
Multiomics Signature Reveals Network Regulatory Mechanisms in a CRC Continuum
 ,
,  , and
, and
Abstract

1. Introduction
2. Results
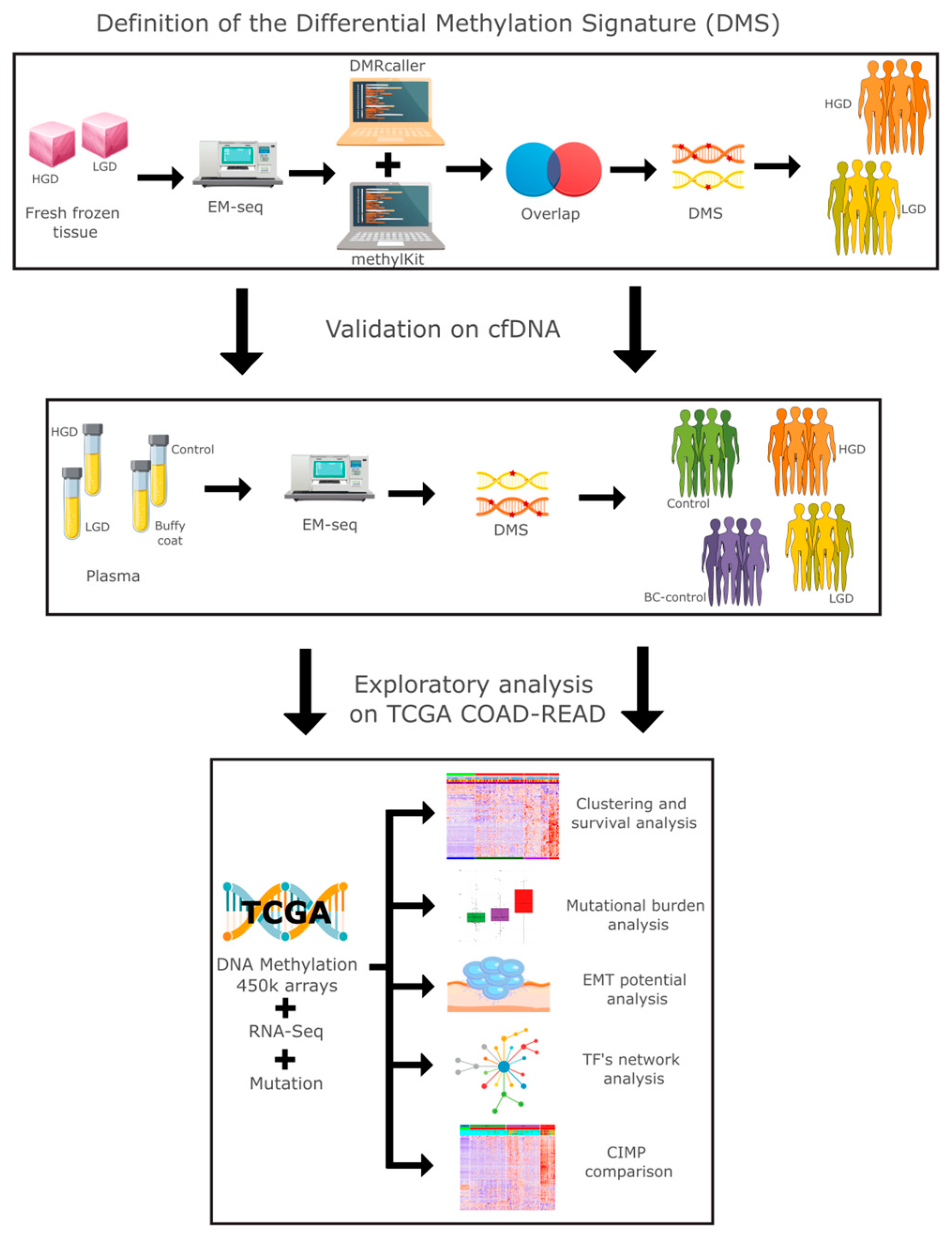
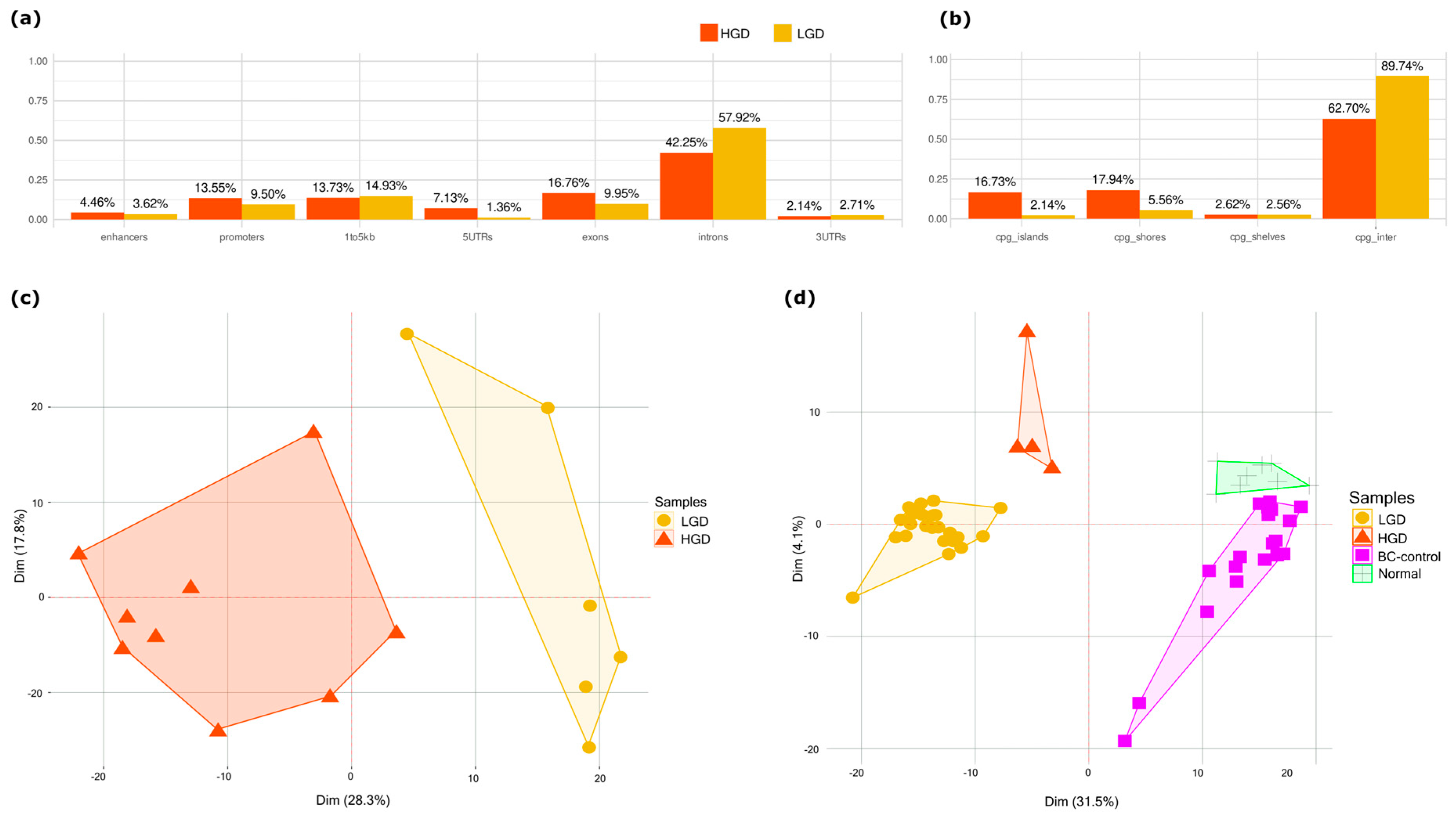
2.1. Methylation Differences Between HGD and LGD Stratifies Dysplasia in Tissue and Plasma
2.2. Functional Enrichment Analysis
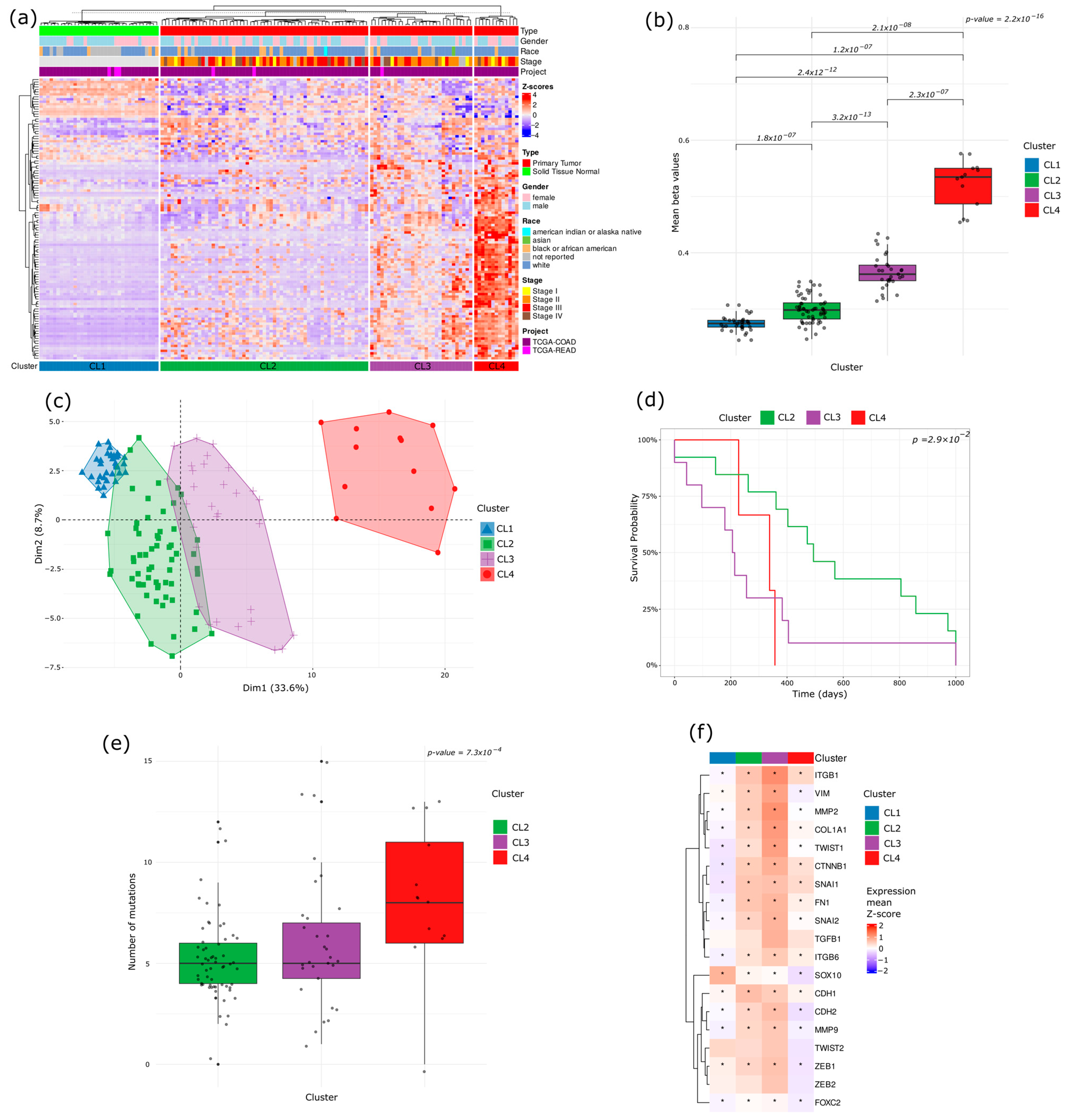
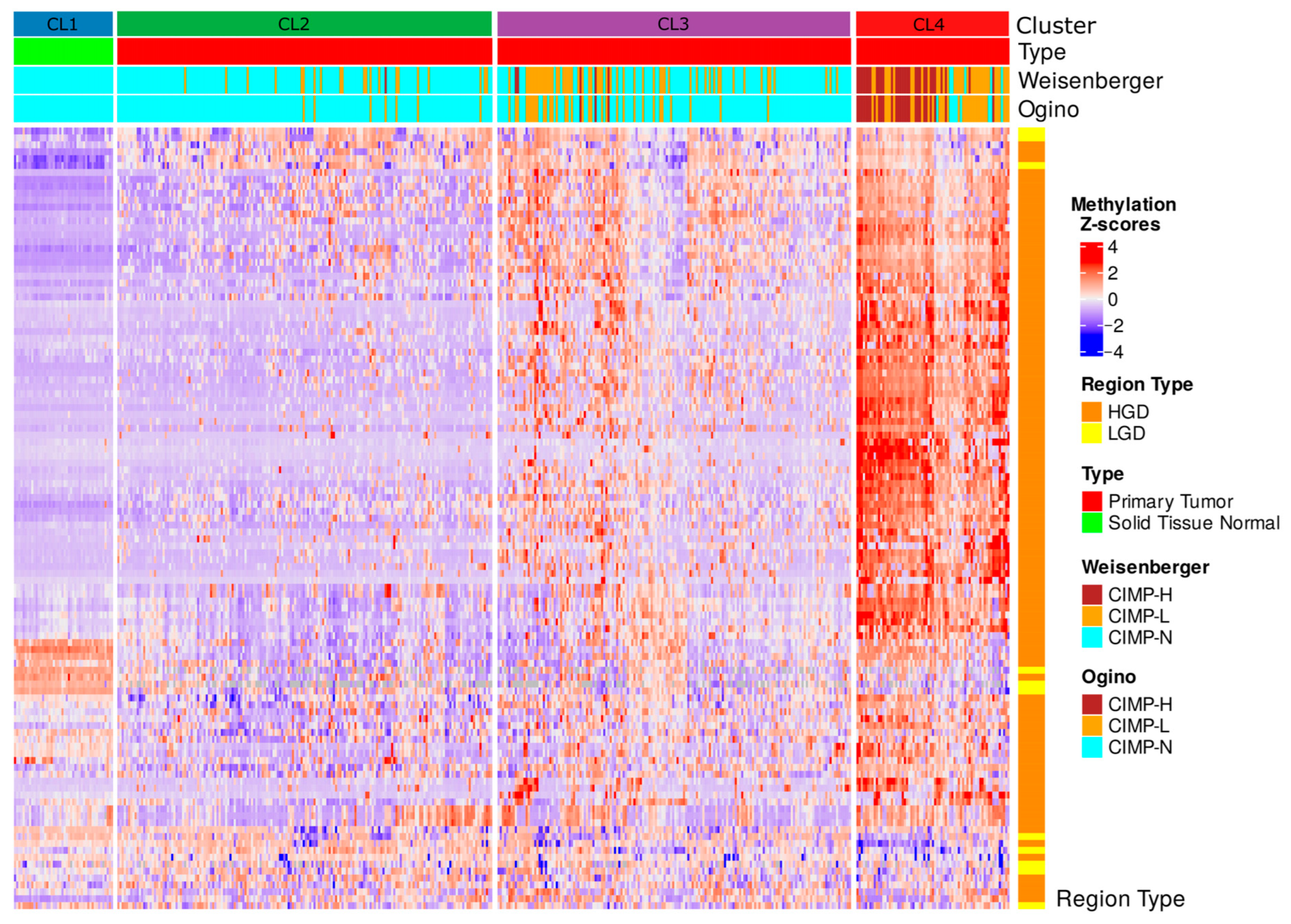
2.3. Methylation-Based Tumor Stratification and Clinical Implications
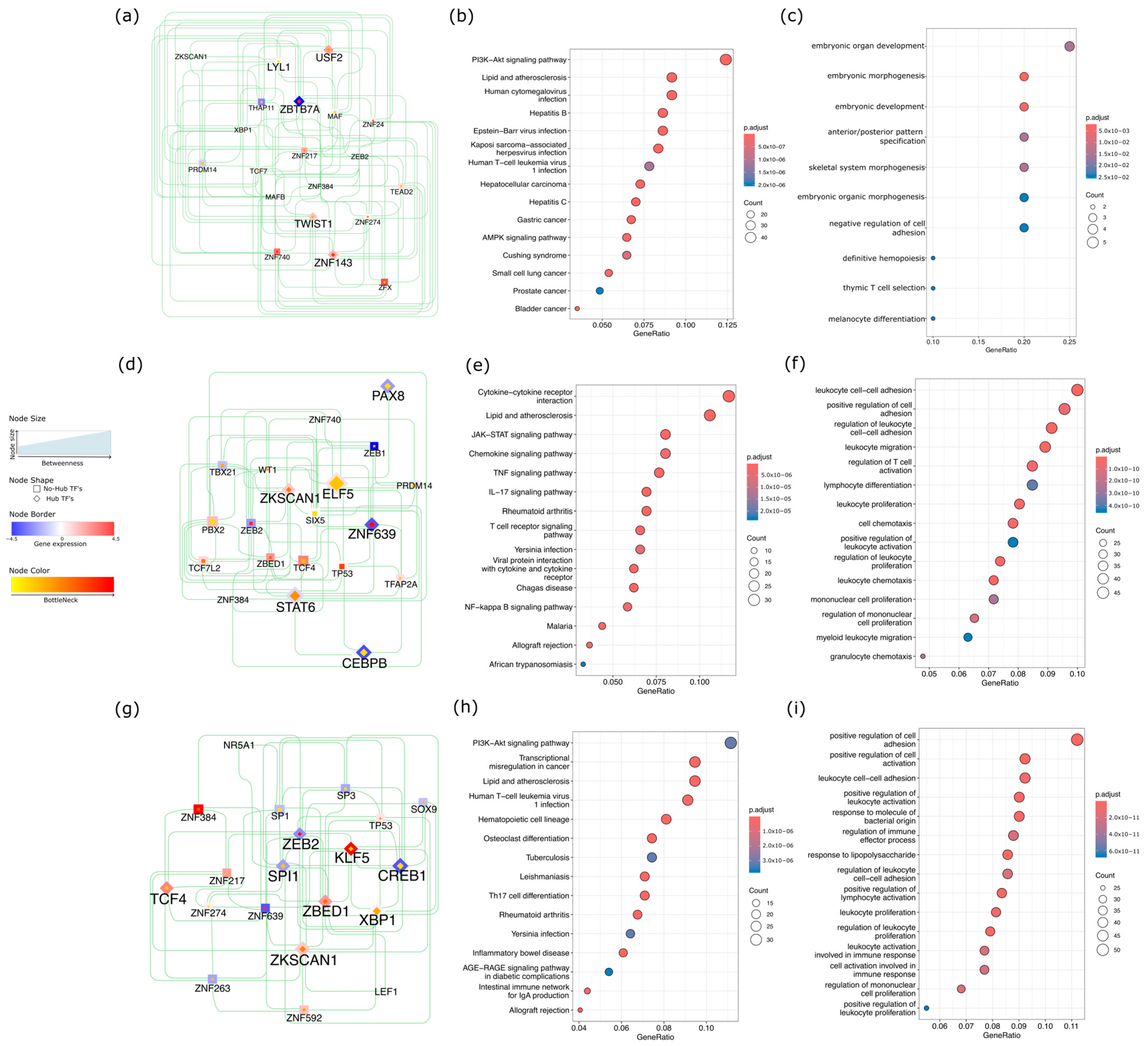
2.4. Network-Level Insights into Transcription Factors Highlight Oncogenic and Immune Divergence Across CRC Subtypes
2.5. CIMP Stratification Reveals Epigenetic Convergence with DMS-Based Clusters
3. Discussion
3.1. Early Epigenetic Dysregulation in Adenomatous Lesions
3.2. cfDNA Reflects Tumor-Specific Methylation Changes
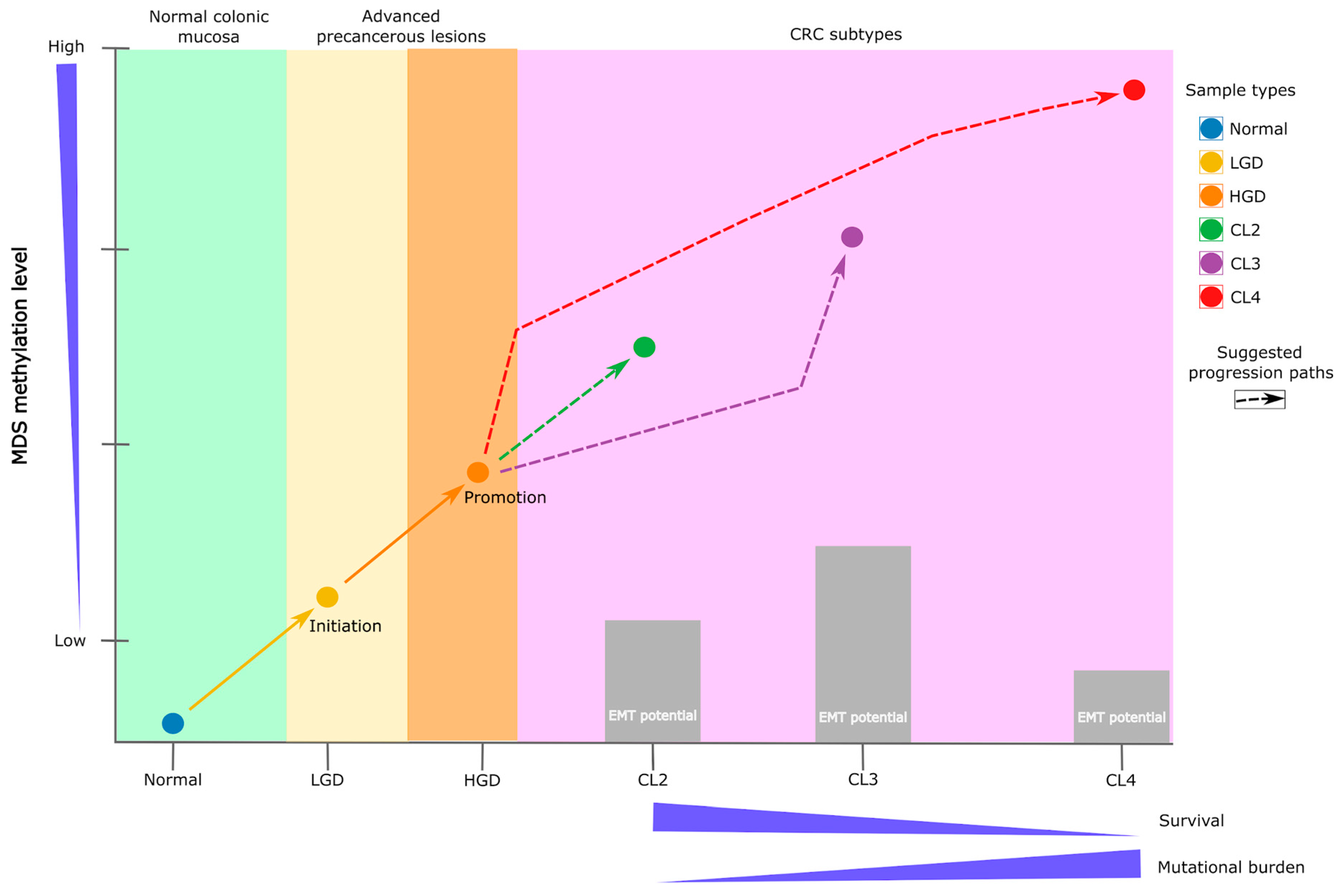
3.3. DMS Reveals a Continuum and a Branching of Epigenetic States
3.4. Transcription Factor Network Rewiring
3.5. DMS vs. Classical CIMP Classification
3.6. Translational Outlook and Study Limitations
4. Materials and Methods
4.1. Sample Preparation and Whole-Genome EM-seq
4.2. Sequence QC and DNA Methylation Analysis
4.3. Integration of TCGA Multimodal Data and Analysis
