

Unraveling TNXB Epigenetic Alterations Through Genome-Wide DNA Methylation Analysis and Their Implications for Colorectal Cancer
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The DNA Methylation Landscape in Colorectal Cancer
2.2. TNXB Gene Is Largely Hypomethylated and Overexpressed in Colorectal Cancer

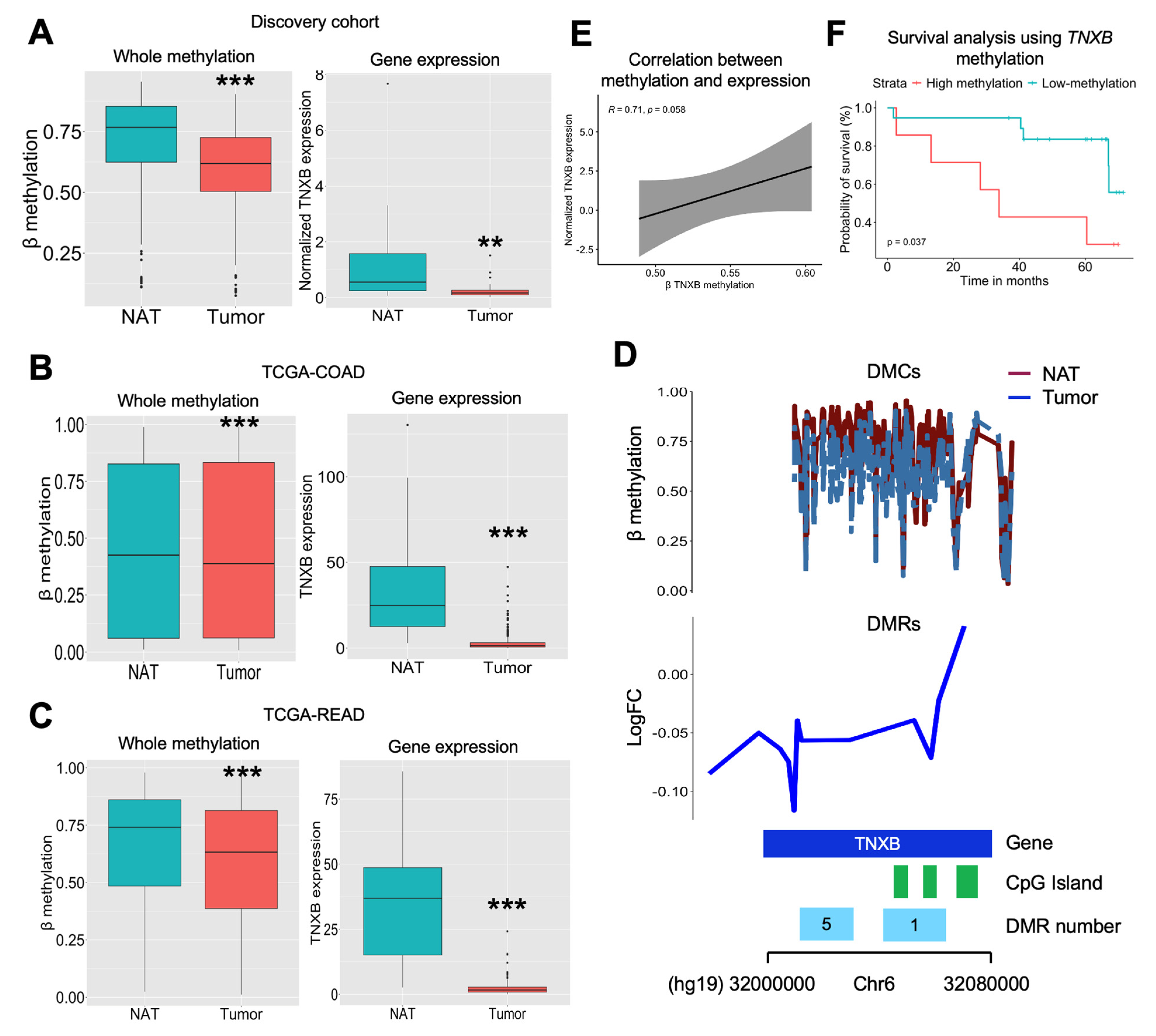
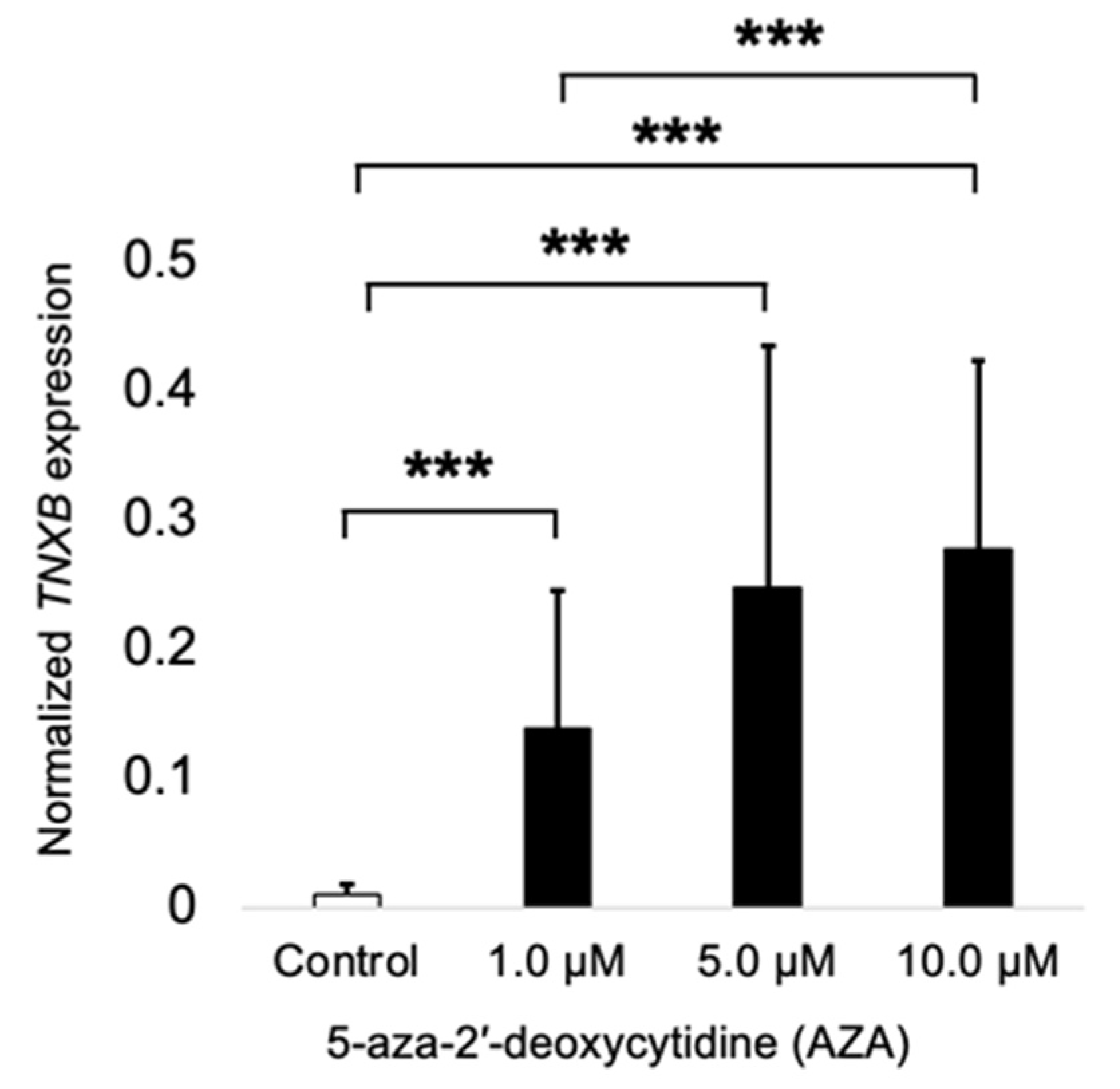
2.3. TNXB Gene Is Found to Be Epigenetically Regulated
3. Discussion
4. Materials and Methods
4.1. Study Design and Participants
4.2. Samples Included in This Study
4.3. DNA and RNA Extraction
4.4. Laboratory Measurements
4.5. Bisulfite Reaction and Genome-Wide DNA Methylation Analysis
4.6. Gene Expression Analysis
4.7. Cell Culture
4.8. DNA Demethylation In Vitro
4.9. Bioinformatic Analysis: DNA Methylation Analysis, TCGA Data and Single-Cell Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Tomasello, G.; Borgonovo, K.; Ghidini, M.; Turati, L.; Dallera, P.; Passalacqua, R.; Sgroi, G.; Barni, S. Prognostic survival associated with left-sided vs right-sided colon cancer a systematic review and meta-analysis. JAMA Oncol. 2017, 3, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Akhavan-Niaki, H.; Samadani, A.A. DNA methylation and cancer development: Molecular mechanism. Cell Biochem. Biophys. 2013, 67, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Boughanem, H.; Martin-Nuñez, G.M.; Torres, E.; Arranz-Salas, I.; Alcaide, J.; Morcillo, S.; Tinahones, F.J.; Crujeiras, A.B.; Macias-Gonzalez, M. Impact of Tumor LINE-1 Methylation Level and Neoadjuvant Treatment and Its Association with Colorectal Cancer Survival. J. Pers. Med. 2020, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Mulero, A.; Crujeiras, A.B.; Izquierdo, A.G.; Torres, E.; Ayers, D.; Casanueva, F.F.; Tinahones, F.J.; Morcillo, S.; Macias-Gonzalez, M. Novel SFRP2 DNA Methylation Profile Following Neoadjuvant Therapy in Colorectal Cancer Patients with Different Grades of BMI. J. Clin. Med. 2019, 8, 1041. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Huh, J.W.; Kim, H.R.; Kim, Y.J. CpG island methylator phenotype is an independent predictor of survival after curative resection for colorectal cancer: A prospective cohort study. J. Gastroenterol. Hepatol. 2017, 32, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, N.; Tierling, S.; Es, H.A.; Varkiani, M.; Mojarad, E.N.; Aghdaei, H.A.; Walter, J.; Totonchi, M. DNA methylation biomarkers in colorectal cancer: Clinical applications for precision medicine. Int. J. Cancer 2022, 151, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Dámaso, E.; González-Acosta, M.; Vargas-Parra, G.; Navarro, M.; Balmaña, J.; Cajal, T.R.Y.; Tuset, N.; Thompson, B.A.; Marín, F.; Fernández, A.; et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers 2020, 12, 1799. [Google Scholar] [CrossRef] [PubMed]
- Baharudin, R.; Ishak, M.; Yusof, A.M.; Saidin, S.; Syafruddin, S.E.; Nazarie, W.F.W.M.; Lee, L.-H.; Ab Mutalib, N.-S. Epigenome-Wide DNA Methylation Profiling in Colorectal Cancer and Normal Adjacent Colon Using Infinium Human Methylation 450K. Diagnostics 2022, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Huang, T.; Ye, G.; Wang, B.; Zhang, X. Methylation of SFRP2 gene as a promising noninvasive biomarker using feces in colorectal cancer diagnosis: A systematic meta-analysis. Sci. Rep. 2016, 6, 33339. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, J.; Bi, H.; Ge, A.; Xia, T.; Liu, Y.; Sun, H.; Li, D.; Zhao, Y. and WIF1 and prognosis of postoperative colorectal cancer patients. BMC Cancer 2019, 19, 1212. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Demond, H.; Brebi, P.; Ili, C.G. Novel Methylation Biomarkers for Colorectal Cancer Prognosis. Biomolecules 2021, 11, 1722. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; Neefs, I.; Ibrahim, J.; Schepers, A.; Pauwels, P.; Peeters, M.; Van Camp, G.; de Beeck, K.O. Epigenome-wide methylation analysis of colorectal carcinoma, adenoma and normal tissue reveals novel biomarkers addressing unmet clinical needs. Clin. Epigenet. 2023, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.W.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Naumov, V.A.; Generozov, E.V.; Zaharjevskaya, N.B.; Matushkina, D.S.; Larin, A.K.; Chernyshov, S.V.; Alekseev, M.V.; Shelygin, Y.A.; Govorun, V.M. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics 2013, 8, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Morcillo, S.; Diaz-Lagares, A.; Sandoval, J.; Castellano-Castillo, D.; Torres, E.; Hervas, D.; Moran, S.; Esteller, M.; Macias-Gonzalez, M.; et al. Identification of an episignature of human colorectal cancer associated with obesity by genome-wide DNA methylation analysis. Int. J. Obes. 2019, 43, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Bañobre, J.; Rodriguez-Casanova, A.; Costa-Fraga, N.; Bao-Caamano, A.; Alvarez-Castro, A.; Carreras-Presas, M.; Brozos-Vazquez, E.; Vidal-Insua, Y.; Vazquez-Rivera, F.; Candamio-Folgar, S.; et al. Noninvasive early detection of colorectal cancer by hypermethylation of the LINC00473 promoter in plasma cell-free DNA. Clin. Epigenet. 2022, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P.; Ward, R.L. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 2009, 46, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome (Review). Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Valcourt, U.; Alcaraz, L.B.; Exposito, J.-Y.; Lethias, C.; Bartholin, L. Tenascin-X: Beyond the architectural function. Cell Adhes. Migr. 2015, 9, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Aubert, A.; Hervieu, V.; El Kholti, N.; Schalkwijk, J.; Verrier, B.; Valcourt, U.; Lambert, E. Loss of Tenascin-X expression during tumor progression: A new pan-cancer marker. Matrix Biol. Plus 2020, 6–7, 100021. [Google Scholar] [CrossRef]
