1. Introduction
Hand, foot, and mouth disease (HFMD) remains a critical public health challenge in China, consistently ranking within the top three Class C infectious diseases in terms of both incidence and mortality [
1,
2,
3]. While the deployment of inactivated EV-71 vaccines has mitigated EV-71-associated HFMD outbreaks [
4,
5], the prevalence of non-EV-A71 pathogens is increasing year by year [
6]. Many enteroviruses can cause HFMD, but, in general, coxsackieviruses are important pathogens. In addition to coxsackievirus group A (CV-A), coxsackievirus B3 (CV-B3) has also emerged as a notable etiological agent of HFMD. CV-B3 infections manifest in various clinical presentations, including blisters and HFMD-like rashes, as well as causing myocarditis and encephalitis in children in particular [
7].
The severity of CV-B infections in younger populations is particularly concerning [
8,
9,
10]. Several lines of epidemiological evidence support this age-specific vulnerability affecting young children, indicating an age-related distribution of CV-B infections, and high mortality rates have been observed in newborns and young children infected with CV-B2 (11.1%), CV-B3 (18.8%), and CV-B4 (40%) [
11]. Numerous clinical studies have consistently reported that CV-B infections in infants are frequently accompanied by severe clinical symptoms and poor outcomes [
6,
9,
11]. Although CV-B3-related myocarditis and encephalitis have been documented in pediatric populations, the molecular mechanisms underlying the age-specific susceptibility to these severe manifestations remain poorly understood. While this heightened vulnerability was traditionally attributed to immature immune systems in young children, this explanation appears to be oversimplified given the paradoxical observation that the same demographic exhibits a markedly reduced susceptibility to other viral pathogens, such as SARS-CoV-2 [
12,
13,
14], suggesting that age-dependent susceptibility patterns may be governed by pathogen-specific factors rather than generalized immune immaturity. From another viewpoint, the coxsackievirus and adenovirus receptor (CAR) serves as the primary cellular entry point for CV-B viruses, and it has been identified as a critical determinant of human infection susceptibility [
1]. The CAR’s pronounced tropism, particularly in cardiac tissues, proves that it is essential not only for the direct infection of target organs but also for viral dissemination from primary infection sites. This tissue-specific distribution may be attributed to the differential expression of four CAR splice variants (CAR1–CAR4), which exhibit distinct expression patterns across tissues and developmental stages.
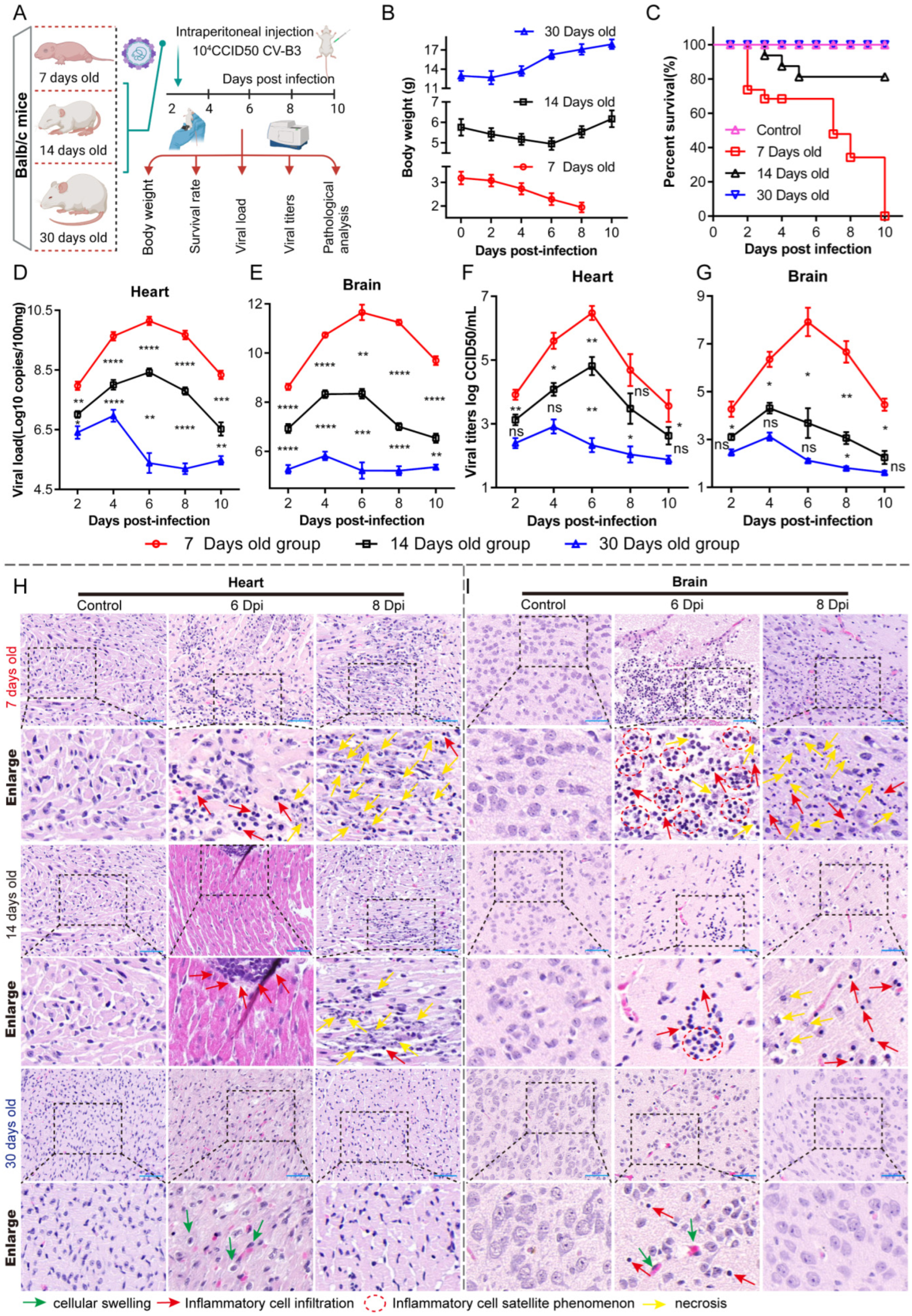
To better understand the CAR’s role in CV-B3 myocarditis and encephalitis pathogenesis, we evaluated CV-B3 infection progression in Balb/c mice across age groups ranging from 7 to 30 days post-birth, with a particular focus on the cardiac system and the central nervous system (CNS). We found that CAR splice variant expression patterns exhibited pronounced tissue tropism with significant age-related variations. During infection, CV-B3 proliferation in young mice (with high CAR1 and CAR2 expression) was accelerated, inducing tissue destruction and inflammation, while it was markedly reduced in 30-day-old mice (with high CAR3 and CAR4 expression). These data demonstrate the underlying susceptibility and severity of CV-B3 infections in the early life of Balb/c mice.