4.4. Clinical Analysis and Epithelial–Mesenchymal Transition Expression Signature (EMTes)
4.5. Transcription Factor Network and Functional Enrichment Analysis
4.6. CIMP Phenotype Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| HGD | High-grade dysplasia |
| LGD | Low-grade dysplasia |
| DMS | Differential methylation signature |
| TCGA | The Cancer Genome Atlas |
| cfDNA | Cell free DNA |
| EM-seq | Enzymatic methyl sequencing |
| TF | Transcription factor |
| EMT | Epithelial–mesenchymal transition |
| CIMP | CpG island methylator phenotype |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; Arends, M.J. Molecular Pathological Classification of Colorectal Cancer—An Update. Virchows Arch. 2024, 484, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Hajebi Khaniki, S.; Shokoohi, F.; Esmaily, H.; Kerachian, M.A. Analyzing Aberrant DNA Methylation in Colorectal Cancer Uncovered Intangible Heterogeneity of Gene Effects in the Survival Time of Patients. Sci. Rep. 2023, 13, 22104. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Wankhede, D.; Edelmann, D.; Kather, J.N.; Tagscherer, K.E.; Roth, W.; Bewerunge-Hudler, M.; Brobeil, A.; Kloor, M.; Bläker, H.; et al. Large-Scale External Validation and Meta-Analysis of Gene Methylation Biomarkers in Tumor Tissue for Colorectal Cancer Prognosis. eBioMedicine 2024, 105, 105223. [Google Scholar] [CrossRef] [PubMed]
