
Targets for CAR Therapy in Multiple Myeloma
 , , , , ,
, , , , ,  ,
,
Abstract

1. Introduction
2. Structural Organization and Biological Fundamentals of CAR Cell Products in Immunotherapy of Blood Cancer Diseases
2.1. Background for CAR Cell Products in Therapy of MM
2.2. Structural Organization of CAR Cell Products and Their Interaction with Tumor Antigen
2.3. Sources for Producing CAR Cell Products
2.4. Safety of CAR Cell Products
2.5. Bioengineering Platforms and State-of-the-Art Strategies for Multi-Specific CAR Therapy in Cancer Immunotherapy
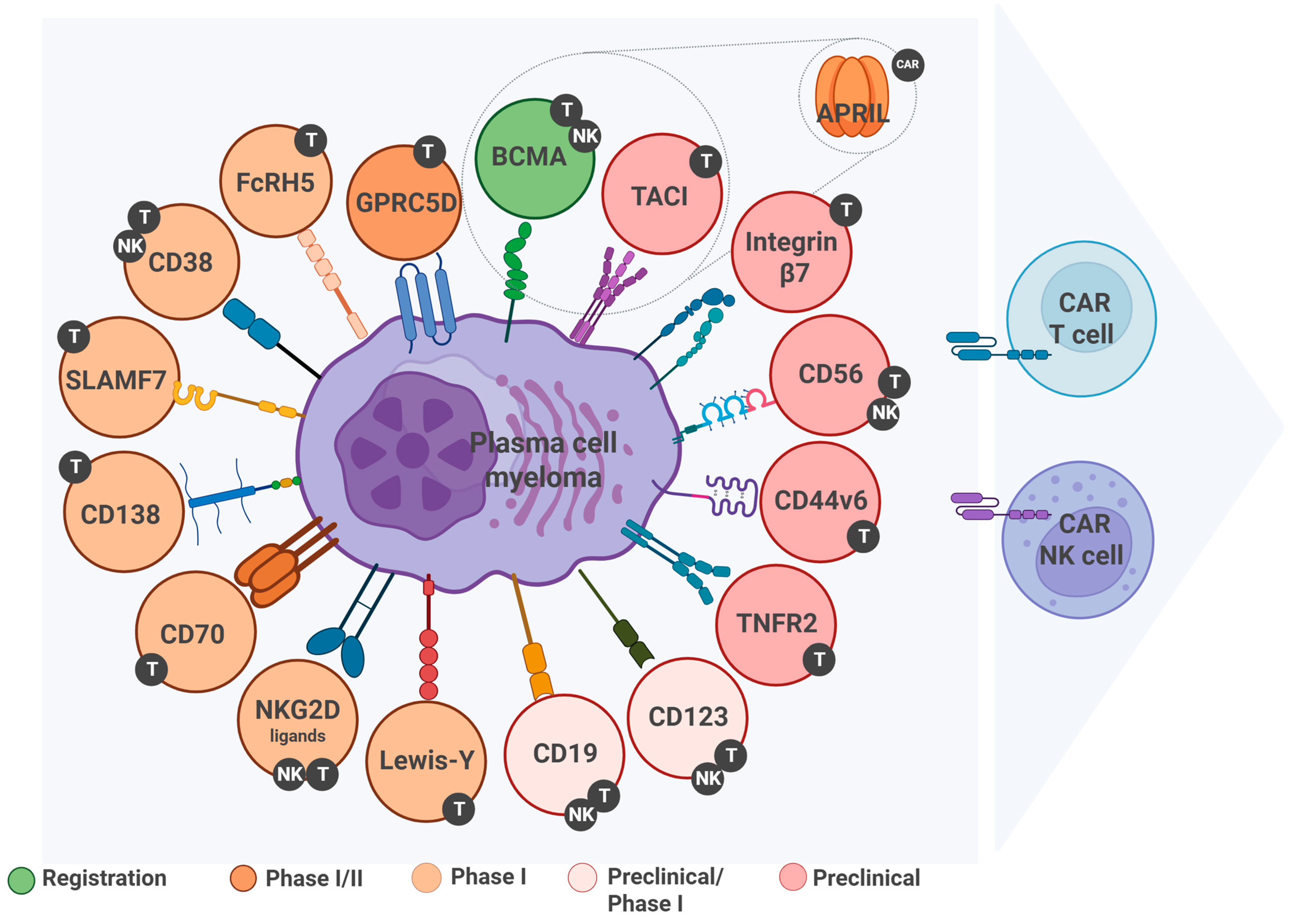
3. Tumor Targets in CAR Therapy for Multiple Myeloma
3.1. B-Cell Maturation Antigen (BCMA)
3.2. G Protein-Coupled Receptor Class C Group 5 Member D (GPRC5D)
3.3. Fc Receptor-Homolog 5 (FcRH5, CD307)
3.4. CD38 as a Therapeutic Target for Cellular Immunotherapy
3.5. Signaling Lymphocytic Activation Molecule Family 7 (SLAMF7)
3.6. CD138 (Syndecan-1) as a Therapeutic Target for Cellular Immunotherapy
3.7. CD70 as a Therapeutic Target for Cellular Immunotherapy
3.8. NKG2D Ligands (NKG2DLs) as a Target for Cellular Therapy of Multiple Myeloma
3.9. CD19 as a Therapeutic Target for Cellular Immunotherapy
3.10. Transmembrane Activator and CAML Interactor (TACI, TNFRSF13B)
3.11. A Proliferation-Inducing Ligand (APRIL)
3.12. Tumor Necrosis Factor Receptor 2 (TNFR2, TNFRSF1B)
3.13. CD44v6 as a Target for Cellular Therapy of Multiple Myeloma
3.14. Alternative Targets for CAR Cell Therapy in Multiple Myeloma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lee, H.; Neri, P.; Bahlis, N.J. BCMA- or GPRC5D-Targeting Bispecific Antibodies in Multiple Myeloma: Efficacy, Safety, and Resistance Mechanisms. Blood 2024, 143, 1211–1217. [Google Scholar] [CrossRef]
- Manier, S.; Ingegnere, T.; Escure, G.; Prodhomme, C.; Nudel, M.; Mitra, M.; Facon, T. Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev. 2022, 54, 100929. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, D.; Li, H.; Niu, T.; Tong, A. Multiple Myeloma: Signaling Pathways and Targeted Therapy. Mol. Biomed. 2024, 5, 18. [Google Scholar] [CrossRef]
- Scalzulli, E.; Grammatico, S.; Vozella, F. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Expert Opin. Investig. Drugs 2018, 27, 199–213. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Varga, C.; Maglio, M.; Ghobrial, I.M. Current Use of Monoclonal Antibodies in the Treatment of Multiple Myeloma. Br. J. Haematol. 2018, 181, 447–460. [Google Scholar] [CrossRef]
- Larocca, A.; Mina, R.; Gay, F.; Bringhen, S. Emerging Drugs and Combinations to Treat Multiple Myeloma. Oncotarget 2017, 8, 60656–60672. [Google Scholar] [CrossRef] [PubMed]
- Gagelmann, N.; Riecken, K.; Wolschke, C.; Berger, C. Development of CAR-T Cell Therapies for Multiple Myeloma. Leukemia 2020, 34, 2317–2332. [Google Scholar] [CrossRef]
- Rasche, L.; Hudecek, M.; Einsele, H. CAR T-Cell Therapy in Multiple Myeloma: Mission Accomplished? Blood 2024, 143, 305–316. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, S.; Maity, R.; Leblay, N.; Ziccheddu, B.; Truger, M.; Chojnacka, M.; Cirrincione, A.; Durante, M.; Tilmont, R.; et al. Mechanisms of Antigen Escape from BCMA- or GPRC5D-Targeted Immunotherapies in Multiple Myeloma. Nat. Med. 2023, 29, 2135–2145. [Google Scholar] [CrossRef]
- Rade, M.; Grieb, N.; Weiss, R.; Sia, J.; Fischer, L.; Born, P.; Boldt, A.; Fricke, S.; Franz, P.; Scolnick, J.; et al. Single-Cell Multiomic Dissection of Response and Resistance to Chimeric Antigen Receptor T Cells Against BCMA in Relapsed Multiple Myeloma. Nat. Cancer 2024, 5, 567–579. [Google Scholar] [CrossRef]
- Parikh, R.H.; Lonial, S. Chimeric Antigen Receptor T-Cell Therapy in Multiple Myeloma: A Comprehensive Review of Current Data and Implications for Clinical Practice. CA Cancer J. Clin. 2023, 73, 275–285. [Google Scholar] [CrossRef]
- Dhakal, B.; Hari, P.N.; Usmani, S.Z. Chimeric Antigen Receptor T Cell Therapy in Multiple Myeloma: Promise and Challenges. Bone Marrow Transpl. 2021, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, C.; Wang, L.; Jia, Y.; Qin, Y. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Front. Immunol. 2022, 13, 1050522. [Google Scholar] [CrossRef] [PubMed]
- Minguet, S.; Maus, M.V.; Schamel, W.W. From TCR Fundamental Research to Innovative Chimeric Antigen Receptor Design. Nat. Rev. Immunol. 2024, 24, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, A.; Salehi, A.; Khosravi, S.; Shariati, Y.; Nasrabadi, N.; Kahrizi, M.S.; Maghsoodi, S.; Mardi, A.; Azizi, R.; Jamali, S.; et al. Recent Findings on Chimeric Antigen Receptor (CAR)-Engineered Immune Cell Therapy in Solid Tumors and Hematological Malignancies. Stem Cell Res. Ther. 2022, 13, 482. [Google Scholar] [CrossRef]
- Fujiwara, K.; Kitaura, M.; Tsunei, A.; Kusabuka, H. Structure of the Signal Transduction Domain in Second-Generation CAR Regulates the Input Efficiency of CAR Signals. Int. J. Mol. Sci. 2021, 22, 2476. [Google Scholar] [CrossRef]
- Hanssens, H.; Meeus, F.; De Veirman, K. The Antigen-Binding Moiety in the Driver’s Seat of CARs. Med. Res. Rev. 2022, 42, 306–342. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Y.; Zhao, X.; Zheng, R.; Zhang, Y.; Meng, R.; Dong, H.; Liang, S.; He, X.; Song, Y.; et al. Chimeric Antigen Receptor with Novel Intracellular Modules Improves Antitumor Performance of T Cells. Signal Transduct. Target. Ther. 2025, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Sferruzza, G.; Yang, L.; Zhou, L. CAR-T and CAR-NK as Cellular Cancer Immunotherapy for Solid Tumors. Cell. Mol. Immunol. 2024, 21, 1089–1108. [Google Scholar] [CrossRef] [PubMed]
- Soldierer, M.; Bister, A.; Haist, C.; Thivakaran, A. Genetic Engineering and Enrichment of Human NK Cells for CAR-Enhanced Immunotherapy of Hematological Malignancies. Front. Immunol. 2022, 13, 847008. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef]