3. Discussion
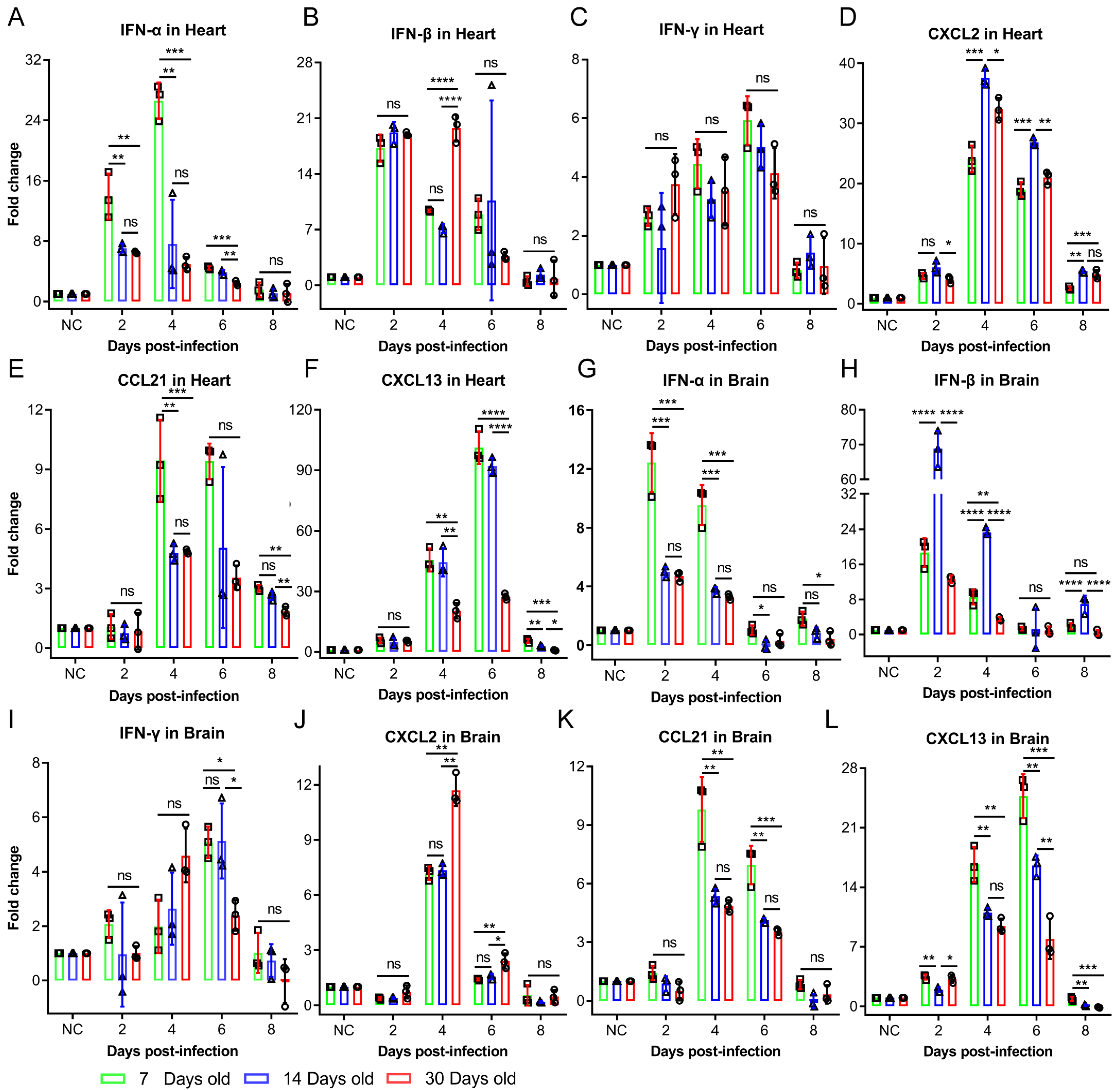
The dynamic interplay between viral infection and host immunity critically determines infection outcomes [
16]. In this study, the intraperitoneal challenge of Balb/c mice with identical CV-B3 doses revealed significantly enhanced susceptibility and pathological severity in 7-day-old mice compared to in 14- and 30-day-old mice. Cytokine profiling demonstrated the robust activation of both innate and adaptive immune responses in younger infected groups—occasionally exceeding the responses in older groups. However, a cytokine analysis alone does not provide sufficient mechanistic insight into the heightened CV-B3 vulnerability and accelerated disease progression observed in neonates. Viral tropism and replication kinetics are governed by multifactorial determinants [