- Galuppini, F.; Fassan, M.; Mastracci, L.; Gafà, R.; Lo Mele, M.; Lazzi, S.; Remo, A.; Parente, P.; D’Amuri, A.; Mescoli, C.; et al. The Histomorphological and Molecular Landscape of Colorectal Adenomas and Serrated Lesions. Pathologica 2021, 113, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of Colorectal Cancer: Biomarker and Therapeutic Potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, J.; Guo, S.; Tao, C.; Zhang, H.; Wang, W.; Zhang, Y.; Zhang, D.; Ding, S.; Zeng, C. Genome-Wide DNA Methylation Profiles of Low- and High-Grade Adenoma Reveals Potential Biomarkers for Early Detection of Colorectal Carcinoma. Clin. Epigenet. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, T.-T.; An, Z.-T.; Wei, L.-F.; Gu, C.; Li, H.; Tian, Y.-Z. CCDC68 Predicts Poor Prognosis in Patients with Colorectal Cancer: A Study Based on TCGA Data. J. Gastrointest. Oncol. 2022, 13, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhuo, C.; Lin, R.; Ke, F.; Wang, M.; Yang, C. Identification and Verification of Key Genes in Colorectal Cancer Liver Metastases Through Analysis of Single-Cell Sequencing Data and TCGA Data. Ann. Surg. Oncol. 2024, 31, 8664–8679. