
DNA Methylation Concurrence, Independent of DNA Methylation Ratios, Is Associated with Chromatin Accessibility and 3D Genome Architecture
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
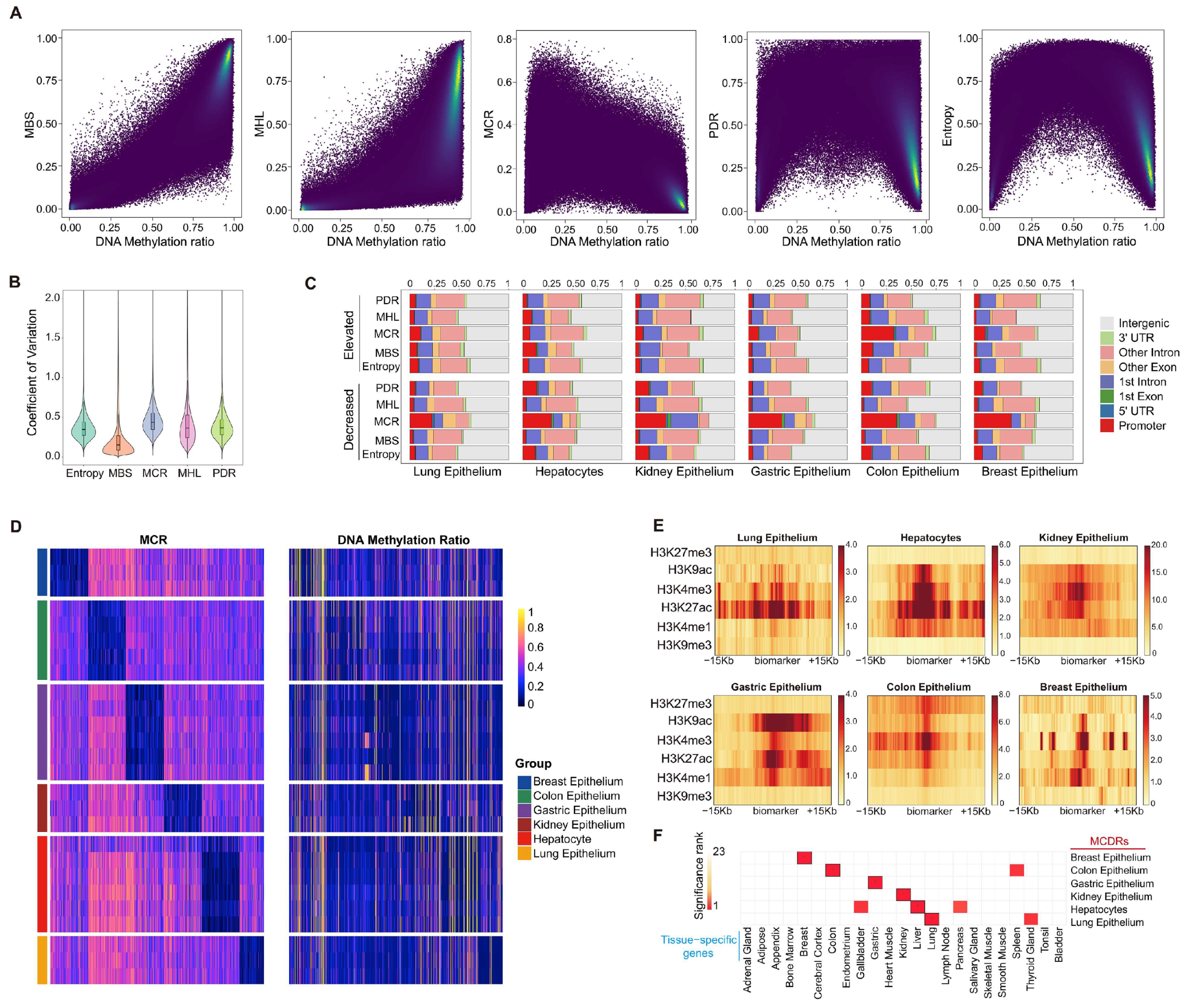
2.1. MCR Represents Tissue-Specific Epigenetic Features Independent of DNA Methylation Ratios
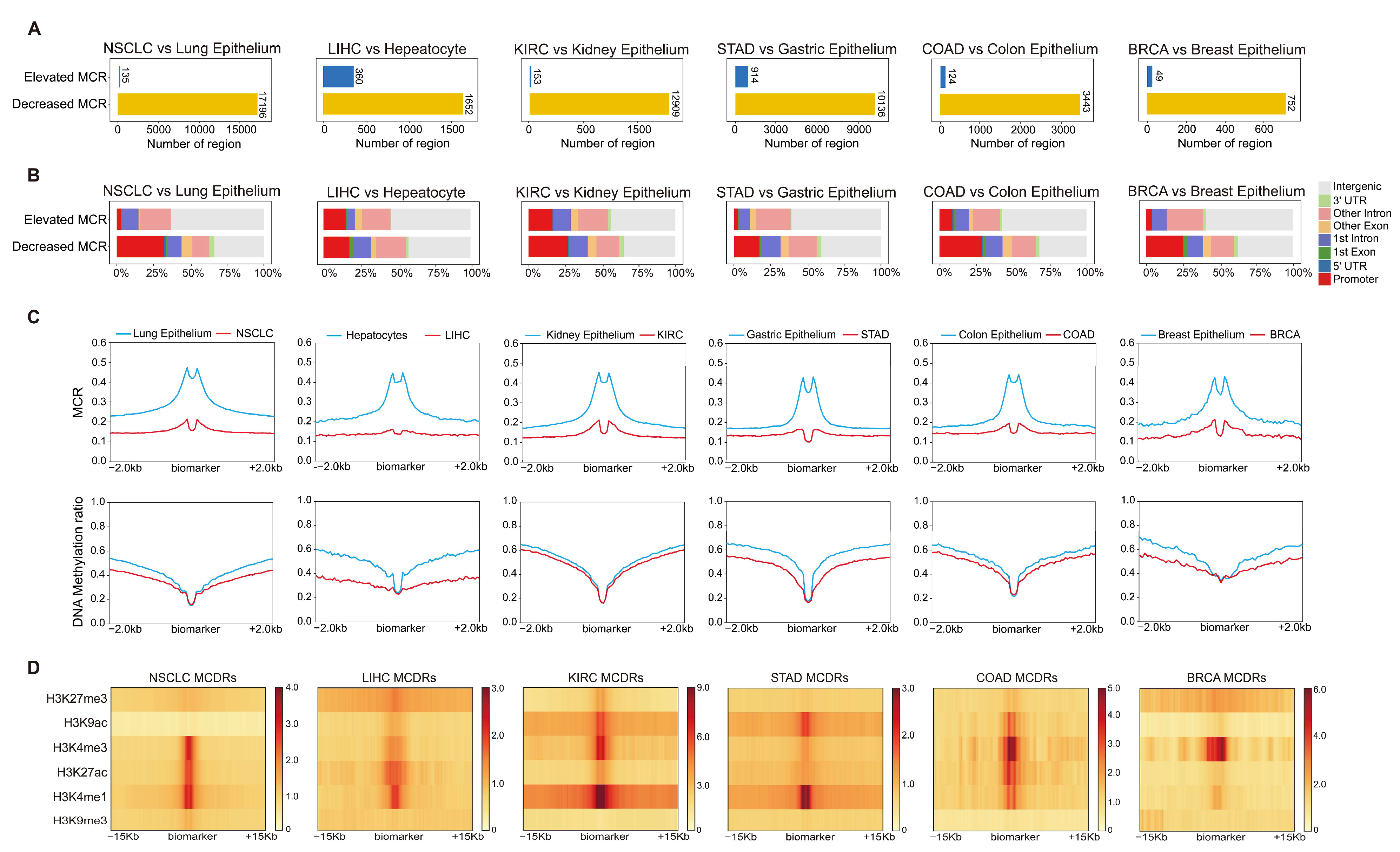
2.2. Disrupted DNA Methylation Concurrence in Tumors Identifies Regulatory Elements
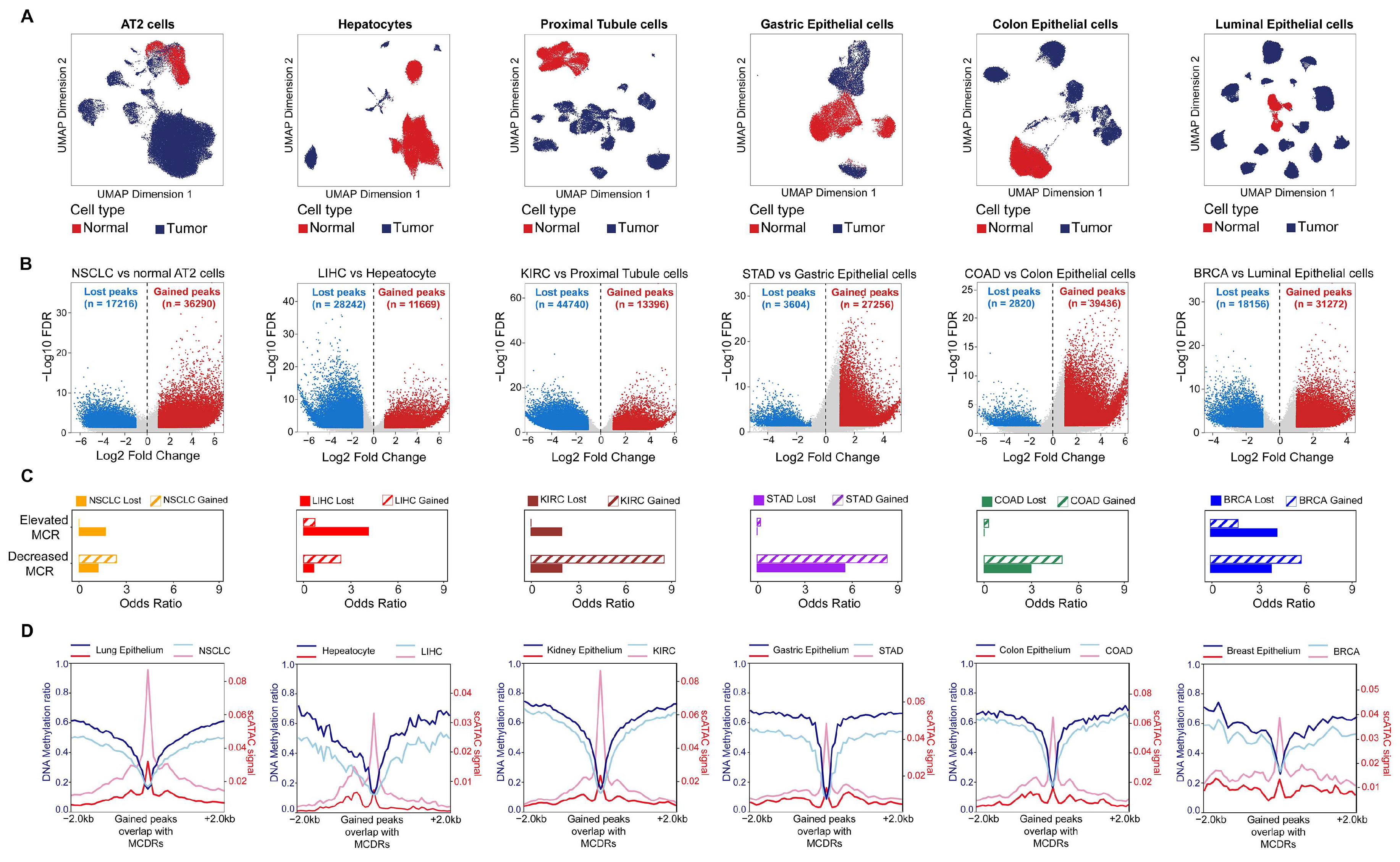
2.3. Decreased DNA Methylation Concurrence Is Associated with the Acquisition of Open Chromatin in Tumors
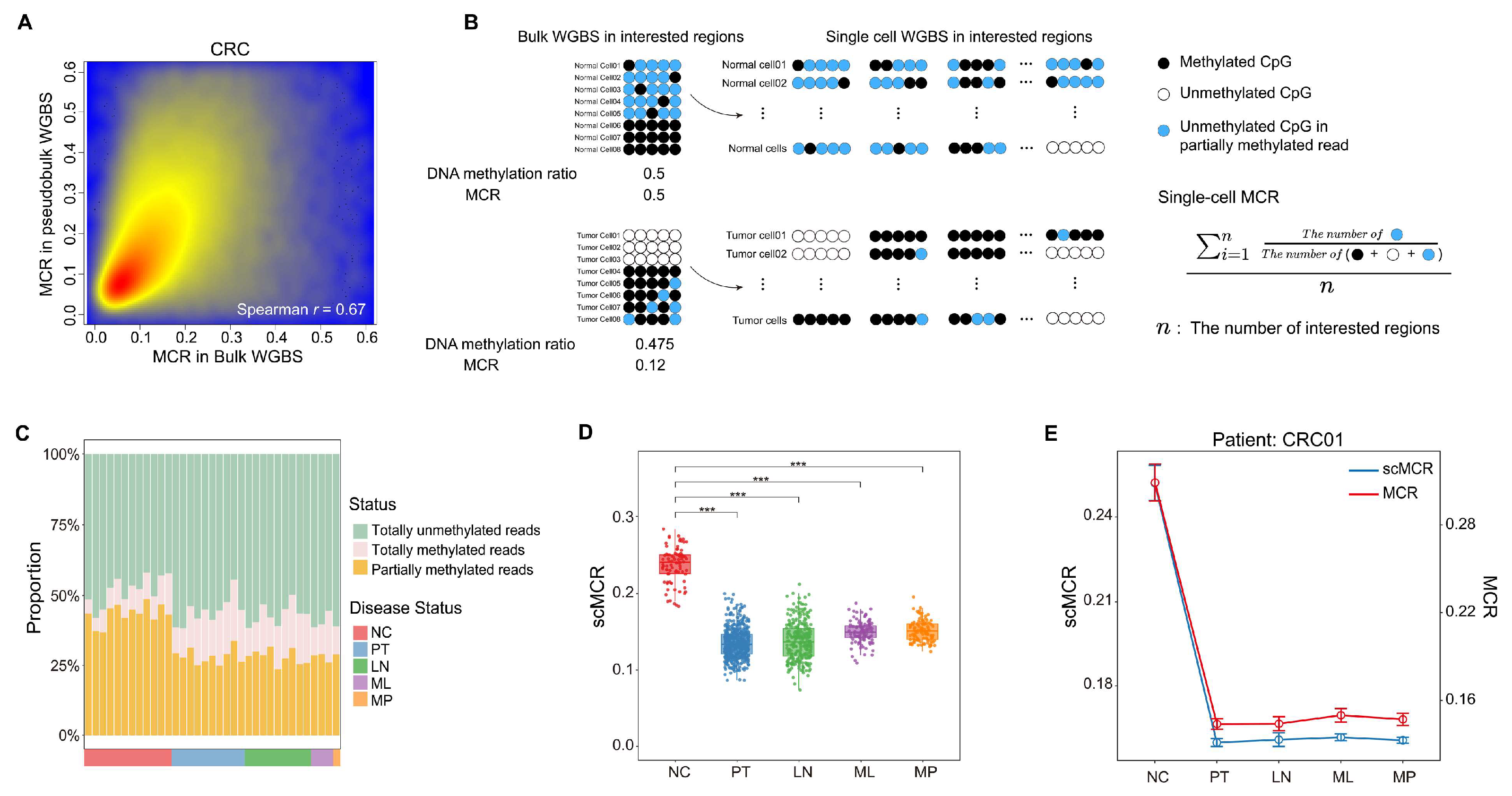
2.4. Dissecting MCR at the Single-Cell Level in Colorectal Cancer
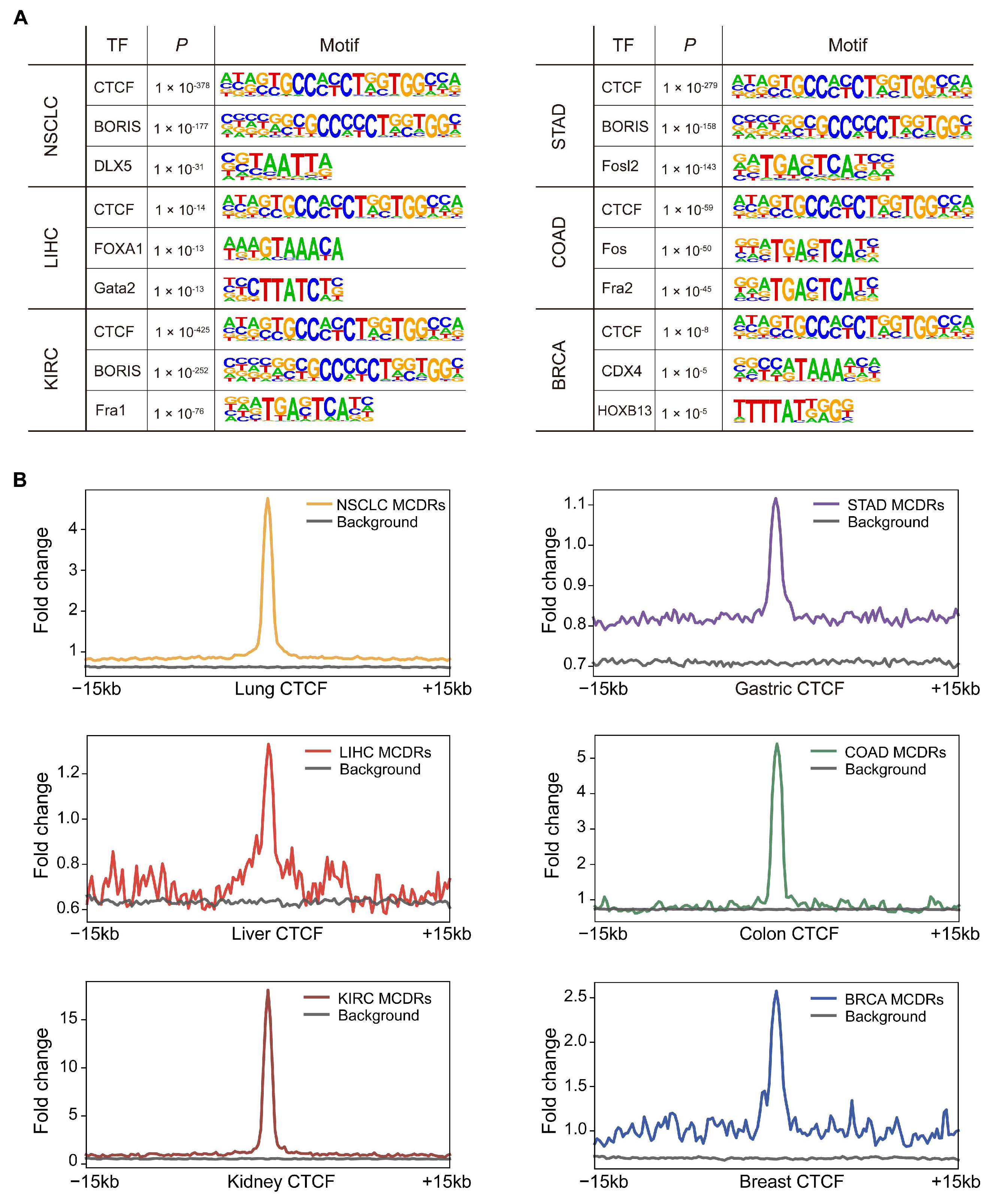
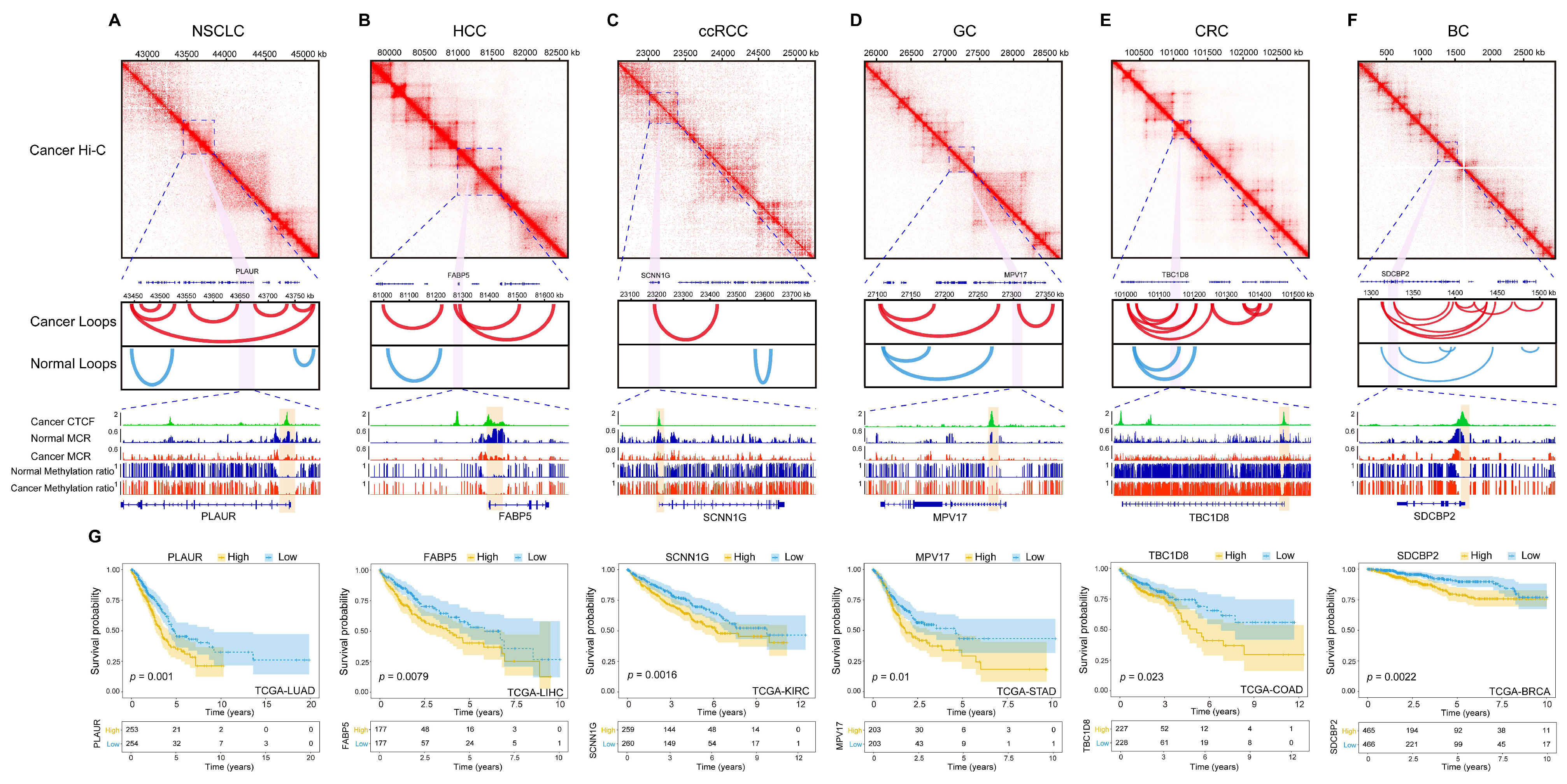
2.5. CTCF Binding at MCR-Decreased Regions Underlies Tumor-Specific Chromatin Reorganization and Gene Regulation
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. DNA Methylation Data Analysis
4.3. Identification of Regions Associated with DNA Methylation Patterns Independent of DNA Methylation Ratios
4.4. Single-Cell ATAC Data Analysis
4.5. Motif Analysis
4.6. Enrichment Analysis of Tissue-Specific Genes
4.7. Enrichment Analysis for Gained and Lost Peak Regions
4.8. ChIP-Seq Data Analysis