- Ruppel, K.E.; Fricke, S.; Köhl, U.; Schmiedel, D. Taking Lessons from CAR-T Cells and Going Beyond: Tailoring Design and Signaling for CAR-NK Cells in Cancer Therapy. Front. Immunol. 2022, 13, 822298. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, P.; Huang, H. Engineering Better Chimeric Antigen Receptor T Cells. Exp. Hematol. Oncol. 2020, 9, 31. [Google Scholar] [CrossRef]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric Antigen Receptor (CAR) Natural Killer (NK)-Cell Therapy: Leveraging the Power of Innate Immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Ramírez-Chacón, A.; Betriu-Méndez, S.; Bartoló-Ibars, A.; González, A.; Martí, M.; Juan, M. Ligand-based CAR-T cell: Different strategies to drive T cells in future new treatments. Front. Immunol. 2022, 13, 932559. [Google Scholar] [CrossRef]
- Balkhi, S.; Zuccolotto, G.; Di Spirito, A.; Rosato, A. CAR-NK Cell Therapy: Promise and Challenges in Solid Tumors. Front. Immunol. 2025, 16, 1574742. [Google Scholar] [CrossRef]
- Martarelli, N.; Capurro, M.; Mansour, G. Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors. Int. J. Mol. Sci. 2024, 25, 7231. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Kong, W.; He, Y. The Affinity of Antigen-Binding Domain on the Antitumor Efficacy of CAR T Cells: Moderate Is Better. Front. Immunol. 2022, 13, 1032403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Wang, Z.; Liu, Y.; Wang, T.; Zhang, M.; Xia, C.; Zhang, F.; Huang, D.; Zhang, L.; et al. Comparison of Seven CD19 CAR Designs in Engineering NK Cells for Enhancing Anti-Tumour Activity. Clin. Transl. Immunol. 2024, 13, e13683. [Google Scholar] [CrossRef]
- Wu, X.; Matosevic, S. Gene-Edited and CAR-NK Cells: Opportunities and Challenges with Engineering of NK Cells for Immunotherapy. Mol. Ther. Oncolytics 2024, 27, 224–238. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, Y.; Cui, J. Manufacturing CAR-NK Against Tumors: Who Is the Ideal Supplier? Chin. J. Cancer Res. 2024, 36, 145–157. [Google Scholar] [CrossRef]
- Mansouri, V.; Yazdanpanah, N.; Rezaei, N. The Immunologic Aspects of Cytokine Release Syndrome and Graft Versus Host Disease Following CAR T Cell Therapy. Int. Rev. Immunol. 2022, 41, 649–668. [Google Scholar] [CrossRef]
- Jørgensen, L.V.; Christensen, E.B.; Barnkob, M.B. The Clinical Landscape of CAR NK Cells. Exp. Hematol. Oncol. 2025, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, B.; Cao, W.; Zhang, W.; Li, T.; Liu, L.; Xu, L.; Gao, F.; Wang, Y.; Wang, F.; et al. Identification of potential resistance mechanisms and therapeutic targets for the relapse of BCMA CAR-T therapy in relapsed/refractory multiple myeloma through single-cell sequencing. Exp. Hematol. Oncol. 2023, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Kumar, S.K. Chimeric Antigen Receptor T-Cells, Bispecific Antibodies, and Antibody-Drug Conjugates for Multiple Myeloma: An Update. Am. J. Hematol. 2022, 97, 803–815. [Google Scholar] [CrossRef]
- Tan, M.S.Y.; Chen, Y.; Smith, E.L. Beyond BCMA: Newer Immune Targets in Myeloma. Blood Adv. 2024, 8, 4433–4445. [Google Scholar] [CrossRef]
- Schavgoulidze, A.; Cazaubiel, T.; Perrot, A. Multiple Myeloma: Heterogeneous in Every Way. Cancers 2021, 13, 1285. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewska, E.; Tota, M.; Kisielewska, M.; Skowron, I. Overcoming Antigen Escape and T-Cell Exhaustion in CAR-T Therapy for Leukemia. Cells 2024, 13, 1596. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-Antigen-Targeted Chimeric Antigen Receptor T Cells for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Timmers, M.; Roex, G.; Wang, Y. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef]
- Lam, N.; Finney, R.; Yang, S.; Choi, S.; Wu, X.; Cutmore, L.; Andrade, J.; Huang, L.; Amatya, C.; Cam, M.; et al. Development of a bicistronic anti-CD19/CD20 CAR construct including abrogation of unexpected nucleic acid sequence deletions. Mol. Ther. Oncolytics 2023, 30, 132–149. [Google Scholar] [CrossRef]
- Bachiller, M.; Barceló-Genestar, N.; Rodriguez-Garcia, A.; Alserawan, L.; Dobaño-López, C.; Giménez-Alejandre, M.; Castellsagué, J.; Colell, S.; Otero-Mateo, M.; Antoñana-Vildosola, A.; et al. ARI0003: Co-Transduced CD19/BCMA Dual-Targeting CAR-T Cells for the Treatment of Non-Hodgkin Lymphoma. Mol. Ther. 2023, 33, 317–335. [Google Scholar] [CrossRef]
- de Oliveira Canedo, G.; Roddie, C.; Amrolia, P.J. Dual-Targeting CAR T Cells for B-Cell Acute Lymphoblastic Leukemia and B-Cell Non-Hodgkin Lymphoma. Blood Adv. 2025, 9, 704–721. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, J.; Jiang, H.; Zhou, M. Strategies to Overcome Antigen Heterogeneity in CAR-T Cell Therapy. Cells 2025, 14, 320. [Google Scholar] [CrossRef]
- Tousley, A.M.; Rotiroti, M.C.; Labanieh, L.; Rysavy, L.W. Co-Opting Signalling Molecules Enables Logic-Gated Control of CAR T Cells. Nature 2023, 617, 763–769. [Google Scholar] [CrossRef]
- Bangayan, N.J.; Wang, L.; Burton Sojo, G.; Noguchi, M.; Cheng, D.; Ta, L.; Gunn, D.; Mao, Z.; Liu, S.; Yin, Q.; et al. Dual-Inhibitory Domain iCARs Improve the Efficiency of the AND-NOT Gate CAR T Strategy. Proc. Natl. Acad. Sci. USA 2023, 120, e2312374120. [Google Scholar] [CrossRef]
- Ebert, L.M.; Yu, W.; Gargett, T.; Brown, M.P. Logic-Gated Approaches to Extend the Utility of Chimeric Antigen Receptor T-Cell Technology. Biochem. Soc. Trans. 2018, 46, 391–401. [Google Scholar] [CrossRef]
- Pievani, A.; Biondi, M.; Tettamanti, S. CARs Are Sharpening Their Weapons. J. Hematol. Oncol. 2024, 12, e008275. [Google Scholar] [CrossRef] [PubMed]
- Alabanza, L.M.; Xiong, Y.; Vu, B.; Webster, B.; Wu, D.; Hu, P.; Zhu, Z.; Dropulic, B.; Dash, P.; Schneider, D. Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β. Front. Immunol. 2022, 13, 832645. [Google Scholar] [CrossRef]
- Tran, T.M.; Thakuri, B.K.C. Armored TGFβRIIDN ROR1-CAR T Cells Reject Solid Tumors and Resist Suppression by Constitutively-Expressed and Treatment-Induced TGFβ1. J. Immunother. Cancer 2024, 12, e008261. [Google Scholar] [CrossRef] [PubMed]
