, Q.; Gao, Y.; Han, Y.; Li, Y.; et al. Cross-Reactive Antibody Response to Monkeypox Virus Surface Proteins in a Small Proportion of Individuals with and without Chinese Smallpox Vaccination History. BMC Biol. 2023, 21, 205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Cheng, X.; Zhu, Y.; Mo, O.; Yu, H.; Zhu, L.; Zhang, J.; Kuang, L.; Gao, Y.; Cao, R.; et al. Multi-Valent mRNA Vaccines against Monkeypox Enveloped or Mature Viron Surface Antigens Demonstrate Robust Immune Response and Neutralizing Activity. Sci. China Life Sci. 2023, 66, 2329–2341. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.B.; Keckler, M.S.; Patel, N.; Davies, D.H.; Felgner, P.; Damon, I.K.; Karem, K.L. Humoral Immunity to Smallpox Vaccines and Monkeypox Virus Challenge: Proteomic Assessment and Clinical Correlations. J. Virol. 2013, 87, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Duke-Cohan, J.S.; Wollenick, K.; Witten, E.A.; Seaman, M.S.; Baden, L.R.; Dolin, R.; Reinherz, E.L. The Heterogeneity of Human Antibody Responses to Vaccinia Virus Revealed through Use of Focused Protein Arrays. Vaccine 2009, 27, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Lorente, E.; Martín-Galiano, A.J.; Barnea, E.; Barriga, A.; Palomo, C.; García-Arriaza, J.; Mir, C.; Lauzurica, P.; Esteban, M.; Admon, A.; et al. Proteomics Analysis Reveals That Structural Proteins of the Virion Core and Involved in Gene Expression Are the Main Source for HLA Class II Ligands in Vaccinia Virus-Infected Cells. J. Proteome Res. 2019, 18, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Couñago, R.M.; Knapp, K.M.; Nakatani, Y.; Fleming, S.B.; Corbett, M.; Wise, L.M.; Mercer, A.A.; Krause, K.L. Structures of Orf Virus Chemokine Binding Protein in Complex with Host Chemokines Reveal Clues to Broad Binding Specificity. Structure 2015, 23, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate Sequence-Based Prediction of Protein Solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A. Thermostability and Aliphatic Index of Globular Proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Crofts, K.F.; Page, C.L.; Swedik, S.M.; Holbrook, B.C.; Meyers, A.K.; Zhu, X.; Parsonage, D.; Westcott, M.M.; Alexander-Miller, M.A. An Analysis of Linker-Dependent Effects on the APC Activation and In Vivo Immunogenicity of an R848-Conjugated Influenza Vaccine. Vaccines 2023, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.A.; Slowinska, K.; Moore, T.; Esser-Kahn, A. Immune Response Modulation of Conjugated Agonists with Changing Linker Length. ACS Chem. Biol. 2016, 11, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant--An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.J.; Hammond, R.A.; Forsberg, E.M.; Geoghegan-Barek, K.M.; Karalius, B.H.; Marsh, H.C.; Rittershaus, C.W. Co-Administration of a CpG Adjuvant (VaxImmune, CPG 7909) with CETP Vaccines Increased Immunogenicity in Rabbits and Mice. Hum. Vaccin. 