4.9. Hi-C Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Razin, A.; Riggs, A.D. DNA Methylation and Gene Function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the Cancer Genome and Epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.A.; Wang, Z.; Wu, X.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. DNA Methylation Biomarkers for Lung Cancer. Tumor Biol. 2012, 33, 287–296. [Google Scholar] [CrossRef]
- Zhu, J.D. The Altered DNA Methylation Pattern and Its Implications in Liver Cancer. Cell Res. 2005, 15, 272–280. [Google Scholar] [CrossRef]
- Shenoy, N.; Vallumsetla, N.; Zou, Y.; Galeas, J.N.; Shrivastava, M.; Hu, C.; Susztak, K.; Verma, A. Role of DNA Methylation in Renal Cell Carcinoma. J. Hematol. Oncol. 2015, 8, 88. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kim, H.S.; Castoro, R.J.; Chung, W.; Estecio, M.R.H.; Kondo, K.; Guo, Y.; Ahmed, S.S.; Toyota, M.; Itoh, F.; et al. Sensitive and Specific Detection of Early Gastric Cancer with DNA Methylation Analysis of Gastric Washes. Gastroenterology 2009, 136, 2149–2158. [Google Scholar] [CrossRef]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA Methylation Epigenotypes in Human Colorectal Cancer. Clin. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, H.; Huang, Y.; Xu, S.; Zhou, Y.; Zhang, W.; Li, J.; Ming, Y.; Wang, X.; Zhao, S.; et al. Genome-Wide Cell-Free DNA Methylation Analyses Improve Accuracy of Non-Invasive Diagnostic Imaging for Early-Stage Breast Cancer. Mol. Cancer 2021, 20, 36. [Google Scholar] [CrossRef]