- Brog, R.A.; Ferry, S.L.; Schiebout, C.T. Superkine IL-2 and IL-33 Armored CAR T Cells Reshape the Tumor Microenvironment and Reduce Growth of Multiple Solid Tumors. Cancer Immunol. Res. 2022, 10, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Carcopino, C.; Erdogan, E.; Henrich, M.; Kobold, S. Armoring chimeric antigen receptor (CAR) T cells as micropharmacies for cancer therapy. Immunooncol Technol. 2024, 24, 100739. [Google Scholar] [CrossRef]
- Glienke, W.; Dragon, A.C.; Zimmermann, K.; Martyniszyn-Eiben, A.; Mertens, M.; Abken, H.; Rossig, C.; Altvater, B.; Aleksandrova, K.; Arseniev, L.; et al. GMP-Compliant Manufacturing of TRUCKs: CAR T Cells Targeting GD2 and Releasing Inducible IL-18. Front. Immunol. 2022, 13, 839783. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 19–34. [Google Scholar] [CrossRef]
- Thomas, S.; Abken, H. CAR T Cell Therapy Becomes CHIC: “Cytokine Help Intensified CAR” T Cells. Front. Immunol. 2023, 14, 1090959. [Google Scholar] [CrossRef]
- Silveira, C.R.F.; Corveloni, A.C.; Caruso, S.R. Cytokines as an Important Player in the Context of CAR-T Cell Therapy for Cancer: Their Role in Tumor Immunomodulation, Manufacture, and Clinical Implications. Front. Immunol. 2022, 13, 947648. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, 84. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, S.; Liu, M.; Chen, C.; Zhang, L.; Zhou, D. Fine-Tuning Through Generations: Advances in Structure and Production of CAR-T Therapy. Cancers 2023, 15, 3476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Nandakumar, K.S.; Cheng, K. Optimization of CAR-T Cell-Based Therapies Using Small-Molecule-Based Safety Switches. J. Med. Chem. 2021, 64, 9577–9591. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T Cells by Hypoxia-Inducible Transcription Amplification (HiTA) System Significantly Enhance Systemic Safety and Retain Antitumor Efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef] [PubMed]
- Wolter, T.; Wang, Y.; Hu, Q. Engineering Strategies to Mitigate Toxicities Associated with CAR-T Cell Therapy. BMEMat 2025, 3, e12109. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, F. Strategies to Overcome Tumor Relapse Caused by Antigen Escape After CAR T Therapy. Mol. Cancer 2025, 24, 126. [Google Scholar] [CrossRef]
- Zhai, Y.; Du, Y.; Li, G.; Yu, M.; Hu, H.; Pan, C.; Wang, D.; Shi, Z.; Yan, X.; Li, X.; et al. Trogocytosis of CAR Molecule Regulates CAR-T Cell Dysfunction and Tumor Antigen Escape. Nat. Commun. 2023, 14, 1708. [Google Scholar] [CrossRef]
- Dai, Z.; Mu, W.; Zhao, Y.; Cheng, J.; Lin, H.; Ouyang, K.; Jia, X.; Liu, J.; Wei, Q.; Wang, M.; et al. T Cells Expressing CD5/CD7 Bispecific Chimeric Antigen Receptors with Fully Human Heavy-Chain-Only Domains Mitigate Tumor Antigen Escape. Nat. Immunol. 2022, 23, 672–685. [Google Scholar] [CrossRef]
- Wei, J.; Mao, Z.; Wang, N.; Huang, L.; Cao, Y.; Sun, W.; Long, X.; Tan, J.; Li, C.; Xiao, Y.; et al. Long-Term Outcomes of Relapsed/Refractory Double-Hit Lymphoma (R/R DHL) Treated with CD19/22 CAR T-Cell Cocktail Therapy. Clin. Transl. Med. 2020, 10, e176. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, B.; Hu, B.; Zhang, W.; Zhu, Q.; Liu, Y.; Wang, S.; Zhang, P.; Yang, Y.; Yang, J.; et al. Sequential Different B-Cell Antigen–Targeted CAR T-Cell Therapy for Pediatric Refractory/Relapsed Burkitt Lymphoma. Blood Adv. 2022, 6, 717–724. [Google Scholar] [CrossRef]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H. CAR-T Cell Combination Therapy: The Next Revolution in Cancer Treatment. J. Cancer Sci. Ther. 2022, 14, 445–452. [Google Scholar] [CrossRef]
- Liu, W.; Liu, W.; Zou, H.; Chen, L.; Huang, W.; Lv, R.; Xu, Y.; Liu, H.; Shi, Y.; Wang, K.; et al. Combinational therapy of CAR T-cell and HDT/ASCT demonstrates impressive clinical efficacy and improved CAR T-cell behavior in relapsed/refractory large B-cell lymphoma. J. Immunother. Cancer 2024, 12, e008857. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target Selection for CAR-T Therapy. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N. A Review on Tumor Heterogeneity and Evolution in Multiple Myeloma: Pathological, Radiological, Molecular Genetics, and Clinical Integration. Virchows Arch. 2020, 476, 19–28. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Lan, H.; Wu, J.; Xiao, Y. CAR-T Cell Therapy in Multiple Myeloma: Current Limitations and Potential Strategies. Front. Immunol. 2023, 14, 1101495. [Google Scholar] [CrossRef]
- Shademan, B.; Karamad, V. CAR T Cells: Cancer Cell Surface Receptors are the Target for Cancer Therapy. Adv. Pharm. Bull. 2021, 12, 476–489. [Google Scholar] [CrossRef]
- Leow, C.C.Y.; Low, M.S.Y. Targeted Therapies for Multiple Myeloma. J. Pers. Med. 2021, 11, 334. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Mackall, C.L. CAR Immune Cells: Design Principles, Resistance, and the Next Generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 81. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/study/NCT03361748 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04162210 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04555551 (accessed on 15 May 2025).
- He, X.; Zhang, X.; Yu, Z.; Meng, J.; Jiang, Y.; Zhao, Y.; Lyu, H.; Bai, X.; Xiao, X.; Zhao, M. Anti-FcRL5 CAR-T exhibits anti-MM activity against EMM after progression of anti-BCMA and anti-GPRC5D CAR-T: A case report. Blood Adv. 2025, 9, 1159–1162. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/study/NCT04351022 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT03958656 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04430530 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT02830724 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04623944 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT02135406 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04318678 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT02159495 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT02623582 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT04599543 (accessed on 15 May 2025).
- Available online: https://clinicaltrials.gov/study/NCT01716364 (accessed on 15 May 2025).