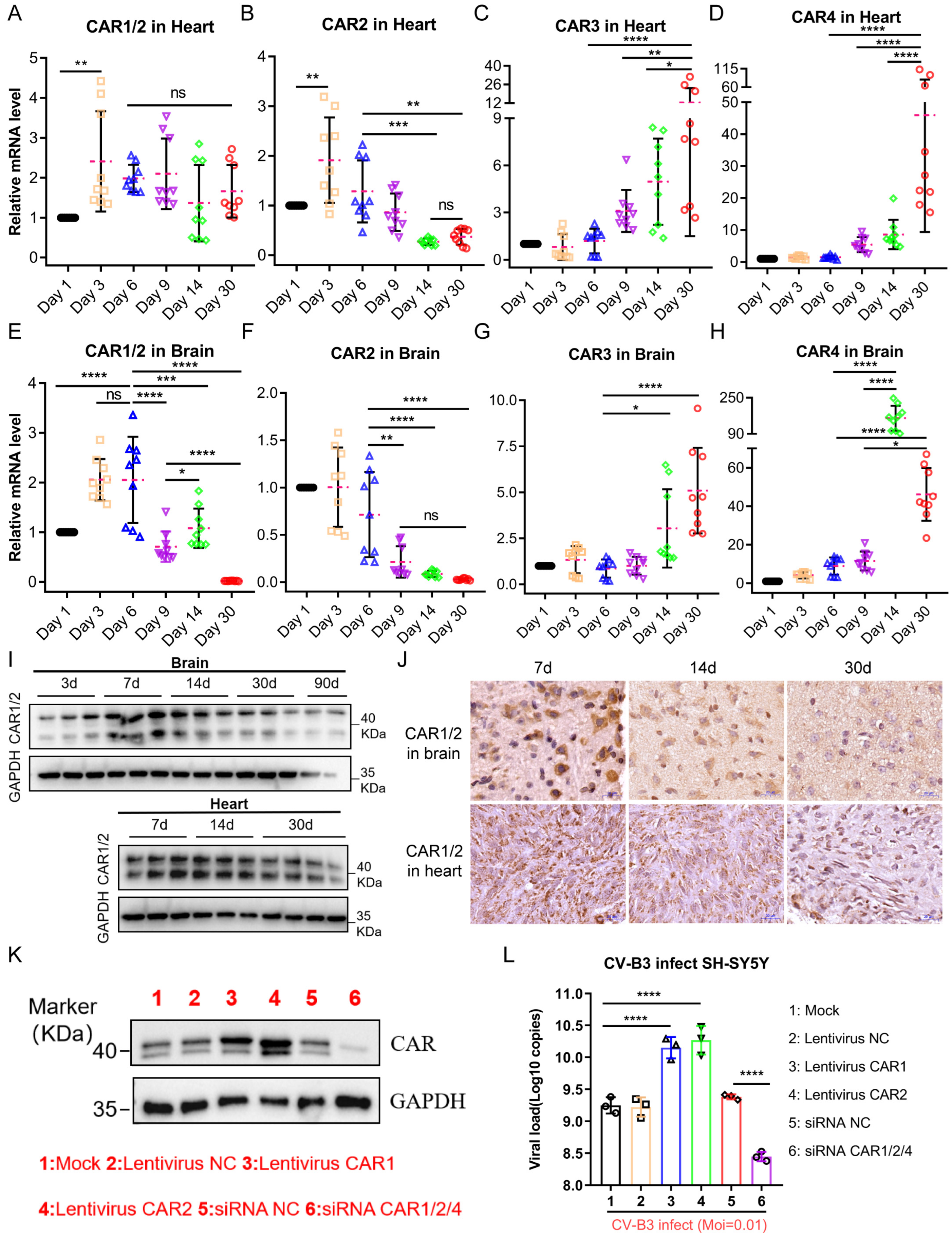
17]. For coxsackieviruses, receptor-mediated entry represents a critical initial step for cellular infection. We therefore quantified CAR expression in the tissues of 7-day-old infected mice, identifying a positive correlation between CAR abundance and early-stage viral loads. Notably, the murine CAR exhibited developmentally regulated isoform expression with tissue-specific distribution patterns. This suggests that age-dependent disparities in CAR expression profiles within target organs (particularly cardiac and CNS tissues) may fundamentally modulate CV-B3 infection efficiency and replication dynamics across developmental stages.