- Hong, Y.; Liu, L.; Feng, Y.; Zhang, Z.; Hou, R.; Xu, Q.; Shi, J. mHapBrowser: A Comprehensive Database for Visualization and Analysis of DNA Methylation Haplotypes. Nucleic Acids Res. 2023, 52, D929–D937. [Google Scholar] [CrossRef]
- Shi, J.; Xu, J.; Chen, Y.E.; Li, J.S.; Cui, Y.; Shen, L.; Li, J.J.; Li, W. The Concurrence of DNA Methylation and Demethylation Is Associated with Transcription Regulation. Nat. Commun. 2021, 12, 5285. [Google Scholar] [CrossRef]
- Liang, N.; Li, B.; Jia, Z.; Wang, C.; Wu, P.; Zheng, T.; Wang, Y.; Qiu, F.; Wu, Y.; Su, J.; et al. Ultrasensitive Detection of Circulating Tumour DNA via Deep Methylation Sequencing Aided by Machine Learning. Nat. Biomed. Eng. 2021, 5, 586–599. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-of-Origin Mapping from Plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intra-Tumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Satpathy, A.T.; Zhou, W.; Ji, H.; Stratton, J.A.; Jaffer, A.; Bahlis, N.; Morrissy, S.; Biernaskie, J.A. Profiling Chromatin Accessibility at Single-Cell Resolution. Genom. Proteom. Bioinform. 2021, 19, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Berman, B.P.; Farnham, P.J. Demystifying the Secret Mission of Enhancers: Linking Distal Regulatory Elements to Target Genes. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 550–573. [Google Scholar] [CrossRef] [PubMed]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA Methylation Atlas of Normal Human Cell Types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-Cell Multiomics Sequencing and Analyses of Human Colorectal Cancer. Science 2018, 362, 1060–1063. [Google Scholar] [CrossRef]
- Andreu, M.J.; Alvarez-Franco, A.; Portela, M.; Gimenez-Llorente, D.; Cuadrado, A.; Badia-Careaga, C.; Tiana, M.; Losada, A.; Manzanares, M. Establishment of 3D Chromatin Structure after Fertilization and the Metabolic Switch at the Morula-to-Blastocyst Transition Require CTCF. Cell Rep. 2022, 41, 111501. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Guo, H.; Vuille, J.A.; Wittner, B.S.; Lachtara, E.M.; Hou, Y.; Lin, M.; Zhao, T.; Raman, A.T.; Russell, H.C.; Reeves, B.A.; et al. DNA Hypomethylation Silences Anti-Tumor Immune Genes in Early Prostate Cancer and CTCs. Cell 2023, 186, 2765–2782.e28. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the Lung Alveolus by an Evolutionarily Conserved Epithelial Progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing Lineage Hierarchies of the Distal Lung Epithelium Using Single-Cell RNA-Seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Español-Suñer, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular Carcinoma Originates from Hepatocytes and Not from the Progenitor/Biliary Compartment. J. Clin. Investig. 2015, 125, 3891–3903. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Primer 2017, 3, 17009. [Google Scholar] [CrossRef]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.