2009, 5, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; del Guercio, M.F.; Maewal, A.; Qiao, L.; Fikes, J.; Chesnut, R.W.; Paulson, J.; Bundle, D.R.; DeFrees, S.; Sette, A. Linear PADRE T Helper Epitope and Carbohydrate B Cell Epitope Conjugates Induce Specific High Titer IgG Antibody Responses. J. Immunol. 2000, 164, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An Overview of Novel Adjuvants Designed for Improving Vaccine Efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.-G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A Potential Protein Adjuvant Derived from Mycobacterium Tuberculosis Rv0652 Enhances Dendritic Cells-Based Tumor Immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef] [PubMed]
- Vemula, S.V.; Amen, O.; Katz, J.M.; Donis, R.; Sambhara, S.; Mittal, S.K. Beta-Defensin 2 Enhances Immunogenicity and Protection of an Adenovirus-Based H5N1 Influenza Vaccine at an Early Time. Virus Res. 2013, 178, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, Y.L.; Jeong, Y.; Jang, Y.-S. Conjugation of Human β-Defensin 2 to Spike Protein Receptor-Binding Domain Induces Antigen-Specific Protective Immunity against Middle East Respiratory Syndrome Coronavirus Infection in Human Dipeptidyl Peptidase 4 Transgenic Mice. Vaccines 2020, 8, 635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, X.; Xie, Q.; Ma, J.; Luo, Y.; Cao, Y.; Chen, F.; Bi, Y. The Potent Adjuvant Effects of Chicken Beta-Defensin-1 When Genetically Fused with Infectious Bursal Disease Virus VP2 Gene. Vet. Immunol. Immunopathol. 2010, 136, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. ISBN 978-1-59259-890-8. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.W. Therapeutic Strategies to Address Monkeypox. Expert. Rev. Anti-Infect. Ther. 2022, 20, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.W.; Harms, T.J.; Reynolds, M.G.; Harrison, L.H. Use of Vaccinia Virus Smallpox Vaccine in Laboratory and Health Care Personnel at Risk for Occupational Exposure to Orthopoxviruses—Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sharff, K.A.; Tandy, T.K.; Lewis, P.F.; Johnson, E.S. Cardiac Events Following JYNNEOS Vaccination for Prevention of Mpox. Vaccine 2023, 41, 3410–3412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-Epitope Vaccines: A Promising Strategy against Tumors and Viral Infections. Cell Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; von Boehmer, L.; Knuth, A.; Ochsenbein, A.F.; et al. Immunologic Response to the Survivin-Derived Multi-Epitope Vaccine EMD640744 in Patients with Advanced Solid Tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A Tale of Two Clades: Monkeypox Viruses. J. Gen. Virol. 2005, 86, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.H.; McCausland, M.M.; Valdez, C.; Huynh, D.; Hernandez, J.E.; Mu, Y.; Hirst, S.; Villarreal, L.; Felgner, P.L.; Crotty, S. Vaccinia Virus H3L Envelope Protein Is a Major Target of Neutralizing Antibodies in Humans and Elicits Protection against Lethal Challenge in Mice. J. Virol. 2005, 79, 11724–11733. [Google Scholar] [CrossRef] [PubMed]