Our analysis revealed an age-dependent decline in the cardiac and cerebral expression of CAR1 and CAR2 splice variants—functional receptors for CV-B3—concomitant with an increased expression of the non-receptor isoforms CAR3 and CAR4 in Balb/c mice. Due to the lack of human samples to verify this phenomenon, there are no data showing whether similar expression patterns exist in human CARs; however, clinical studies using CV-B3 as an oncolytic agent have reported marked CAR upregulation in tumor tissues versus low expression in normal adult tissues [
18,
19], suggesting the potential conservation of this developmental regulation. This phenomenon is consistent with the role of CAR as an adhesion molecule, essential for intercellular communication during development. The upregulation of CAR4, characterized by an incomplete extracellular domain but a complete intracellular domain, may serve to maintain intercellular signaling functions as the organism matures. Consequently, this phenomenon could be explained by the CAR gene’s involvement in the organism’s developmental biology through variable splicing. However, this regulatory phenomenon impacts CV-B3 infection differently in Balb/c mice of various ages. In younger mice, the high CAR abundance facilitates rapid viral proliferation, allowing for significant “primitive accumulation” during the early “window period” of infection when the immune system is not yet fully activated. In contrast, older Balb/c mice exhibit slower viral proliferation due to their lower CAR abundance.
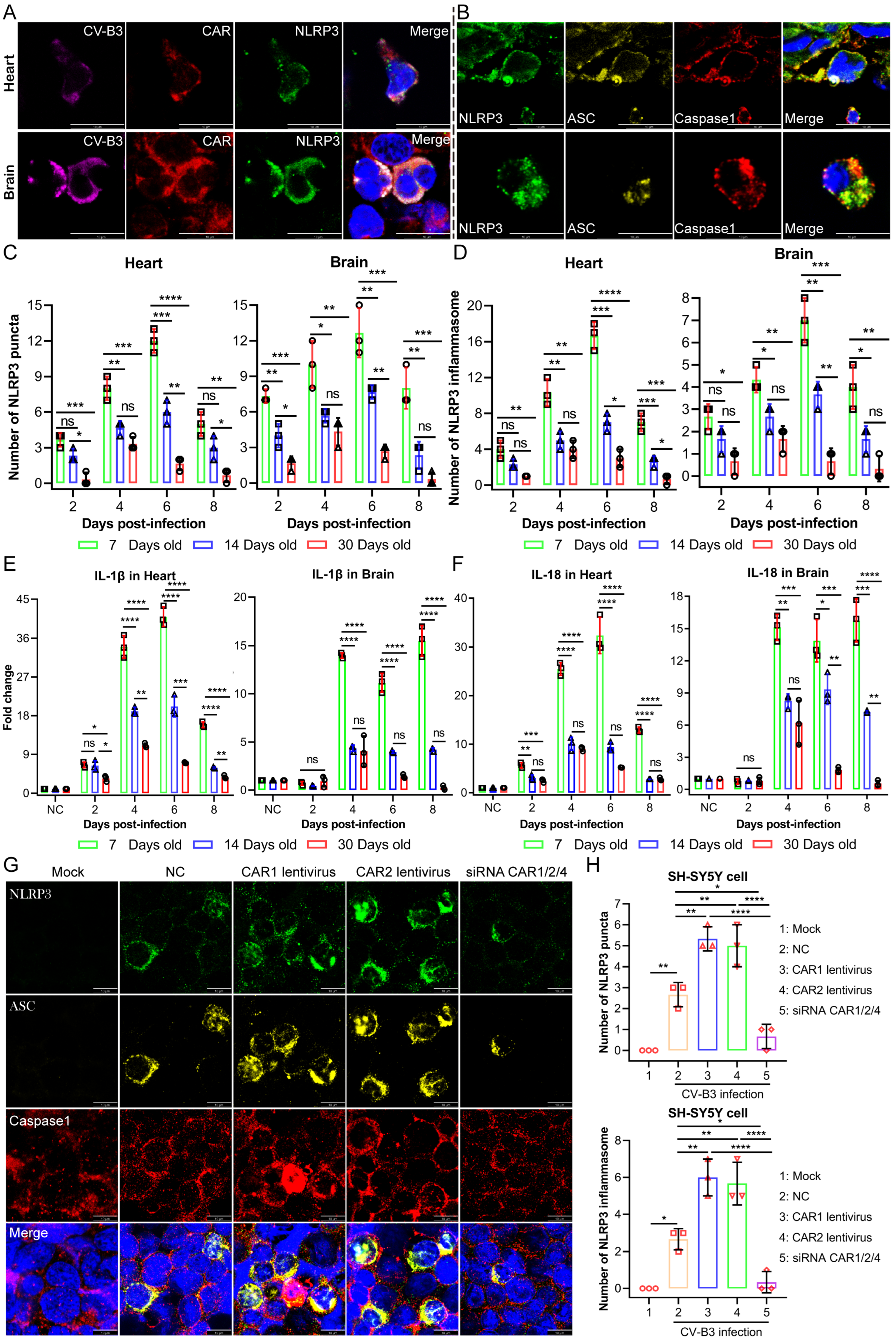
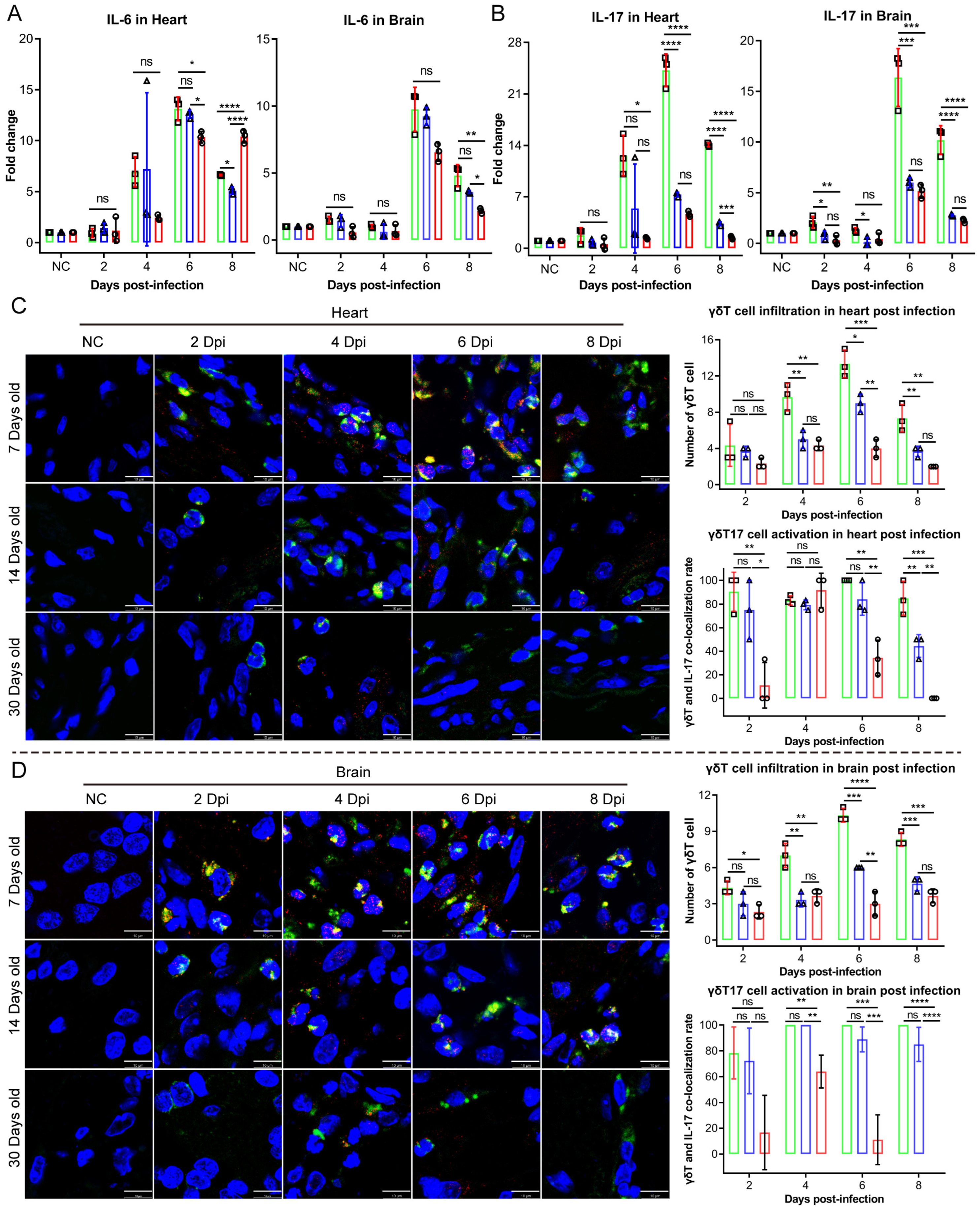
Upon the detection of endogenous danger signals and exogenous pathogens, the NLRP3 inflammasome—a critical component of innate immunity—rapidly activates to mediate the maturation of the proinflammatory cytokines IL-1β and IL-18. This process induces pyroptotic cell death, releasing inflammatory mediators that recruit additional innate immune cells and establish localized inflammatory responses [
20]. While moderate NLRP3 activation supports host defense against microbial threats, excessive activation drives dysregulated inflammation and cytokine storms, which potentiate inflammatory pathologies [
21]. In our study, moderate NLRP3 inflammasome activation exerted protective antiviral effects without causing significant tissue damage in the 14- and 30-day-old infected mice groups. Conversely, the 7-day-old infected mice group exhibited NLRP3 hyperactivation, which provided antiviral benefits while simultaneously provoking excessive inflammatory pathology. These findings establish NLRP3 inflammasome dysregulation as a critical determinant of CV-B3-induced tissue damage. In addition to IL-1β and IL-18, serum levels of the proinflammatory cytokine IL-17 were significantly elevated in the 7-day-old infected group. IL-17 is primarily produced by Th17, NK, and γδT17 cells. γδT17 cells have been implicated in the pathogenesis of multiple viral infections. In a hepatitis B virus infection model, these cells exacerbated disease progression by inducing the recruitment of immunosuppressive polymorphonuclear neutrophils (PMNs), thereby promoting CD8
+ T cell exhaustion [
22]. Similarly, skin-resident γδT17 cells were associated with inflammatory tissue pathology in an epicutaneous vaccinia virus infection model [