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Li, X.; Su, X.; Wen, H.; Chen, T.; Wu, H.; Liu, M. The Role of Multiple Metabolic Genes in Predicting the Overall Survival of Colorectal Cancer: A Study Based on TCGA and GEO Databases. PLoS ONE 2021, 16, e0251323. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium; Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; Ahn, K.S.; Ahn, S.-M.; Aikata, H.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Shibue, T.; Dempster, J.M.; Hahn, W.C.; Vazquez, F. The Present and Future of the Cancer Dependency Map. Nat. Rev. Cancer 2025, 25, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-T.; Shah, R.H.; Tegay, D.; Onel, K. Precision Oncology: Lessons Learned and Challenges for the Future. Cancer Manag. Res. 2019, 11, 7525–7536. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive Review of Targeted Therapy for Colorectal Cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yu, X.; Weisenberger, D.J.; Lu, T.; Liang, G. A Multi-Omics Overview of Colorectal Cancer to Address Mechanisms of Disease, Metastasis, Patient Disparities and Outcomes. Cancers 2023, 15, 2934. [Google Scholar] [CrossRef] [PubMed]
- Nazemalhosseini Mojarad, E.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Tao, K. Somatic Mutations in Colorectal Cancer Are Associated with the Epigenetic Modifications. J. Cell Mol. Med. 2020, 24, 11828–11836. [Google Scholar] [CrossRef] [PubMed]