-M.; Loudon, K.W.; et al. Single-Cell Transcriptomes from Human Kidneys Reveal the Cellular Identity of Renal Tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef]
- Tan, P.; Yeoh, K.-G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162.e3. [Google Scholar] [CrossRef]
- Joanito, I.; Wirapati, P.; Zhao, N.; Nawaz, Z.; Yeo, G.; Lee, F.; Eng, C.L.P.; Macalinao, D.C.; Kahraman, M.; Srinivasan, H.; et al. Single-Cell and Bulk Transcriptome Sequencing Identifies Two Epithelial Tumor Cell States and Refines the Consensus Molecular Classification of Colorectal Cancer. Nat. Genet. 2022, 54, 963–975. [Google Scholar] [CrossRef]
- Molyneux, G.; Geyer, F.C.; Magnay, F.-A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; MacKay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 Basal-like Breast Cancers Originate from Luminal Epithelial Progenitors and Not from Basal Stem Cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef]
- Sayaman, R.W.; Miyano, M.; Carlson, E.G.; Senapati, P.; Zirbes, A.; Shalabi, S.F.; Todhunter, M.E.; Seewaldt, V.E.; Neuhausen, S.L.; Stampfer, M.R.; et al. Luminal Epithelial Cells Integrate Variable Responses to Aging into Stereotypical Changes that Underlie Breast Cancer Susceptibility. eLife 2024, 13, e95720. [Google Scholar] [CrossRef]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-Based Chromatin Interactions Enable Regulated Gene Expression within Preexisting Architectural Compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef]