- Barrow, T.M.; Klett, H.; Toth, R.; Böhm, J.; Gigic, B.; Habermann, N.; Scherer, D.; Schrotz-King, P.; Skender, S.; Abbenhardt-Martin, C.; et al. Smoking is associated with hypermethylation of the APC 1A promoter in colorectal cancer: The ColoCare Study. J. Pathol. 2017, 243, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.G.; Boughanem, H.; Diaz-Lagares, A.; Arranz-Salas, I.; Esteller, M.; Tinahones, F.J.; Casanueva, F.F.; Macias-Gonzalez, M.; Crujeiras, A.B. DNA methylome in visceral adipose tissue can discriminate patients with and without colorectal cancer. Epigenetics 2021, 17, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-S.; Fu, L.-H.; Yan, H.-C.; Cheng, L.-Y.; Ru, H.-M.; Mo, S.; Wei, C.-Y.; Li, D.-M.; Mo, X.-W.; Tang, W.-Z.; et al. Proteomics and liquid biopsy characterization of human EMT-related metastasis in colorectal cancer. Front. Oncol. 2022, 12, 790096. [Google Scholar] [CrossRef] [PubMed]
- Urh, K.; Zidar, N.; Boštjančič, E. Bioinformatics Analysis of RNA-seq Data Reveals Genes Related to Cancer Stem Cells in Colorectal Cancerogenesis. Int. J. Mol. Sci. 2022, 23, 13252. [Google Scholar] [CrossRef] [PubMed]
- Carr, N.J.; Robin, L.H. WHO Classification of Tumors of the Digestive System, 4th ed.; National Institutes of Health (NIH): Bethesda, MD, USA, 2010. [Google Scholar]
- Bairaktari, E.T.; Seferiadis, K.I.; Elisaf, M.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. J. Cardiovasc. Pharmacol. Ther. 1972, 10, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Boughanem, H.; Cabrera-Mulero, A.; Millán-Gómez, M.; Garrido-Sánchez, L.; Cardona, F.; Tinahones, F.J.; Moreno-Santos, I.; Macías-González, M. C/EBP-α and PPAR-γ2 genes and their association with obesityrelated insulin resistance. Genes 2019, 10, 706. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Protocol Mycoplasma Detection Test. Available online: https://projects.iq.harvard.edu/files/hlalab/files/mycoplasm-test_hla.pdf (accessed on 23 November 2022).