- Drexler, I.; Staib, C.; Kastenmuller, W.; Stevanović, S.; Schmidt, B.; Lemonnier, F.A.; Rammensee, H.-G.; Busch, D.H.; Bernhard, H.; Erfle, V.; et al. Identification of Vaccinia Virus Epitope-Specific HLA-A*0201-Restricted T Cells and Comparative Analysis of Smallpox Vaccines. Proc. Natl. Acad. Sci. USA 2003, 100, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Zhang, Y.; Tarke, A.; Sidney, J.; Rubiro, P.; Reina-Campos, M.; Filaci, G.; Dan, J.M.; Scheuermann, R.H.; Sette, A. Defining Antigen Targets to Dissect Vaccinia Virus and Monkeypox Virus-Specific T Cell Responses in Humans. Cell Host Microbe 2022, 30, 1662–1670.e4. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Song, Q.; Li, J.; Dunmall, L.S.C.; Jiang, Y.; Qin, B.; Wang, J.; Guo, H.; Cheng, Z.; Wang, Z.; et al. Redirecting Anti-Vaccinia Virus T Cell Immunity for Cancer Treatment by AAV-Mediated Delivery of the VV B8R Gene. Mol. Ther.-Oncolytics 2022, 25, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Flesch, I.E.A.; Tscharke, D.C. Vaccinia Virus CD8+ T-Cell Dominance Hierarchies Cannot Be Altered by Prior Immunization with Individual Peptides. J. Virol. 2009, 83, 9008–9012. [Google Scholar] [CrossRef] [PubMed]
- Baur, K.; Brinkmann, K.; Schweneker, M.; Pätzold, J.; Meisinger-Henschel, C.; Hermann, J.; Steigerwald, R.; Chaplin, P.; Suter, M.; Hausmann, J. Immediate-Early Expression of a Recombinant Antigen by Modified Vaccinia Virus Ankara Breaks the Immunodominance of Strong Vector-Specific B8R Antigen in Acute and Memory CD8 T-Cell Responses. J. Virol. 2010, 84, 8743–8752. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.N.; Koopmans, M.; Otter, A.; Grobusch, M.P.; Jokelainen, P.; Atkinson, B.; Cunha, F.; Valdoleiros, S.R.; Preda, V.G.; Fusco, F.M.; et al. Implications of the 2023–2024 MPXV Clade I Outbreak in the Democratic Republic of Congo to Global Public Health. Clin. Microbiol. Infect. 2024, 30, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Kugelman, J.R.; Johnston, S.C.; Mulembakani, P.M.; Kisalu, N.; Lee, M.S.; Koroleva, G.; McCarthy, S.E.; Gestole, M.C.; Wolfe, N.D.; Fair, J.N.; et al. Genomic Variability of Monkeypox Virus among Humans, Democratic Republic of the Congo. Emerg. Infect. Dis. 2014, 20, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Vakaniaki, E.H.; Kacita, C.; Kinganda-Lusamaki, E.; O’Toole, Á.; Wawina-Bokalanga, T.; Mukadi-Bamuleka, D.; Amuri-Aziza, A.; Malyamungu-Bubala, N.; Mweshi-Kumbana, F.; Mutimbwa-Mambo, L.; et al. Sustained Human Outbreak of a New MPXV Clade I Lineage in the Eastern Democratic Republic of the Congo. Nat. Med. 2024, 30, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Gooyit, M.; Miranda, P.O.; Wenthur, C.J.; Ducime, A.; Janda, K.D. Influencing Antibody-Mediated Attenuation of Methamphetamine CNS Distribution through Vaccine Linker Design. ACS Chem. Neurosci. 2017, 8, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Azmi, F.; Ahmad Fuaad, A.A.H.; Skwarczynski, M.; Toth, I. Recent Progress in Adjuvant Discovery for Peptide-Based Subunit Vaccines. Hum. Vaccin. Immunother. 2014, 10, 778–796. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, G.; Chaudhary, K.; Agrawal, P.; Raghava, G.P.S. Computer-Aided Prediction of Antigen Presenting Cell Modulators for Designing Peptide-Based Vaccine Adjuvants. J. Transl. Med. 2018, 16, 181. [Google Scholar] [CrossRef] [PubMed]