23]. With γδT17 cells being the predominant source following CV-B3 infection, Vγ4
+ γδT17 cells have been reported as pathogenic mediators in coxsackievirus B3 infection, triggering acute pancreatitis through IL-17 production [
15,
24]. In healthy organisms, γδT17 cells predominantly reside in peripheral blood, skin, and mucosal-associated lymphoid tissues. Following infection or tissue injury, these cells rapidly infiltrate affected sites. The cell adhesion molecule coxsackievirus and adenovirus receptor (CAR) facilitates immune cell migration to infection/injury loci, and the CAR directly engages junctional adhesion molecule-like protein (JAML) on γδT17 cells, thereby inducing IL-17 release [
25,
26,
27,
28]. Based on prior studies, we aimed to assess whether CAR abundance influences the recruitment and activation of γδT17 cells in Balb/c mice of varying ages. Our findings showed that the 7-day-old infected group exhibited the highest CAR abundance, resulting in a significantly increased infiltration and activation of γδT17 cells in cardiac and brain tissues compared to in those of the 14- and 30-day-old mice groups. Overall, CAR abundance critically influences susceptibility to coxsackievirus B3 (CV-B3) infection and the heightened incidence of severe illness in younger individuals. During the early life stages, elevated CAR levels facilitate rapid viral replication during the initial, rate-limiting step of infection. This triggers an immune response characterized by NLRP3 inflammasome overactivation and a substantial production of proinflammatory cytokines, including IL-1β and IL-18. Furthermore, the CAR promotes the migration and activation of γδT17 cells, resulting in increased IL-17 production within cardiac and brain tissues. While the enhanced expression and secretion of these proinflammatory cytokines mediate partial antiviral effects, leading to a decline in viral load and titers, they simultaneously contribute to significant inflammatory pathology. Given that the CAR functions as a universal viral receptor for CV-B and adenovirus—pathogens that are prevalent in younger populations—this study provides novel insights into the heightened susceptibility to CV-B infection during early life and its associated severe disease rates.
This study has several limitations. First, rapid age-dependent changes in CAR alternative splicing occur postnatally, whereas viral-mediated knockdown or overexpression requires extended timeframes for implementation. Second, current viral delivery tools lack tissue specificity for cardiac and brain compartments in neonatal Balb/c mice, impeding targeted CAR manipulation in these critical organs. Finally, as the CAR plays essential physiological roles in cell adhesion and intercellular signaling, constitutive global knockout via gene editing may induce confounding developmental effects. Consequently, we were unable to definitively establish this mechanism in vivo using conventional approaches. Further mechanistic validation could be achieved using cardiac and brain organoids engineered for CAR overexpression or knockdown, coupled with an analysis of CVB3 viral kinetics post-infection.
4. Materials and Methods
4.1. Animal Models