- Li, C.; Cao, W.; Que, Y.; Wang, Q.; Xiao, Y.; Gu, C.; Wang, D.; Wang, J.; Jiang, L.; Xu, H.; et al. A Phase I Study of Anti-BCMA CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma and Plasma Cell Leukemia. Cancer Med. 2021, 10, 346. [Google Scholar] [CrossRef]
- Tannoury, M.; Garnier, D.; Susin, S.A.; Bauvois, B. Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next? Cancers 2022, 14, 6026. [Google Scholar] [CrossRef]
- Makarova, A.O.; Svirshchevskaya, E.V.; Titov, M.M. Prospects for the Use of Antibody-Drug Conjugates in Cancer Therapy. Russ. Chem. Rev. 2025, 94, 5597. [Google Scholar] [CrossRef]
- Sierro-Martínez, B.; Escamilla-Gómez, V. Next-generation BCMA-targeted Chimeric Antigen Receptor CARTemis-1: The Impact of Manufacturing Procedure on CAR T-cell Features. Cell. Oncol. 2025, 48, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Abd-Aziz, N.; Poh, C.L. Development of Peptide-Based Vaccines for Cancer. J. Oncol. 2022, 2022, 9749363. [Google Scholar] [CrossRef] [PubMed]
- Roex, G.; Gordon, K.S.; Lion, E. Expanding the CAR Toolbox with High Throughput Screening Strategies for CAR Domain Exploration: A Comprehensive Review. J. Immunother. Cancer 2025, 13, e010658. [Google Scholar] [CrossRef] [PubMed]
- Utley, A.; Lipchick, B.; Lee, K.P.; Nikiforov, M.A. Targeting Multiple Myeloma Through the Biology of Long-lived Plasma Cells. Cancers 2020, 12, 2117. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Wang, M.; Zhou, R. From Molecular Design to Clinical Translation: Dual-targeted CAR-T Strategies in Cancer Immunotherapy. Int. J. Biol. Sci. 2025, 21, 2676–2691. [Google Scholar] [CrossRef]
- Petrucci, M.T.; Bringhen, S.; Entrala Cerezo, C. Optimizing Treatment Sequencing in Multiple Myeloma: A Novel Model to Predict Survival Outcomes. Acta Haematol. 2024, 143, 2432815. [Google Scholar] [CrossRef]
- Krejci, M.; Adam, Z.; Krejci, M.; Pour, L.; Sandecka, V.; Stork, M. CAR-T cells for the treatment of relapsed/refractory multiple myeloma in 2022: Efficacy and toxicity. Neoplasma 2022, 69, 1008–1018. [Google Scholar] [CrossRef]
- Natrajan, K.; Kaushal, M.; George, B.; Kanapuru, B.; Theoret, M.R. FDA Approval Summary: Ciltacabtagene Autoleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2024, 30, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Strassl, I.; Podar, K. The Preclinical Discovery and Clinical Development of Ciltacabtagene Autoleucel (Cilta-cel) for the Treatment of Multiple Myeloma. Expert Opin. Drug Discov. 2024, 19, 15–25. [Google Scholar] [CrossRef]
- Abebe, E.C.; Shiferaw, M.Y. Ciltacabtagene Autoleucel: The Second Anti-BCMA CAR T-Cell Therapeutic Armamentarium of Relapsed or Refractory Multiple Myeloma. Front. Immunol. 2022, 13, 991092. [Google Scholar] [CrossRef]
- van den Berg, J.; Holbro, A. EBMT-EHA 6th European CAR T-Cell Meeting 2024: Advancing Treatments and New Frontiers. Onco-Hema Heal. Times 2024, 20, 34–39. [Google Scholar] [CrossRef]
- Raab, M.S.; Cohen, Y.C.; Schjesvold, F.; Aardalen, K.; Oka, A.; Spencer, A.; Wermke, M.; Souza, A.D.; Kaufman, J.L.; Cafro, A.M.; et al. Preclinical Discovery and Initial Clinical Data of WVT078, a BCMA× CD3 Bispecific Antibody. Leukemia 2023, 37, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Coffey, D.G.; Ataca Atilla, P.; Atilla, E.; Landgren, O. Single-Cell Analysis of the Multiple Myeloma Microenvironment after γ-Secretase Inhibition and CAR T-Cell Therapy. Blood 2025, 135, 1156–1168. [Google Scholar] [CrossRef]
- Yu, T.; Jiao, J.H.; Wu, M.F. CAR-T Cells in the Treatment of Multiple Myeloma: An Encouraging Cell Therapy. Front. Immunol. 2025, 16, 1499590. [Google Scholar] [CrossRef]
- Tang, C.; Zhang, Y. Potential alternatives to αβ-T cells to prevent graft-versus-host disease (GvHD) in allogeneic chimeric antigen receptor (CAR)-based cancer immunotherapy: A comprehensive review. Pathol Res Pract. 2024, 262, 155518. [Google Scholar] [CrossRef]
- Lonez, C.; Breman, E. Allogeneic CAR-T Therapy Technologies: Has the Promise Been Met? Cells 2024, 13, 146. [Google Scholar] [CrossRef]
- Ryszkiewicz, P.; Malinowska, B.; Schlicker, E. Polypharmacology: New Drugs in 2023–2024. Pharmacol. Rep. 2025, 77, 134–142. [Google Scholar] [CrossRef]
- Rodriguez-Otero, P.; van de Donk, N.W.C.J. GPRC5D as a Novel Target for the Treatment of Multiple Myeloma: A Narrative Review. Leukemia 2024, 38, 767–779. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, Y.; Chen, C.; Li, Z.; Xu, K.; Zhao, K. Targeting GPRC5D for Multiple Myeloma Therapy. Leukemia 2024, 38, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Ming, X.; Zheng, R.; Zhu, X.; Xiao, Y. Application of GPRC5D Targeting Therapy in Relapsed Refractory Multiple Myeloma. Cancer Med. 2025, 14, 70764. [Google Scholar] [CrossRef]
- Nakashima, T.; Kagoya, Y. Current Progress of CAR-T-Cell Therapy for Patients with Multiple Myeloma. Int. J. Hematol. 2024, 109, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Robat-Jazi, B.; Mahalleh, M.; Dashti, M.; Nejati, N.; Ahmadpour, M.; Alinejad, E.; Mohammadi, S.; Lorestani, P.; Hamidieh, A.A.; Habibi, M.A.; et al. A Systematic Review and Meta-analysis on the Safety and Efficacy of CAR T Cell Therapy Targeting GPRC5D in Patients with Multiple Myeloma: A New Insight in Cancer Immunotherapy. Anticancer Agents Med. Chem. 2025, 25, 1017–1028. [Google Scholar] [CrossRef]
- Zhang, M.; Wei, G.; Zhou, L.; Zhou, J.; Chen, S.; Zhang, W.; Wang, D.; Luo, X.; Cui, J.; Huang, S.; et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): A first-in-human, single-center, single-arm, phase 1 trial. Lancet Haematol. 2023, 10, e107–e116. [Google Scholar] [CrossRef]
- Xia, J.; Sun, Q.; Zhou, D.; Li, H.; Wang, Y.; Qi, Y.; Cao, J.; Yan, Z.; Li, D.; Cheng, H.; et al. Anti-GPRC5D CAR T-cell therapy as a salvage treatment in patients with progressive multiple myeloma after anti-BCMA CAR T-cell therapy: A single-centre, single-arm, phase 2 trial. Lancet Haematol. 2025, 12, e365–e375. [Google Scholar] [CrossRef]
- Vera-Cruz, S.; Jornet Culubret, M.; Konetzki, V.; Alb, M. Cellular Therapies for Multiple Myeloma: Engineering Hope. Cancers 2024, 16, 3867. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Luo, Y.; Pal, A.S.; Ding, H.; Lai, Y.; Guan, S.; Lan, W.; Li, W. A GPRC5D and BCMA bispecific CAR-T product demonstrated dual antigen targeting capability and efficacy against multiple myeloma cells bearing BCMA. J Immunother Cancer. 2024, 12 (Suppl. S2), A284. [Google Scholar] [CrossRef]
- Zhou, J.; Luo, X.; Wu, K.; Shi, F.; Lei, B.; Xu, Y.; Zhou, J.; He, X. Abstract 3183: GPRC5D and BCMA Bi-Specific CAR-T: Optimized CAR Design to Mitigate Antigen Escape and Elicit Deep and Durable Response in Heterogeneous Multiple Myeloma. Cancer Res. 2025, 85 (Suppl. S1), 3183. [Google Scholar] [CrossRef]
- Carlson, S.; Lin, T.L.; Larson, S.M. Allogeneic Chimeric Antigen Receptors (CARs) as an “Off-the-Shelf” Therapy in Multiple Myeloma. Immunotherapy 2025, 17, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, T.; Wang, Q.; Gong, Y.; Gao, F.; Zhou, F.; Cao, Z. Abstract 6122: Off-the-Shelf BCMA/GPRC5D Dual Targeted CAR-NK Cell Therapy Combined with Daratumumab in Treating Multiple Myeloma. Cancer Res. 2025, 85 (Suppl. S1), 6122. [Google Scholar] [CrossRef]