- Halajian, E.A.; LeBlanc, E.V.; Gee, K.; Colpitts, C.C. Activation of TLR4 by Viral Glycoproteins: A Double-Edged Sword? Front. Microbiol. 2022, 13, 1007081. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of Loops in Protein Structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher Sequence Analysis Tools Framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M. Contacts-Based Prediction of Binding Affinity in Protein–Protein Complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef] [PubMed]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [PubMed]
- Lopéz-Blanco, J.R.; Garzón, J.I.; Chacón, P. iMod: Multipurpose Normal Mode Analysis in Internal Coordinates. Bioinformatics 2011, 27, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Szilárd, P.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2015; Volume 8759, pp. 3–27. [Google Scholar]
- Ragone, C.; Manolio, C.; Cavalluzzo, B.; Mauriello, A.; Tornesello, M.L.; Buonaguro, F.M.; Castiglione, F.; Vitagliano, L.; Iaccarino, E.; Ruvo, M.; et al. Identification and Validation of Viral Antigens Sharing Sequence and Structural Homology with Tumor-Associated Antigens (TAAs). J. Immunother. Cancer 2021, 9, e002694. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, P.; Castiglione, F.; Mastrostefano, E.; Di Biase, I.; Di Biase, S.; Palmieri, G.; Prisco, A. In-Silico Evaluation of Adenoviral COVID-19 Vaccination Protocols: Assessment of Immunological Memory up to 6 Months after the Third Dose. Front. Immunol. 2022, 13, 998262. [Google Scholar] [CrossRef] [PubMed]
- Masirika, L.M.; Udahemuka, J.C.; Schuele, L.; Ndishimye, P.; Otani, S.; Mbiribindi, J.B.; Marekani, J.M.; Mambo, L.M.; Bubala, N.M.; Boter, M.; et al. Ongoing Mpox Outbreak in Kamituga, South Kivu Province, Associated with Monkeypox Virus of a Novel Clade I Sub-Lineage, Democratic Republic of the Congo, 2024. Euro Surveill. 2024, 29, 2400106. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. Identifying Candidate Subunit Vaccines Using an Alignment-Independent Method Based on Principal Amino Acid Properties. Vaccine 2007, 25, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. Bioinformatic Approach for Identifying Parasite and Fungal Candidate Subunit Vaccines. TOVACJ 2008, 1, 22–26. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity Prediction by Descriptor Fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of Interferon-Gamma Inducing MHC Class-II Binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Kumar, A.; Kaur, J. Strategies for Optimization of Heterologous Protein Expression in E. Coli: Roadblocks and Reinforcements. Int. J. Biol. Macromol. 