Balb/c mice, a well-established model for CV-B3 infection studies, were utilized in this study. To analyze age-dependent infection characteristics, 7-, 14-, and 30-day-old mice were selected. The animals were procured from the Laboratory Animal Center, Institute of Medical Biology, Chinese Academy of Medical Sciences. All experimental procedures received prior approval from the Institutional Animal Care and Use Committee of IMBCAMS (Ethical Approval Code: DWSP202308001; date: 8 August 2023) and strictly adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
4.2. Procedure for Animal Experiments
Mice from each age cohort were randomly allocated to two experimental groups: (1) a PBS control group (50 μL, intraperitoneal injection) and (2) a CV-B3-infected group (104 CCID50 of CV-B3 in 50 μL PBS, administered via an intraperitoneal injection). Body weights were recorded using an electronic analytical balance at 2, 4, 6, 8, and 10 days post-infection. Survival rates were analyzed using a GraphPad Prism 8.0.1 survival curve analysis. At predetermined time points (2, 4, 6, 8, and 10 dpi), randomly selected mice were euthanized for organ collection. The harvested tissues were subjected to further analyses, including viral titer determination, viral load quantification, and HE staining.
4.3. Virus Strains
The CV-B3 strain HX03/YN/2021 (GenBank accession: PQ001506.1), originally isolated from a clinical rectal swab specimen obtained in Yunnan Province, China, was serially passaged three times in Vero cells to generate the P3 working stock. Virus-containing supernatants were harvested at peak cytopathic effect (>90% CPE). Cellular debris was removed via centrifugation at 2000× g for 30 min at 4 °C. Clarified supernatants were concentrated 5-fold using centrifugal filters with a 100 kDa molecular weight cut-(Merck Millipore, Darmstadt, Germany). Viral stocks were titrated by performing a 50% tissue culture infectious dose (TCID50) assay on Vero cells, aliquoted, and cryopreserved at −80 °C in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
4.4. HE Staining
At 2, 4, 6, 8, and 10 days post-infection, hearts and brains were harvested from the infected Balb/c mice. The tissues were immediately fixed in 10 volumes of 10% neutral buffered formalin at 4 °C for 24 h to ensure complete penetration. After fixation, the specimens were dehydrated through a graded ethanol series (70% → 95% → 100%), cleared in xylene, and infiltrated with paraffin using an automated tissue processor (Leica, Wetzlar, Germany, ASP300). The paraffin-embedded tissues were sectioned at 4 μm thickness with a rotary microtome (Leica RM2245). The tissue sections were deparaffinized in xylene (5 min, 2 times), rehydrated through a graded ethanol series (100% → 95% → 70%; 2 min per concentration), and rinsed in distilled water. Nuclear staining was performed with hematoxylin (Solarbio, Beijing, China, G1120; 8 min), followed by differentiation in 1% acid ethanol (30 s) and bluing in 0.2% ammonia water (1 min), and the sections were then rinsed under running tap water for 30 min. Cytoplasmic counterstaining was conducted with eosin (Solarbio, G1100; 1 min), followed by dehydration through graded ethanol (95% → 100%; 1 min per concentration) and clearing in xylene (5 min, 2 times). Finally, the sections were mounted with neutral balsam. Whole-slide imaging was performed using a Pannoramic MIDI digital slide scanner (3D HISTECH, Thermo Fisher Scientific, Waltham, MA, USA). A histopathological analysis was conducted using CaseViewer 2.4.0.