- Yu, Z.; Li, H.; Lu, Q.; Zhang, Z.; Tong, A.; Niu, T. Fc Receptor-Like 5 (FCRL5)-Directed CAR-T Cells Exhibit Antitumor Activity Against Multiple Myeloma. Signal Transduct. Target. Ther. 2024, 9, 16. [Google Scholar] [CrossRef]
- Jiang, D.; Huang, H.; Qin, H.; Tang, K.; Shi, X.; Zhu, T.; Gao, Y.; Zhang, Y.; Tian, X.; Fu, J.; et al. Chimeric Antigen Receptor T Cells Targeting FcRH5 Provide Robust Tumour-Specific Responses in Murine Xenograft Models of Multiple Myeloma. Nat. Commun. 2023, 14, 3642. [Google Scholar] [CrossRef]
- Miller, K.C.; Hashmi, H.; Rajeeve, S. Beyond BCMA: The Next Wave of CAR T Cell Therapy in Multiple Myeloma. Front. Oncol. 2024, 14, 1398902. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ren, Q.; Liu, X.; Guo, X.; Song, Y. Bispecific Antibodies Targeting BCMA, GPRC5D, and FcRH5 for Multiple Myeloma Therapy: Latest Updates from ASCO 2023 Annual Meeting. J. Hematol. Oncol. 2023, 16, 92. [Google Scholar] [CrossRef]
- Narra, R.K.; Peshin, S.; Dhakal, B. Novel Approaches of Cellular Therapy in Multiple Myeloma: Focus on Chimeric Antigen Receptor T-Cells. Acta Haematol. 2024, 148, 330–345. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 746–754. [Google Scholar] [CrossRef]
- Waldschmidt, J.M.; Rasche, L.; Kortüm, K.M. Bispecific Antibody Constructs in Multiple Myeloma: Affinities, Dosing Strategies, and Future Perspectives. Clin. Lymphoma Myeloma Leuk. 2024, 24, 267–277. [Google Scholar] [CrossRef]
- O’Neill, N.; van de Donk, N.W.C.J. T-Cell Redirecting Bispecific Antibodies in Multiple Myeloma: Current Landscape and Future Directions. EJHaem 2023, 4, 811–822. [Google Scholar] [CrossRef]
- He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. [Google Scholar] [CrossRef] [PubMed]
- Honikel, M.M.; Olejniczak, S.H. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules 2022, 12, 1303. [Google Scholar] [CrossRef]
- Fu, B.; Liu, R.; Gao, G.; Lin, Z.; He, A. Mechanisms and Salvage Treatments in Patients with Multiple Myeloma Relapsed Post-BCMA CAR-T Cell Therapy. Front. Immunol. 2024, 15, 1433774. [Google Scholar] [CrossRef]
- Mishra, A.K.; Gupta, A.; Dagar, G.; Das, D.; Chakraborty, A.; Haque, S.; Prasad, C.P.; Singh, A.; Bhat, A.A.; Macha, M.A.; et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines 2023, 11, 1721. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Deng, Q.W.; Zhao, Y.J. The Calcium Signaling Enzyme CD38—A Paradigm for Membrane Topology Defining Distinct Protein Functions. Cell Calcium 2022, 99, 102424. [Google Scholar] [CrossRef]
- Adolph, T.E.; Meyer, M.; Jukic, A.; Tilg, H. Heavy Arch: From Inflammatory Bowel Diseases to Metabolic Disorders. Gut 2024, 73, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Choi, Y.M.; Rah, S.Y.; Park, D.R.; Park, S.A.; Chung, Y.J.; Park, S.M.; Park, J.K.; Jang, K.Y.; Kim, U.H. Seminal CD38 Is a Pivotal Regulator for Fetomaternal Tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 1559–1564. [Google Scholar] [CrossRef]
- Encinas, C.; Lozano, V.; Hlavacek, P.; Llinares, J.; Toribio-Castelló, S.; Carcedo, D.; Asís-Montalt, J.; Martínez-López, J. Cost-Effectiveness of Elranatamab Versus Current Therapies for the Management of Patients with Triple-Class Exposed, Relapsed and Refractory Multiple Myeloma, Including Other Bispecific and Physician’s Choice of Treatment in Spain. Oncol. Ther. 2025, 13, 381–395. [Google Scholar] [CrossRef]
- Hashmi, H.; Husnain, M.; Khan, A. CD38-Directed Therapies for Management of Multiple Myeloma. Immunotherapy 2021, 13, 273–285. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Sierro-Martínez, B. Overcoming Chimeric Antigen Receptor (CAR) Modified T-Cell Therapy Limitations in Multiple Myeloma. Front. Immunol. 2020, 11, 1128. [Google Scholar] [CrossRef]
- Deng, H.; Wang, Q.; Tong, X.; Cui, Z. Recent Advances of CAR-T Cells in Acute Myeloid Leukemia. Ther. Adv. Hematol. 2025, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, J.; Guo, J.; Li, X.; Wang, S.; Xie, Y.; Jiang, H.; Wang, Y.; Wang, M.; Hu, M.; et al. All-Trans Retinoic Acid Improves NSD2-Mediated RARα Phase Separation and Efficacy of Anti-CD38 CAR T-Cell Therapy in Multiple Myeloma. J. Immunother. Cancer 2023, 11, e006325. [Google Scholar] [CrossRef] [PubMed]
- Sheykhhasan, M.; Ahmadieh-Yazdi, A. CAR T Therapies in Multiple Myeloma: Unleashing the Future. Cancer Gene Ther. 2024, 5, 210–222. [Google Scholar] [CrossRef]
- Keller, A.L.; Reiman, L.T.; Perez de Acha, O.; Parzych, S.E.; Forsberg, P.A.; Kim, P.S.; Bisht, K.; Wang, H.; Van de Velde, H.; Sherbenou, D.W.; et al. Ex Vivo Efficacy of SAR442257 Anti-CD38 Trispecific T-cell Engager in Multiple Myeloma Relapsed After Daratumumab and BCMA-targeted Therapies. Cancer Res Commun. 2024, 4, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A Bispecific CAR-T Cell Therapy Targeting BCMA and CD38 in Relapsed or Refractory Multiple Myeloma. Leukemia 2021, 35, 667–678. [Google Scholar] [CrossRef]
- De Novellis, D.; Fontana, R.; Giudice, V.; Serio, B. Innovative Anti-CD38 and Anti-BCMA Targeted Therapies in Multiple Myeloma: Mechanisms of Action and Resistance. Int. J. Mol. Sci. 2022, 24, 645. [Google Scholar] [CrossRef]
- Bruno, B.; Wäsch, R.; Engelhardt, M.; Gay, F. European Myeloma Network Perspective on CAR T-Cell Therapies for Multiple Myeloma. J. Clin. Oncol. 2021, 39, 2681–2692. [Google Scholar] [CrossRef]
- Zamagni, E.; Tacchetti, P.; Pantani, L.; Cavo, M. Anti-CD38 and Anti-SLAMF7: The Future of Myeloma Immunotherapy. Crit. Rev. Oncol. Hematol. 2018, 132, 131–142. [Google Scholar] [CrossRef]
- Veillette, A.; Guo, H. CS1, a SLAM Family Receptor Involved in Immune Regulation, Is a Therapeutic Target in Multiple Myeloma. Crit. Rev. Oncol. Hematol. 2013, 88, 168–177. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Chen, Z.; Xia, L. Emerging Roles of SLAMF7 in Immune Cells and Related Diseases. Innate Immun. 2025, 31, 267–279. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T Cells Eliminate Myeloma and Confer Selective Fratricide of SLAMF7+ Normal Lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Kostelecka, K.; Brylinski, L.; Wolinski, F.; Smyk, M. The Potential Role of CAR-T in Multiple Myeloma Treatment: A Review. Cent. Eur. J. Oncol. 2024, 12, 133–144. [Google Scholar] [CrossRef]
- Wang, H.; Pan, W. Challenges of Chimeric Antigen Receptor-T/Natural Killer Cell Therapy in the Treatment of Solid Tumors: Focus on Colorectal Cancer and Evaluation of Combination Therapies. Mol. Cell. Biochem. 2023, 478, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A First-in-Human Clinical Trial with SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther. 2021, 28, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Wu, J.; Kang, S.S.; Kang, Y. SLAMF7 as a Promising Immunotherapeutic Target in Multiple Myeloma Treatments. Curr. Oncol. 2023, 30, 573. [Google Scholar] [CrossRef]
- Hassan, N.; Efing, J.; Kiesel, L.; Bendas, G.; Götte, M. The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans. Cancers 2023, 15, 1524. [Google Scholar] [CrossRef]