2018, 106, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Lajmi, A.R.; Wallace, T.R.; Shin, J.A. Short, Hydrophobic, Alanine-Based Proteins Based on the Basic Region/Leucine Zipper Protein Motif: Overcoming Inclusion Body Formation and Protein Aggregation during Overexpression, Purification, and Renaturation. Protein Expr. Purif. 2000, 18, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Shortle, D.; Stites, W.E.; Meeker, A.K. Contributions of the Large Hydrophobic Amino Acids to the Stability of Staphylococcal Nuclease. Available online: https://pubs.acs.org/doi/pdf/10.1021/bi00487a007 (accessed on 23 June 2024).
- Bui, H.-H.; Sidney, J.; Li, W.; Fusseder, N.; Sette, A. Development of an Epitope Conservancy Analysis Tool to Facilitate the Design of Epitope-Based Diagnostics and Vaccines. BMC Bioinform. 2007, 8, 361. [Google Scholar] [CrossRef] [PubMed]
- Zaru, R.; Orchard, S.; UniProt Consortium. UniProt Tools: BLAST, Align, Peptide Search, and ID Mapping. Curr. Protoc. 2023, 3, e697. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting Population Coverage of T-Cell Epitope-Based Diagnostics and Vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; McCabe, A.; dos Santos, E.J.M.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P.S. ToxinPred2: An Improved Method for Predicting Toxicity of Proteins. Brief. Bioinform. 2022, 23, bbac174. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Heo, L.; Seok, C. Effective Protein Model Structure Refinement by Loop Modeling and Overall Relaxation. Proteins 2016, 84 (Suppl. 1), 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for Checking the Quality of Protein Structures Solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of Protein-Structure Coordinates. urn:isbn 2012, F, 684–687. [Google Scholar] [CrossRef]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical Quality of Protein Structure Coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Recognition of Errors in Three-Dimensional Structures of Proteins. Proteins 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Rarey, M. Computational Methods for Biomolecular Docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Trellet, M.E.; Jiménez-García, B.; Schaarschmidt, J.J.; Giulini, M.; Reys, V.; Koukos, P.I.; Rodrigues, J.P.G.L.M.; Karaca, E.; van Zundert, G.C.P.; et al. The HADDOCK2.4 Web Server for Integrative Modeling of Biomolecular Complexes. Nat. Protoc. 2024, 19, 3219–3241. [Google Scholar] [CrossRef] [PubMed]
- Lopes, P.E.M.; Guvench, O.; MacKerell, A.D. Current Status of Protein Force Fields for Molecular Dynamics Simulations. Methods Mol. Biol. 2015, 1215, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D. Empirical Force Fields for Biological Macromolecules: Overview and Issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain Fluctuations and Elastic Constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]

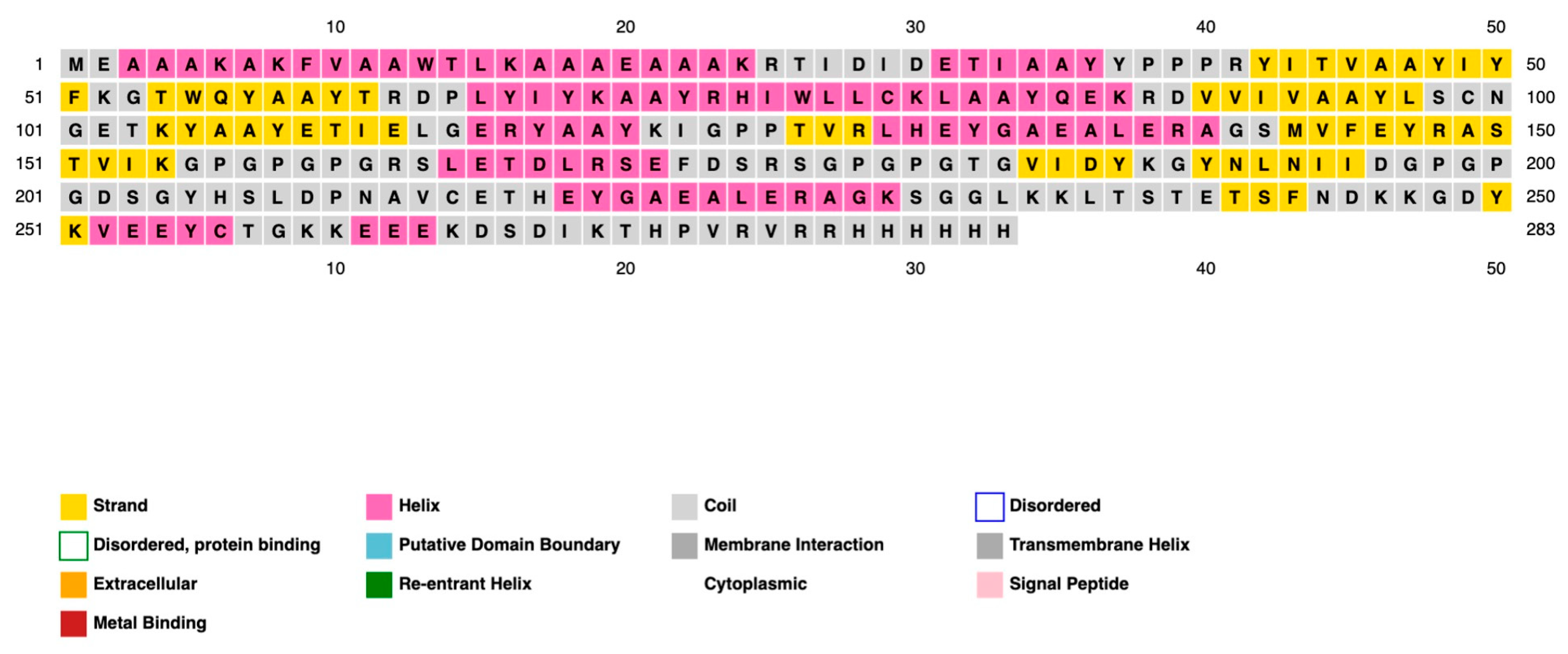




| Structural Protein | |||
| Protein Name | Product | Antigenicity | Allergenicity |
| A14L | IMV membrane protein | 0.5019 | Non-allergen |
| A15L | Phosphorylated IMV membrane protein | 0.4759 | Non-allergen |
| A15.5L | IV and IMV membrane protein | 0.7480 | Non-allergen |