- Bagci, B.; Sari, M.; Karadayi, K.; Turan, M.; Ozdemir, O.; Bagci, G. KRAS, BRAF Oncogene Mutations and Tissue Specific Promoter Hypermethylation of Tumor Suppressor SFRP2, DAPK1, MGMT, HIC1 and P16 Genes in Colorectal Cancer Patients. Cancer Biomark. 2016, 17, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Dai, W.; Wang, H.; Lan, X.; Ma, C.; Su, Z.; Xiang, W.; Han, L.; Luo, W.; Zhang, L.; et al. Early Detection and Prognosis Prediction for Colorectal Cancer by Circulating Tumour DNA Methylation Haplotypes: A Multicentre Cohort Study. eClinicalMedicine 2022, 55, 101717. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Tsui, S.K.-W.; Wu, H.; Wei, Y. Pan-Cancer Analysis of Differential DNA Methylation Patterns. BMC Med. Genom. 2020, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Guan, X.; Cui, L.; Ruan, Y.; Chen, Z.; Zou, H.; Lan, Y.; Liu, C.; Zhang, Y. Epigenetics and Immunotherapy in Colorectal Cancer: Progress and Promise. Clin. Epigenet. 2024, 16, 123. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological Classification of Pancreatic Ductal Adenocarcinoma That Predicts Molecular Subtypes and Correlates with Clinical Outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The Genome-Wide Mutational Consequences of DNA Hypomethylation. Sci. Rep. 2023, 13, 6874. [Google Scholar] [CrossRef] [PubMed]