- Steiner, N.; Gunsilius, E. CAR-T Cells in Multiple Myeloma: Current Status. Memo—Mag. Eur. Med. Oncol. 2020, 13, 158–165. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J.; Usmani, S.Z.; Yong, K. CAR T-Cell Therapy for Multiple Myeloma: State of the Art and Prospects. Lancet Haematol. 2021, 8, e446–e461. [Google Scholar] [CrossRef]
- Ding, L.; Hu, Y.; Huang, H. Novel Progresses of Chimeric Antigen Receptor (CAR) T Cell Therapy in Multiple Myeloma. Stem Cell Investig. 2021, 8, 33–45. [Google Scholar] [CrossRef]
- Riccardi, F.; Tangredi, C.; Dal Bo, M.; Toffoli, G. Targeted Therapy for Multiple Myeloma: An Overview on CD138-Based Strategies. Front. Oncol. 2024, 14, 1370854. [Google Scholar] [CrossRef]
- Kasap, C.; Izgutdina, A.; Patiño-Escobar, B.; Kang, A. Targeting High-Risk Multiple Myeloma Genotypes with Optimized Anti-CD70 CAR-T Cells. Nat. Commun. 2024, 15, 1234. [Google Scholar] [CrossRef]
- Kasap, C.; Izgutdina, A.; Patiño Escobar, B.; Lin, Y.-T.; Geng, H.; Wicaksono, G.; Dupere-Richer, D.; Ramos, E.; Barpanda, A.; Vijayanarayanan, A.; et al. Discovery and Development of CD70 as a Cellular Therapy Target in High-Risk Multiple Myeloma. Blood 2023, 142 (Suppl. S1), 465. [Google Scholar] [CrossRef]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B. CD70-Specific CAR T Cells Have Potent Activity Against Acute Myeloid Leukemia Without HSC Toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef]
- Wu, G.; Guo, S.; Luo, Q.; Wang, X.; Deng, W.; Ouyang, G.; Pu, J.J.; Lei, W.; Qian, W. Preclinical Evaluation of CD70-Specific CAR T Cells Targeting Acute Myeloid Leukemia. Front. Immunol. 2023, 14, 1093750. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Reimers, M.A.; Garmezy, B.; Xu, W.; Hoimes, C.J.; Weinstein, M.; Cullingford, E.L.; Ma, A.; Williams, L.M.; Dar, H.; et al. A Phase 1/2, Open-Label, Multicenter, Dose Escalation and Cohort Expansion Study of the Safety and Efficacy of Anti-CD70 Allogeneic CRISPR-Cas9–Engineered T Cells (CTX131) in Adult Patients with Relapsed or Refractory Solid Tumors. J. Clin. Oncol. 2024, 42 (Suppl. S15), TPS2676. [Google Scholar] [CrossRef]
- Iyer, S.P.; Sica, R.A.; Ho, P.J.; Prica, A.; Zain, J.; Foss, F.M.; Hu, B.; Beitinjaneh, A.; Weng, W.K.; Kim, Y.H.; et al. Safety and Activity of CTX130, a CD70-Targeted Allogeneic CRISPR-Cas9-Engineered CAR T-Cell Therapy, in Patients with Relapsed or Refractory T-Cell Malignancies (COBALT-LYM): A Single-Arm, Open-Label, Phase 1, Dose-Escalation Study. Lancet Oncol. 2024, 25, 110–122. [Google Scholar] [CrossRef]
- Lin, P.; Silva, F.C.R.; Gilbert, A.L.; Acharya, S. CD70 CAR NK Cells in the Treatment of Multiple Myeloma. Blood 2023, 142, 3463. [Google Scholar] [CrossRef]
- Guo, S.; Lei, W.; Jin, X.; Liu, H.; Wang, J.Q.; Deng, W.; Qian, W. CD70-Specific CAR NK Cells Expressing IL-15 for the Treatment of CD19-Negative B-Cell Malignancy. Blood Adv. 2024, 8, 2635–2645. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Emerging Role of Chimeric Antigen Receptor-Natural Killer Cells for the Treatment of Hematologic Malignancies. Cancers 2025, 17, 1454. [Google Scholar] [CrossRef]
- Olejarz, W.; Basak, G. Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers 2023, 15, 5765. [Google Scholar] [CrossRef]
- Boneva, E.; Shivarov, V.; Ivanova, M. A Concise Review of the Role of the NKG2D Receptor and Its Ligands in Cancer. Immuno 2025, 5, 9. [Google Scholar] [CrossRef]
- Tan, G.; Spillane, K.M.; Maher, J. The Role and Regulation of the NKG2D/NKG2D Ligand System in Cancer. Biology 2023, 12, 1079. [Google Scholar] [CrossRef]
- Prajapati, K.; Perez, C.; Rojas, L.B.P.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8+ T Cells: An Opportunity for Immunotherapy. Cell. Mol. Immunol. 2018, 15, 470–479. [Google Scholar] [CrossRef]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The Cellular Immune System in Myelomagenesis: NK Cells and T Cells in the Development of Myeloma and Their Uses in Immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dai, K.; Saliu, M.A.; Salisu, M.D.; Gan, J.; Afolabi, L.O.; Yan, D.; Zhang, G.; Liu, M.; Wan, X.; et al. Sodium valproate enhances efficacy of NKG2D CAR-T cells against glioblastoma. Front. Immunol. 2025, 15, 1519777. [Google Scholar] [CrossRef]
- Trabolsi, A.; Arumov, A.; Schatz, J.H. Bispecific Antibodies and CAR-T Cells: Dueling Immunotherapies for Large B-Cell Lymphomas. Blood Cancer J. 2024, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Dada, R. Redefining Precision Management of R/R Large B-Cell Lymphoma: Novel Antibodies Take on CART and BMT in the Quest for Future Treatment Strategies. Cells 2023, 12, 1858. [Google Scholar] [CrossRef]
- Moreno-Cortes, E.; Forero-Forero, J.V. Chimeric Antigen Receptor T Cell Therapy in Oncology–Pipeline at a Glance: Analysis of the ClinicalTrials.gov Database. Leuk. Res. 2021, 104, 106567. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chng, W.J. CAR T-Cell Therapy in Multiple Myeloma: More Room for Improvement. Blood Cancer J. 2021, 11, 84. [Google Scholar] [CrossRef]
- Rejeski, K.; Jain, M.D.; Smith, E.L. Mechanisms of Resistance and Treatment of Relapse After CAR T-Cell Therapy for Large B-Cell Lymphoma and Multiple Myeloma. Transplant. Cell Ther. 2023, 29, 418–428. [Google Scholar] [CrossRef]
- Rubinstein, P.G.; Galvez, C.; Ambinder, R.F. Hematopoietic Stem Cell Transplantation and Cellular Therapy in Persons Living with HIV. Curr. Opin. Infect. Dis. 2024, 37, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Almotiri, A. CAR T-Cell Therapy in Acute Myeloid Leukemia. Saudi Med. J. 2024, 45, 1007–1019. [Google Scholar] [CrossRef]
- Salehi-Shadkami, H.; Sedghi, M.; Tavoosi, S. Effective Targeting of CD19 Positive Primary B-ALL Cells Using CAR-NK Cells Generated with mRNA-LNPs. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kang, L.; Zhang, J.; Li, M.; Xu, N.; Qi, W.; Tan, J.; Lou, X.; Yu, Z.; Sun, J.; Wang, Z.; et al. Characterization of Novel Dual Tandem CD19/BCMA Chimeric Antigen Receptor T Cells to Potentially Treat Multiple Myeloma. Biomark. Res. 2020, 8, 14. [Google Scholar] [CrossRef]
- Jiang, H.; Dong, B.; Gao, L.; Liu, L.; Ge, J.; He, A.; Du, J.; Li, L.; Lu, J.; Chen, X.; et al. Clinical Results of a Multicenter Study of the First-in-Human Dual BCMA and CD19 Targeted Novel Platform Fast CAR-T Cell Therapy for Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. S1), 25–26. [Google Scholar] [CrossRef]
- Du, J.; Fu, W.; Jiang, H.; Dong, B.; Gao, L.; Liu, L.; Ge, J.; He, A.; Li, L.; Lu, J.; et al. P869: Updated Results of a Phase I, Open-Label Study of BCMA/CD19 Dual-Targeting FasTCAR-T GC012F for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). HemaSphere 2023, 7 (Suppl. S3), e84060bf. [Google Scholar] [CrossRef]
- Steinhardt, M.; Einsele, H. Advances in CD19-Targeting CAR-T Cell Therapies for Multiple Myeloma. Expert Opin. Biol. Ther. 2025, 25, 229–240. [Google Scholar] [CrossRef]