- Kitamura, Y.; Takahashi, K.; Maezawa, S.; Munakata, Y.; Sakashita, A.; Katz, S.P.; Kaplan, N.; Namekawa, S.H. CTCF-Mediated 3D Chromatin Sets up the Gene Expression Program in the Male Germline. Nat. Struct. Mol. Biol. 2025, 32, 1227–1240. [Google Scholar] [CrossRef]
- Deng, S.; Feng, Y.; Pauklin, S. 3D Chromatin Architecture and Transcription Regulation in Cancer. J. Hematol. Oncol. 2022, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sunkel, B.D.; Ray, W.C.; Stanton, B.Z. Chromatin Structure in Cancer. BMC Mol. Cell Biol. 2022, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo-Sánchez, A.; Noordermeer, D.; Greenberg, M.V.C. The Impact of DNA Methylation on CTCF-Mediated 3D Genome Organization. Nat. Struct. Mol. Biol. 2024, 31, 404–412. [Google Scholar] [CrossRef]
- Johnson, N.D.; Wiener, H.W.; Smith, A.K.; Nishitani, S.; Absher, D.M.; Arnett, D.K.; Aslibekyan, S.; Conneely, K.N. Non-Linear Patterns in Age-Related DNA Methylation May Reflect CD4+ T Cell Differentiation. Epigenetics 2017, 12, 492–503. [Google Scholar] [CrossRef]
- Achinger-Kawecka, J.; Stirzaker, C.; Portman, N.; Campbell, E.; Chia, K.-M.; Du, Q.; Laven-Law, G.; Nair, S.S.; Yong, A.; Wilkinson, A.; et al. The Potential of Epigenetic Therapy to Target the 3D Epigenome in Endocrine-Resistant Breast Cancer. Nat. Struct. Mol. Biol. 2024, 31, 498–512. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Loyfer, N.; Rosenski, J.; Kaplan, T. Wgbstools: A Computational Suite for DNA Methylation Sequencing Data Representation, Visualization, and Analysis. bioRxiv 2024, 5, 593132. [Google Scholar] [CrossRef]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR Is a Scalable Software Package for Integrative Single-Cell Chromatin Accessibility Analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, L.; Kumar, A.; Zatzman, M.; Salcedo, A.; Ravindra, N.; Shams, S.; Louie, B.H.; Bagdatli, S.T.; Myers, M.A.; Sarmashghi, S.; et al. Single-Cell Chromatin Accessibility Reveals Malignant Regulatory Programs in Primary Human Cancers. Science 2024, 385, eadk9217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Z.; Hong, Y.; Ding, Y.; Liu, L.; Gao, S.; Fang, H.; Shi, J. A DNA Methylation Haplotype Block Landscape in Human Tissues and Preimplantation Embryos Reveals Regulatory Elements Defined by Comethylation Patterns. Genome Res. 2023, 33, 2041–2052. [Google Scholar] [CrossRef]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-Specific Gene Enrichment Analysis. Bioinforma. Oxf. Engl. 2019, 35, 1966–1967. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Bock, C. LOLA: Enrichment Analysis for Genomic Region Sets and Regulatory Elements in R and Bioconductor. Bioinformatics 2016, 32, 587–589. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinforma. Oxf. Engl. 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Wolff, J.; Backofen, R.; Grüning, B. Loop Detection Using Hi-C Data with HiCExplorer. GigaScience 2022, 11, giac061. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Yang, Y.; Cui, D.; Li, J. DNA Methylation Concurrence, Independent of DNA Methylation Ratios, Is Associated with Chromatin Accessibility and 3D Genome Architecture. Int. J. Mol. Sci. 2025, 26, 7199. https://doi.org/10.3390/ijms26157199
Zhang G, Yang Y, Cui D, Li J. DNA Methylation Concurrence, Independent of DNA Methylation Ratios, Is Associated with Chromatin Accessibility and 3D Genome Architecture. International Journal of Molecular Sciences. 2025; 26(15):7199. https://doi.org/10.3390/ijms26157199
Chicago/Turabian StyleZhang, Guian, Yixian Yang, Dan Cui, and Jia Li. 2025. "DNA Methylation Concurrence, Independent of DNA Methylation Ratios, Is Associated with Chromatin Accessibility and 3D Genome Architecture" International Journal of Molecular Sciences 26, no. 15: 7199. https://doi.org/10.3390/ijms26157199
APA StyleZhang, G., Yang, Y., Cui, D., & Li, J. (2025). DNA Methylation Concurrence, Independent of DNA Methylation Ratios, Is Associated with Chromatin Accessibility and 3D Genome Architecture. International Journal of Molecular Sciences, 26(15), 7199. https://doi.org/10.3390/ijms26157199