- Edrei, Y.; Levy, R.; Kaye, D.; Marom, A.; Radlwimmer, B.; Hellman, A. Methylation-Directed Regulatory Networks Determine Enhancing and Silencing of Mutation Disease Driver Genes and Explain Inter-Patient Expression Variation. Genome Biol. 2023, 24, 264. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, P.D.; Chatterji, U. Transcriptional Co-Activators: Emerging Roles in Signaling Pathways and Potential Therapeutic Targets for Diseases. Signal Transduct. Target. Ther. 2023, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing Multi-Omics Data within and across 32 Cancer Types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shi, J.; Luo, J.; Zhai, H.; Liu, X.; Wu, Z.; Yan, C.; Luo, H. Deep Learning Approach for Cancer Subtype Classification Using High-Dimensional Gene Expression Data. BMC Bioinform. 2022, 23, 430. [Google Scholar] [CrossRef] [PubMed]
- Gohari, K.; Kazemnejad, A.; Mohammadi, M.; Eskandari, F.; Saberi, S.; Esmaieli, M.; Sheidaei, A. A Bayesian Latent Class Extension of Naive Bayesian Classifier and Its Application to the Classification of Gastric Cancer Patients. BMC Med. Res. Methodol. 2023, 23, 190. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Shrestha, S.; Draghici, S.; Nguyen, T. PINSPlus: A Tool for Tumor Subtype Discovery in Integrated Genomic Data. Bioinformatics 2019, 35, 2843–2846. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, D.; Lal, A.; Wang, B.; Batzoglou, S.; Sidow, A. Multi-Omic Tumor Data Reveal Diversity of Molecular Mechanisms That Correlate with Survival. Nat. Commun. 2018, 9, 4453. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, R.; Li, D.; Wang, Z. Subtype-GAN: A Deep Learning Approach for Integrative Cancer Subtyping of Multi-Omics Data. Bioinformatics 2021, 37, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Shakyawar, S.K.; Sajja, B.R.; Patel, J.C.; Guda, C. iCluF: An Unsupervised Iterative Cluster-Fusion Method for Patient Stratification Using Multiomics Data. Bioinform. Adv. 2024, 4, vbae015. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, K.; Lingasamy, P.; Khatun, M.; Sudhakaran, S.L.; Salumets, A.; Sundararajan, V.; Modhukur, V. Artificial Intelligence in Cancer Epigenomics: A Review on Advances in Pan-Cancer Detection and Precision Medicine. Epigenet. Chromatin 2025, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.R.; Nevins, S.A.; Chen, D.C.; Chiu, R.; Horning, A.M.; Guha, T.K.; Laquindanum, R.; Mills, M.; Chaib, H.; Ladabaum, U.; et al. Single-Cell Analyses Define a Continuum of Cell State and Composition Changes in the Malignant Transformation of Polyps to Colorectal Cancer. Nat. Genet. 