- Xu, H.; Guan, C.; Xu, P.; Zhou, D.; Xu, Y.; Chen, B.; Bai, H. Clinical Efficacy and Safety of Combined Anti-BCMA and Anti-CD19 CAR-T Cell Therapy for Relapsed/Refractory Multiple Myeloma: A Systematic Review and Meta-Analysis. Front. Oncol. 2024, 14, 1355643. [Google Scholar] [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-BCMA CAR T Cell Therapy Followed by Lenalidomide Maintenance after Autologous Stem-Cell Transplantation for High-Risk Newly Diagnosed Multiple Myeloma. Am. J. Hematol. 2022, 97, 238–246. [Google Scholar] [CrossRef]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-Cell Therapy in Multiple Myeloma: Can We Do Better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Shi, M.; Wang, J.; Huang, H.; Liu, D.; Cheng, H. Bispecific CAR T Cell Therapy Targeting BCMA and CD19 in Relapsed/Refractory Multiple Myeloma: A Phase I/II Trial. Nat. Commun. 2024, 15, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Lo Tartaro, D.; De Biasi, S.; Nasi, M.; Borella, R.; Fidanza, L.; Neroni, A.; Troiano, L.; Franceschi, C.; et al. A Comprehensive Analysis of Cytokine Network in Centenarians. Int. J. Mol. Sci. 2023, 24, 2719. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xie, Y.; Wang, X.; Yang, L.; Geng, H.; Yi, Z.; Zhang, Y.; Ma, L.; Chen, F. Targeting BCMA in Multiple Myeloma: Designs, Challenges, and Future Directions. Cancer Immunol. Immunother. 2025, 74, 77. [Google Scholar] [CrossRef]
- Larson, R.C.; Castano, A.; Bouffard, A.A.; Kann, M.C.; Schmidts, A.; Gallagher, K.M.; Maus, M.V. Abstract 556: Novel anti-TACI single and dual-targeting CAR T cells overcome BCMA antigen loss in multiple myeloma. Cancer Res. 2022, 82 (Suppl. S12), 556. [Google Scholar] [CrossRef]
- Ohmine, K.; Uchibori, R. Novel Immunotherapies in Multiple Myeloma. Int. J. Hematol. 2022, 115, 799–810. [Google Scholar] [CrossRef]
- Larson, R.C.; Kann, M.C.; Graham, C.; Mount, C.W.; Castano, A.P.; Lee, W.-H.; Bouffard, A.A.; Takei, H.N.; Almazan, A.J.; Scarfó, I.; et al. Anti-TACI Single and Dual-Targeting CAR T Cells Overcome BCMA Antigen Loss in Multiple Myeloma. Nat. Commun. 2023, 14, 7509. [Google Scholar] [CrossRef]
- Cornelis, R.; Chang, H.D.; Radbruch, A. Keeping Up with the Stress of Antibody Production: BAFF and APRIL Maintain Memory Plasma Cells. Curr. Opin. Immunol. 2021, 71, 97–102. [Google Scholar] [CrossRef]
- Eslami, M.; Schuepbach-Mallepell, S.; Diana, D.; Willen, L.; Kowalczyk-Quintas, C.; Desponds, C.; Peter, B.; Vigolo, M.; Renevey, F.; Donzé, O.; et al. Unique and Redundant Roles of Mouse BCMA, TACI, BAFF, APRIL, and IL-6 in Supporting Antibody-Producing Cells in Different Tissues. Proc. Natl. Acad. Sci. USA 2024, 121, e2404309121. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Rationale for Targeting and Current Therapeutic Approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Camviel, N.; Wolf, B.; Croce, G.; Gfeller, D. Both APRIL and Antibody-Fragment-Based CAR T Cells for Myeloma Induce BCMA Downmodulation by Trogocytosis and Internalization. Front. Immunol. 2022, 13, 1002500. [Google Scholar] [CrossRef]
- Ullah, M.; Mackay, F. The BAFF-APRIL System in Cancer. Cancers 2023, 15, 1791. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, A.; Ormhøj, M.; Choi, B.D.; Taylor, A.O.; Bouffard, A.A.; Scarfò, I.; Larson, R.C.; Frigault, M.J.; Gallagher, K.; Castano, A.P.; et al. Rational Design of a Trimeric APRIL-Based CAR-Binding Domain Enables Efficient Targeting of Multiple Myeloma. Blood Adv. 2019, 3, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Medler, J.; Wajant, H. Tumor Necrosis Factor Receptor-2 (TNFR2): An Overview of an Emerging Drug Target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gao, C.; Huang, Y.; Qiao, Y.; Xu, H.; Liu, S.; Wu, H. Unraveling the Breast Cancer Tumor Microenvironment: Crucial Factors Influencing Natural Killer Cell Function and Therapeutic Strategies. Int. J. Biol. Sci. 2025, 21, 2606–2628. [Google Scholar] [CrossRef]
- Yang, Y.; Islam, M.S.; Hu, Y.; Chen, X. TNFR2: Role in Cancer Immunology and Immunotherapy. ImmunoTargets Ther. 2021, 10, 103–122. [Google Scholar] [CrossRef]
- Li, L.; Ye, R.; Li, Y.; Pan, H.; Han, S.; Lu, Y. Targeting TNFR2 for Cancer Immunotherapy: Recent Advances and Future Directions. J. Transl. Med. 2024, 22, 812. [Google Scholar] [CrossRef]
- Moatti, A.; Cohen, J.L. The TNF-α/TNFR2 Pathway: Targeting a Brake to Release the Anti-Tumor Immune Response. Front. Cell Dev. Biol. 2021, 9, 725473. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, D.; Gao, Y.; Xu, Q.; Zhou, Y.; Ni, Z.; Na, M. Immunosuppressive Regulatory Cells in Cancer Immunotherapy: Restrain or Modulate? Hum. Cell 2024, 37, 931–943. [Google Scholar] [CrossRef]
- Ba, H.; Dai, Z.; Zhang, Z.; Zhang, P.; Yin, B.; Wang, J.; Li, Z.; Zhou, X. Antitumor Effect of CAR-T Cells Targeting Transmembrane Tumor Necrosis Factor Alpha Combined with PD-1 mAb on Breast Cancers. Front. Immunol. 2023, 11, e003837. [Google Scholar] [CrossRef]
- Takahashi, H.; Yoshimatsu, G.; Faustman, D.L. The Roles of TNFR2 Signaling in Cancer Cells and the Tumor Microenvironment and the Potency of TNFR2 Targeted Therapy. Cells 2022, 11, 1952. [Google Scholar] [CrossRef]
- Dawson, N.A.J.; Rosado-Sánchez, I.; Novakovsky, G.E.; Fung, V.C.W.; Huang, Q.; McIver, E.; Sun, G.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci. Transl. Med. 2020, 12, eaaz3866. [Google Scholar] [CrossRef] [PubMed]
- D’Accardo, C.; Porcelli, G.; Mangiapane, L.R.; Modica, C.; Pantina, V.D.; Roozafzay, N.; Di Franco, S.; Gaggianesi, M.; Veschi, V.; Lo Iacono, M.; et al. Cancer cell targeting by CAR-T cells: A matter of stemness. Front. Mol. Med. 2022, 2, 1055028. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-Targeted T Cells Mediate Potent Antitumor Effects Against Acute Myeloid Leukemia and Multiple Myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, K.; Hackert, T.; Zöller, M. CD44/CD44v6: A Reliable Companion in Cancer-Initiating Cell Maintenance and Tumor Progression. Front. Cell Dev. Biol. 2018, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- George, L.L.; Deshpande, S.R.; Cortese, M.J.; Kendall, E.K.; Chattaraj, A.; Shah, Z.; Zhao, J.; Anwer, F. Emerging Targets and Cellular Therapy for Relapsed Refractory Multiple Myeloma: A Systematic Review. Clin. Lymphoma Myeloma Leuk. 2021, 21, 741–751. [Google Scholar] [CrossRef]
- Gupta, S.; Master, S.; Graham, C. Extramedullary Multiple Myeloma: A Patient-Focused Review of the Pathogenesis of Bone Marrow Escape. World J. Oncol. 2022, 13, 311–319. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef]
- Lu, L.; Xie, M.; Yang, B.; Zhao, W.B.; Cao, J. Enhancing the Safety of CAR-T Cell Therapy: Synthetic Genetic Switch for Spatiotemporal Control. Sci. Adv. 2024, 10, eadj6251. [Google Scholar] [CrossRef]
- Ciulean, S.I.; Fischer, J.; Quaiser, A.; Bach, C.; Abken, H.; Tretbar, U.S.; Fricke, S.; Köhl, U.; Schmiedel, D.; Grunwald, T. Harnessing anti-CD44v6 CAR-NK cells for effective head and neck squamous cell carcinoma treatment. J. Immunother. Cancer 2024, 12 (Suppl. S2), A250. [Google Scholar] [CrossRef]