- Kazemiha, V.M.; Shokrgozar, M.A.; Arabestani, M.R.; Moghadam, M.S.; Azari, S.; Maleki, S.; Amanzadeh, A.; Tehrani, M.J.; Shokri, F. PCR-based detection and eradication of mycoplasmal infections from various mammalian cell lines: A local experience. Cytotechnology 2009, 61, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [PubMed]
- Phipson, B.; Maksimovic, J.; Oshlack, A. MissMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 2016, 32, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas—Citing TCGA—NCI. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/using-tcga/citing-tcga (accessed on 28 November 2022).
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Cho, M.; Wang, X. OncoDB: An interactive online database for analysis of gene expression and viral infection in cancer. Nucleic Acids Res. 2022, 50, D1334–D1339. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; Van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.-W.; et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2011. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilo, J.; Rego-Calvo, A.; García-Flores, L.-A.; Arranz-Salas, I.; Alvarez-Mancha, A.I.; Izquierdo, A.G.; Crujeiras, A.B.; Alcaide, J.; Ortega-Castan, M.; Boughanem, H.; et al. Unraveling TNXB Epigenetic Alterations Through Genome-Wide DNA Methylation Analysis and Their Implications for Colorectal Cancer. Int. J. Mol. Sci. 2025, 26, 7197. https://doi.org/10.3390/ijms26157197
Pilo J, Rego-Calvo A, García-Flores L-A, Arranz-Salas I, Alvarez-Mancha AI, Izquierdo AG, Crujeiras AB, Alcaide J, Ortega-Castan M, Boughanem H, et al. Unraveling TNXB Epigenetic Alterations Through Genome-Wide DNA Methylation Analysis and Their Implications for Colorectal Cancer. International Journal of Molecular Sciences. 2025; 26(15):7197. https://doi.org/10.3390/ijms26157197
Chicago/Turabian StylePilo, Jesús, Alejandro Rego-Calvo, Libia-Alejandra García-Flores, Isabel Arranz-Salas, Ana Isabel Alvarez-Mancha, Andrea G. Izquierdo, Ana B. Crujeiras, Julia Alcaide, Maria Ortega-Castan, Hatim Boughanem, and et al. 2025. "Unraveling TNXB Epigenetic Alterations Through Genome-Wide DNA Methylation Analysis and Their Implications for Colorectal Cancer" International Journal of Molecular Sciences 26, no. 15: 7197. https://doi.org/10.3390/ijms26157197
APA StylePilo, J., Rego-Calvo, A., García-Flores, L.-A., Arranz-Salas, I., Alvarez-Mancha, A. I., Izquierdo, A. G., Crujeiras, A. B., Alcaide, J., Ortega-Castan, M., Boughanem, H., & Macías-González, M. (2025). Unraveling TNXB Epigenetic Alterations Through Genome-Wide DNA Methylation Analysis and Their Implications for Colorectal Cancer. International Journal of Molecular Sciences, 26(15), 7197. https://doi.org/10.3390/ijms26157197