4.5. Western Blotting Assay
Immediately after euthanasia, the heart and brain tissues (20 mg) obtained from the Balb/c mice were flash-frozen in liquid nitrogen and stored at −80 °C. For protein extraction, the tissues were minced on ice and homogenized in 300 μL ice-cold RIPA lysis buffer, which was supplemented with 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail. Following complete lysis in RIPA buffer, tissue/cell homogenates were centrifuged at 12,000× g for 30 min at 4 °C, and supernatants were quantified using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Equal amounts of total protein were denatured in 5× SDS loading buffer at 95 °C for 10 min, then the denatured samples were resolved via SDS-PAGE on 12% gels and transferred to 0.22 μm PVDF membranes (Merck Millipore, IPFL00010) using a Trans-Blot® SD Semi-Dry Transfer System (Bio-Rad, Hercules, CA, USA), and the membranes were blocked with 2% BSA in PBST (0.1% Tween-20) for 1 h at 25 °C. Primary antibodies were diluted in blocking buffer and incubated overnight at 4 °C (Rb mAb to CAR/CXADR (ABclonal, Wuhan, China, A22607), Mouse mAb to CV-B3 Merck MAB948, Rb mAb to NLRP3 (Abcam, Cambridge, UK, Ab210491), Rb mAb to Caspase-1(Proteintech, Chicago, IL, USA, 22915-1-AP), Mouse mAb to ASC (SANTA Cruz, CA, USA, sc-514414), Mouse mAb to HRP&GAPDH (Proteintech, HRP-60004). After three washes with PBST (5 min each time), the membranes were incubated with HRP-conjugated secondary antibodies (Goat Anti-Rabbit IgG H&L (HRP) Abcam ab6721, Goat Anti-Mouse IgG H&L (HRP) Abcam ab6789) for 1 h at 25 °C. Protein bands were visualized using BIO-RAD ChemiDoc.
4.6. Immunohistochemistry (IHC)
After deparaffinization in xylene and rehydration through graded ethanol, antigen retrieval was performed in pH 6.0 citrate buffer (95 °C, 20 min). Endogenous peroxidase activity was quenched with 3% H2O2 (25 °C, 10 min). Sections were blocked with 5% normal goat serum (25 °C, 30 min) and incubated with rabbit anti-CAR/CXADR monoclonal antibody (GeneTex, Irvine, CA, USA, GTX118382; 1:300) at 4 °C overnight, followed by response enhancer treatment (Solarbio, SP0021) and secondary antibody incubation (Solarbio, SP0021; 25 °C, 30 min). Immunoreactivity was visualized using DAB substrate (Solarbio, DA1010; 5 min at 25 °C). Nuclei were counterstained with hematoxylin (Solarbio, G1120; 8 min), followed by differentiation in 0.5% acid ethanol (30 s) and bluing in tap water substitute (1 min). Following dehydration through graded ethanol and xylene clearance, the sections were mounted with neutral balsam. Whole-slide images were acquired at 20× magnification using a Pannoramic MIDI slide scanner (3D HISTECH) and analyzed using CaseViewer 2.4.0.
4.7. Immunofluorescence (IF)
The NLRP3 inflammasome is a multiprotein complex that serves as a critical platform for innate immune activation. In quiescent cells, NLRP3 exhibits low expression levels and diffuse cytoplasmic localization. Upon stimulation, NLRP3 undergoes transcriptional upregulation and oligomerizes into discrete punctate foci. These structures subsequently recruit the adaptor protein ASC and pro-Caspase-1, culminating in the assembly of the functional NLRP3 inflammasome complex. The formation of NLRP3 puncta represents a morphological hallmark of inflammasome activation, providing the structural basis for Caspase-1 autocatalysis. Activated pro-Caspase-1 then proteolytically processes the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their bioactive forms (IL-1β and IL-18), which are secreted extracellularly. Concurrently, Caspase-1 triggers pyroptosis—a lytic, proinflammatory mode of programmed cell death. Crucially, the co-localization of NLRP3 puncta with ASC and Caspase-1 constitutes definitive evidence of functional inflammasome assembly and signifies the initiation of a potent inflammatory cascade within the affected cell.