- Ciulean, I.S.; Fischer, J.; Quaiser, A.; Bach, C.; Abken, H.; Tretbar, U.S.; Fricke, S.; Koehl, U.; Schmiedel, D.; Grunwald, T.; et al. CD44v6 Specific CAR-NK Cells for Targeted Immunotherapy of Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2023, 14, 1290488. [Google Scholar] [CrossRef]
- Porcellini, S.; Asperti, C.; Corna, S.; Cicoria, E.; Valtolina, V.; Stornaiuolo, A.; Valentinis, B.; Bordignon, C.; Traversari, C. CAR T Cells Redirected to CD44v6 Control Tumor Growth in Lung and Ovary Adenocarcinoma Bearing Mice. Front. Immunol. 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Lodewijk, I.; Dueñas, M.; Paramio, J.M. CD44v6, STn & O-GD2: Promising Tumor Associated Antigens Paving the Way for New Targeted Cancer Therapies. Front. Immunol. 2023, 14, 1272681. [Google Scholar] [CrossRef]
- Carrabba, M.G.; Casucci, M.; Hudecek, M.; Quintarelli, C.; Briones, J.; Hajek, R.; Sierra, J.; Locatelli, F.; Einsele, H.; Bordignon, C.; et al. Phase I–IIa Clinical Trial to Assess Safety and Efficacy of MLM-CAR44.1, a CD44v6 Directed CAR-T in Relapsed/Refractory Acute Myeloid Leukemia (AML) and Multiple Myeloma (MM). Blood 2018, 132 (Suppl. S1), 5790. [Google Scholar] [CrossRef]
- Van der Schans, J.J.; Van de Donk, N.W.C.J.; Mutis, T. Dual Targeting to Overcome Current Challenges in Multiple Myeloma CAR T-Cell Treatment. Front. Oncol. 2020, 10, 1362. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The Activated Conformation of Integrin β7 Is a Novel Multiple Myeloma–Specific Target for CAR T Cell Therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef]
- Savani, M.; Oluwole, O.; Dholaria, B. New Targets for CAR T Therapy in Hematologic Malignancies. Semin. Hematol. 2021, 58, 208–215. [Google Scholar] [CrossRef]
- Cottini, F.; Rodriguez, J.; Hughes, T.; Sharma, N.; Guo, L.; Lozanski, G.; Liu, B.; Cocucci, E.; Yang, Y.; Benson, D.; et al. Redefining CD56 as a Biomarker and Therapeutic Target in Multiple Myeloma. Mol. Cancer Res. 2022, 20, 1083–1095. [Google Scholar] [CrossRef]
- Koumpis, E.; Tassi, I.; Malea, T.; Papathanasiou, K.; Papakonstantinou, I.; Serpanou, A.; Tsolas, E.; Kapsali, E.; Vassilakopoulos, T.P.; Papoudou-Bai, A.; et al. CD56 Expression in Multiple Myeloma: Correlation with Poor Prognostic Markers but No Effect on Outcome. Pathol. Res. Pract. 2021, 225, 153567. [Google Scholar] [CrossRef]
- Sahara, N.; Takeshita, A. Prognostic Significance of Surface Markers Expressed in Multiple Myeloma: CD56 and Other Antigens. Leuk. Lymphoma 2004, 45, 61–65. [Google Scholar] [CrossRef]
- Hosen, N. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Int. J. Hematol. 2020, 112, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M. Dual targeting of multiple myeloma stem cells and myeloid-derived suppressor cells for treatment of chemotherapy-resistant multiple myeloma. Front. Oncol. 2021, 11, 760382. [Google Scholar] [CrossRef]
- Ho, M.; Xiao, A.; Yi, D.; Zanwar, S.; Bianchi, G. Treating multiple myeloma in the context of the bone marrow microenvironment. Curr. Oncol. 2022, 29, 8975–9005. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a therapeutic target in the treatment of hematological malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.; De Angelis, B.; Del Bufalo, F.; Ciccone, R.; Donsante, S.; Volpe, G.; Manni, S.; Guercio, M.; Pezzella, M.; Iaffaldano, L.; et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J. Hematol. Oncol. 2022, 15, 163. [Google Scholar] [CrossRef]
- Peinert, S.; Prince, H.M.; Guru, P.M.; Kershaw, M.H.; Smyth, M.J.; Trapani, J.A.; Gambell, P.; Harrison, S.; Scott, A.M.; Smyth, F.E.; et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010, 17, 678–686. [Google Scholar] [CrossRef]
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res. Ther. 2021, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Song, Y.; Liu, D. Clinical trials of CAR-T cells in China. J. Hematol. Oncol. 2017, 10, 166. [Google Scholar] [CrossRef]
- Smith, L.; Di Stasi, A. Safety switches used for cellular therapies. In Gene and Cellular Immunotherapy for Cancer; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–15. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target | Expression | Expression in Normal Tissues | Risk of Off-Tumor Toxicity | Development Stage | Clinical Trial ID (If Available) | References |
|---|---|---|---|---|---|---|
| BCMA | High and stable | Plasma cells | Low | Registration (ide-cel, cilta-cel) | NCT03361748 and NCT04162210 | [80,81] |
| GPRC5D | High | Squamous epithelium | Low | Phase I/II | NCT04555551 | [82] |
| FcRH5 | Moderately high | Few | Low | Case Report | ChiCTR2000041025 * | [83] |
| CD38 | High | Immune and epith. cells | Average | Phase I | NCT04351022 | [84] |
| SLAMF7 | Moderate | NK, DC, and T cells | Average | Phase I | NCT03958656 | [85] |
| CD138 | High | Epithelium | Average | Phase I | NCT04430530 | [86] |
| CD70 | Moderate | Activ. immune cells | Average | Phase I | NCT02830724 | [87] |
| NKG2D ligands | Heterogenous | Minimal | Low | Phase I | NCT04623944 | [88] |
| CD19 | Residual (subclones) | B cells | Average | Preclinical/Phase I | NCT02135406 | [89] |
| TACI | Variable | B cells | Low | Preclinical | ||
| APRIL | No data available | No data available | No data available | Preclinical | ||
| TNFR2 | Moderate | Tregs and stroma | Average/high | Preclinical | ||
| CD44v6 | Variable | Keratinocytes | High | Preclinical | ||
| Alternative targets for CAR cell therapy in multiple myeloma | ||||||
| CD56 | 70% of cases | NK cells | Average | Preclinical | ||
| Integrin β7 | Prevalently | Minimal | Low | Preclinical | ||
| CD123 | Low | Plasm. DC | Average | Preclinical/Phase I (AML) | NCT04318678, NCT02159495, NCT02623582, and NCT04599543 (only AML, not MM) | [90,91,92,93] |
| Lewis-Y | ≈50% | Low | Average | Phase I | NCT01716364 | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezborodova, O.A.; Trunova, G.V.; Nemtsova, E.R.; Khokhlova, V.A.; Venediktova, J.B.; Morozova, N.B.; Vorontsova, M.S.; Plyutinskaya, A.D.; Zharova, E.P.; Shegai, P.V.; et al. Targets for CAR Therapy in Multiple Myeloma. Int. J. Mol. Sci. 2025, 26, 6051. https://doi.org/10.3390/ijms26136051
Bezborodova OA, Trunova GV, Nemtsova ER, Khokhlova VA, Venediktova JB, Morozova NB, Vorontsova MS, Plyutinskaya AD, Zharova EP, Shegai PV, et al. Targets for CAR Therapy in Multiple Myeloma. International Journal of Molecular Sciences. 2025; 26(13):6051. https://doi.org/10.3390/ijms26136051
Chicago/Turabian StyleBezborodova, Olga A., Galina V. Trunova, Elena R. Nemtsova, Varvara A. Khokhlova, Julia B. Venediktova, Natalia B. Morozova, Maria S. Vorontsova, Anna D. Plyutinskaya, Elena P. Zharova, Peter V. Shegai, and et al. 2025. "Targets for CAR Therapy in Multiple Myeloma" International Journal of Molecular Sciences 26, no. 13: 6051. https://doi.org/10.3390/ijms26136051
APA StyleBezborodova, O. A., Trunova, G. V., Nemtsova, E. R., Khokhlova, V. A., Venediktova, J. B., Morozova, N. B., Vorontsova, M. S., Plyutinskaya, A. D., Zharova, E. P., Shegai, P. V., & Kaprin, A. D. (2025). Targets for CAR Therapy in Multiple Myeloma. International Journal of Molecular Sciences, 26(13), 6051. https://doi.org/10.3390/ijms26136051