| A30L | IMV surface protein (envelope protein) | 0.6212 | Non-allergen |
| A40L | CD47-like putative membrane protein | 0.4361 | Non-allergen |
| B6R | EEV type-I membrane glycoprotein | 0.5786 | Non-allergen |
| B16R | IFN-alpha/beta-receptor-like secreted glycoprotein | 0.5407 | Non-allergen |
| B19R | Serine protease inhibitor-like SPI-1 protein | 0.7784 | Non-allergen |
| B21R | Putative membrane-associated glycoprotein | 0.5246 | Non-allergen |
| C2L | Serine protease inhibitor-like protein SPI-3 | 0.5702 | Non-allergen |
| F7R | Membrane protein | 0.4334 | Non-allergen |
| H3L | IMV heparin binding surface protein | 0.4538 | Non-allergen |
| L5L | Llate 16 kDa putative membrane protein | 0.7559 | Non-allergen |
| M1R | Myristyl protein | 0.6339 | Non-allergen |
| M5R | Putative membrane protein | 0.4468 | Non-allergen |
| Nonstructural Protein | |||
| Protein Name | Product | Antigenicity | Allergenicity |
| A28L | A-type inclusion protein | 0.4602 | Non-allergen |
| D3R | Secreted epidermal growth factor-like protein | 0.4947 | Non-allergen |
| D10L | Host-range | 0.5965 | Non-allergen |
| E4R | Uracil-DNA glycosylase | 0.5759 | Non-allergen |
| F1L | Poly-A polymerase catalytic subunit VP55 | 0.4419 | Non-allergen |
| F3L | Double-stranded RNA binding protein | 0.4127 | Non-allergen |
| F7R | Membrane protein | 0.4334 | Non-allergen |
| F8L | DNA polymerase | 0.4103 | Non-allergen |
| J3R/J1L | Chemokine-binding protein | 0.7563 | Non-allergen |
| (a) | |||||||
| Protein | CTL Epitope | Antigenic Score | Allergenicity | Toxicity | Class I Immunogenicity Score | Binding Affinity | Human Proteome |
| A40L | RHIWLLCKL | 0.5947 | NA | NT | 0.0734 | Strong Binding | NH |
| B6R | LSCNGETKY | 0.8019 | NA | NT | 0.0100 | Strong Binding | NH |
| B9R | KIGPPTVRL | 1.1705 | NA | NT | 0.0907 | Strong Binding | NH |
| C2L | IYFKGTWQY | 0.8395 | NA | NT | 0.0098 | Strong Binding | NH |
| TRDPLYIYK | 0.4908 | NA | NT | 0.0919 | Strong Binding | NH | |
| F8L | RTIDIDETI | 1.0682 | NA | NT | 0.3232 | Strong Binding | NH |
| YPPPRYITV | 0.4807 | NA | NT | 0.1672 | Strong Binding | NH | |
| ETIELGERY | 1.4370 | NA | NT | 0.2798 | Strong Binding | NH | |
| H3L | QEKRDVVIV | 1.4396 | NA | NT | 0.1551 | Strong Binding | NH |
| (b) | |||||||
| Protein | HTL Epitope | Antigenicity | Allergenicity | Toxicity | IFN-γ Epitope | Binding Affinity | Human Proteome |
| B6R | DSGYHSLDPNAVCET | 0.9057 | NA | NT | Positive | Strong Binding | NH |
| E4R | TGVIDYKGYNLNIID | 1.4293 | NA | NT | Positive | Strong Binding | NH |
| F8L | SMVFEYRASTVIKGP | 0.5161 | NA | NT | Positive | Strong Binding | NH |
| RSLETDLRSEFDSRS | 0.9620 | NA | NT | Positive | Strong Binding | NH | |
| (c) | |||||||
| Protein | LBL Epitope | Antigenicity | Allergenicity | Toxicity | Human Proteome | ||
| B6R | LTSTETSFND | 1.5197 | NA | NT | NH | ||
| B9R | GDYKVEEYCTG | 0.9854 | NA | NT | NH | ||
| H3L J3R/J1L | KSGGL | 2.1517 | NA | NT | NH | ||
| EEEKDSDIKTHPV | 0.6156 | NA | NT | NH | |||
| Properties | Adjuvant | ||||