Following dewaxing in xylene (2 times, each time 5 min) and rehydration through graded ethanol (100% → 95% → 70%; 2 min at each concentration), tissue sections were subjected to antigen retrieval in citrate buffer (10 mM, pH 6.0; 95 °C, 20 min), endogenous peroxidase blockade with 3% H2O2 (10 min, 25 °C), and permeabilization with 0.3% Triton X-100 (15 min, 25 °C) prior to blocking in 5% BSA (1 h, 25 °C). The primary antibodies were incubated overnight at 4 °C: Mouse anti-CV-B3 VP1 (Merck MAB948; 1:300), Rabbit anti-CAR/CXADR (ABclonal A22607; 1:500), Rat anti-NLRP3 (Thermo Fisher Scientific, MA5-23919; 1:500), Rabbit anti-Caspase-1 (Proteintech 22915-1-AP; 1:500), Mouse anti-ASC (Santa Cruz, sc-514414; 1:500), Armenian hamster anti-γδTCR (BioLegend, San Diego, CA, USA, 118108; 1:500), and Rat anti-IL-17A (BioLegend 506916; 1:500). Following PBST washes, species-matched secondary antibodies were applied for 1 h at 25 °C in darkness: Goat anti-rat IgG-Alexa Fluor 488 (Abcam ab150157; 1:500), Goat anti-mouse IgG-Alexa Fluor 555 (Abcam ab150118; 1:1000), Donkey anti-rabbit IgG-Alexa Fluor 647 (Abcam ab150063; 1:1000), and Goat anti-Armenian hamster IgG-Alexa Fluor 647 (Abcam ab173004; 1:500). Nuclei were counterstained with DAPI, slices were mounted, and confocal images were acquired using a Leica TCS SP8 laser confocal microscope.
4.8. ELISA Analysis
Cardiac and cerebral tissues were minced in 2 mL microtubes using sterile ophthalmic scissors, enzyme-free stainless-steel beads were added, and homogenization was performed with 100 μL ice-cold PBS in a homogenizer (60 Hz, 10 × 30 s cycles with 30 s ice-cooling intervals between cycles). Homogenates were centrifuged at 10,000× g for 10 min at 4 °C, and supernatants were collected. IFN-α, IFN-β, IFN-γ, CXCL2, CCL21, and CXCL13 concentrations were quantified using commercial ELISA kits, according to the manufacturers’ protocols.
4.9. Quantitative Real-Time PCR
Viral RNA was extracted using TRIzol (absin, Shanghai, China, abs9331) following the manufacturer’s protocol, including chloroform phase separation and isopropanol precipitation. The RNA purity and concentration were verified using Thermo Fisher Scientific NanoDrop™ spectrophotometry (A260/A280 > 1.8). TaqMan probe-based qPCR for CV-B3 viral load was performed with a One Step PrimeScript™ RT-PCR Kit (Takara, Ōtsu, Shiga, Japan, RR064A) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s procedure. The primer set is shown in
Table 1. Viral copy numbers were calculated as a ratio with respect to the standard control (the formula used to calculate the standard copy number was as follows: copies/mL = 6.02 × 10
23 (copies/mol) × concentration of standard (g/mL)/RNA base number × 340 g/mol).
Viral load standards were prepared as described below: a 852 bp fragment (
Supplementary Table S1) encompassing the CV-B3 (GenBank accession: PQ001506.1) VP1 gene was cloned into the pUC57 vector. The recombinant plasmid was linearized with ECORI and purified using 1.2% agarose gel and a PCR Clean-up Kit (Macherey-Nagel, Düren, Germany, 740609). In vitro transcription was performed with a T7 kit (TaKaRa, Code No. 6140), followed by DNase I treatment to purify the RNA. Ten-fold serial dilutions (10
7 to 10
0 copies/μL) of the purified RNA standard were prepared, and each dilution was analyzed in quadruplicate using a One Step PrimeScript™ RT-qPCR Kit (Takara, RR064A) on an ABI 7500 Real-Time PCR System (Applied Biosystems). The standard curve was generated by plotting the quantification cycle (Cq) values against the log
10 input copy numbers. A linear regression analysis yielded the following equation (
Supplementary Figure S1A): y = −3.3609x + 43.855 (where y = Cq and x = log
10 (copies/μL), with slope = −3.3609 and intercept = 43.855).
The total RNA was extracted from the Balb/c mice heart and brain tissues using TRIzol reagent. Quantification of the relevant expression levels of the four splice variants of the Balb/c mouse CAR gene was performed using the comparative ΔΔCq method with SYBR Green-based real-time PCR (Takara, RR066A). Gene expression levels were normalized to the endogenous reference gene GAPDH, and fold changes were calculated relative to the control group. The primer sequences are provided in
Table 1.
4.10. Virus Titer Assay
The heart and brain tissues were placed in 1.5 mL tubes, minced with fine surgical scissors, and suspended in 100 μL of 1× PBS. After tight sealing, the samples were stored at −80 °C overnight. The frozen tissues were thawed at 25 °C and subjected to three freeze–thaw cycles with thorough grinding. Lysates were centrifuged at 10,000× g for 10 min at 4 °C. Supernatants containing virus particles released during the freeze–thaw cycles were serially diluted (10−1 to 10−7) in MEM maintenance medium supplemented with 2% FBS. Aliquots (100 μL) of each dilution were incubated with 104 Vero cells/well in 96-well plates at 37 °C under 5% CO2. CPEs were monitored daily for 7 days, with the experimental endpoint defined as the timepoint showing no further CPE progression. Viral titers were calculated as TCID50/mL using the Reed–Muench method.
4.11. Statistical Analysis
All statistical analyses were conducted and all plots were constructed using GraphPad Prism software. All experimental data are presented as the mean ± standard deviation (SD). A minimum of three independent biological replicates were analyzed per experimental group (n ≥ 3). A one-way ANOVA with Tukey’s multiple comparisons test was performed, with statistical significance defined as p < 0.05. p-values are reported as follows: GP: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), 0.0001 (****).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}