|---|---|---|---|---|---|
| Cholera Toxin B Subunit (CTB) | PADRE | RS09 | 50S Ribosomal L7/L12 | Beta-Defensin | |
| Antigenicity (VaxiJen 2.0) | 0.5979 | 0.6419 | 0.6340 | 0.5566 | 0.6321 |
| Allergenicity (AllergenFP v.1.0) | Non-allergen | Non-allergen | Non-allergen | Non-allergen | Non-allergen |
| Toxicity (ToxinPred V.2) | Non-Toxin | Non-Toxin | Non-Toxin | Non-Toxin | Toxic |
| Number of amino acids | 373 | 283 | 277 | 400 | 330 |
| Molecular weight (Da) | 41,756.08 | 31,458.34 | 30,802.50 | 43,551.20 | 37,049.07 |
| Theoretical isoelectric point (pI) | 6.92 | 7.17 | 6.64 | 5.60 | 8.83 |
| The estimated half-life1 | 30 h | 30 h | 30 h | 30 h | 30 h |
| The estimated half-life2 | >20 h | >20 h | >20 h | >20 h | >20 h |
| The estimated half-life3 | >10 h | >10 h | >10 h | >10 h | >10 h |
| Aliphatic index | 72.57 | 68.76 | 67.76 | 79.17 | 67.18 |
| Instability index | 34.40 | 30.64 | 34.02 | 29.25 | 36.61 |
| Grand average of hydropathicity (GRAVY) | −0.566 | −0.586 | −0.642 | −0.387 | −0.632 |
| Solubility | 0.83 | 0.93 | 0.87 | 0.98 | 0.72 |
| Signal Peptide | No signal peptide | No signal peptide | No signal peptide | No signal peptide | No signal peptide |
| Tool | Parameter | PADRE Vaccine-TLR4 Complex |
|---|---|---|
| ClusPro 2.0 | Center | −1375.4 |
| Lowest Energy | −1375.4 | |
| HADDOCK 2.4 server | KD (M) at 25 °C | 2.00 × 10−14 |
| ΔG (kcal mol−1) | −18.7 | |
| HADDOCK score | −808.4 ± 7.9 | |
| Cluster size | 20 | |
| RMSD from the overall lowest-energy structure | 0.7 ± 0.4 | |
| Van der Waals energy | −426.0 ± 3.7 | |
| Electrostatic energy | −1417.1 ± 28.0 | |
| Desolvation energy | −99.1 ± 6.6 | |
| Restraints violation energy | 0.0 ± 0.0 | |
| Buried surface area | 12,977.8 ± 131.7 | |
| Z-score | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puagsopa, J.; Jumpalee, P.; Dechanun, S.; Choengchalad, S.; Lohasupthawee, P.; Sutjaritvorakul, T.; Meksiriporn, B. Development of a Broad-Spectrum Pan-Mpox Vaccine via Immunoinformatic Approaches. Int. J. Mol. Sci. 2025, 26, 7210. https://doi.org/10.3390/ijms26157210
Puagsopa J, Jumpalee P, Dechanun S, Choengchalad S, Lohasupthawee P, Sutjaritvorakul T, Meksiriporn B. Development of a Broad-Spectrum Pan-Mpox Vaccine via Immunoinformatic Approaches. International Journal of Molecular Sciences. 2025; 26(15):7210. https://doi.org/10.3390/ijms26157210
Chicago/Turabian StylePuagsopa, Japigorn, Panuwid Jumpalee, Sittichoke Dechanun, Sukanya Choengchalad, Pana Lohasupthawee, Thanawat Sutjaritvorakul, and Bunyarit Meksiriporn. 2025. "Development of a Broad-Spectrum Pan-Mpox Vaccine via Immunoinformatic Approaches" International Journal of Molecular Sciences 26, no. 15: 7210. https://doi.org/10.3390/ijms26157210
APA StylePuagsopa, J., Jumpalee, P., Dechanun, S., Choengchalad, S., Lohasupthawee, P., Sutjaritvorakul, T., & Meksiriporn, B. (2025). Development of a Broad-Spectrum Pan-Mpox Vaccine via Immunoinformatic Approaches. International Journal of Molecular Sciences, 26(15), 7210. https://doi.org/10.3390/ijms26157210