2022, 54, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Mwambi, H.; Mboya, I.B.; Elbashir, M.K.; Omolo, B. A Stacking Ensemble Deep Learning Approach to Cancer Type Classification Based on TCGA Data. Sci. Rep. 2021, 11, 15626. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.A.; Tamada, Y.; Tanaka, Y.; Ikeguchi, M.; Higashihara, K.; Okuno, Y. Novel Cancer Subtyping Method Based on Patient-Specific Gene Regulatory Network. Sci. Rep. 2021, 11, 23653. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Wang, H.; Han, J.; An, J.; Yang, Z.; Mo, X. Multiomics Integration and Machine Learning Reveal Prognostic Programmed Cell Death Signatures in Gastric Cancer. Sci. Rep. 2024, 14, 31060. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wong, C.-J.; Kaz, A.M.; Dzieciatkowski, S.; Carter, K.T.; Morris, S.M.; Wang, J.; Willis, J.E.; Makar, K.W.; Ulrich, C.M.; et al. Differences in DNA Methylation Signatures Reveal Multiple Pathways of Progression from Adenoma to Colorectal Cancer. Gastroenterology 2014, 147, 418–429.e8. [Google Scholar] [CrossRef] [PubMed]
- Durso, D.F.; Bacalini, M.G.; do Valle, Í.F.; Pirazzini, C.; Bonafé, M.; Castellani, G.; Faria, A.M.C.; Franceschi, C.; Garagnani, P.; Nardini, C. Aberrant Methylation Patterns in Colorectal Cancer: A Meta-Analysis. Oncotarget 2017, 8, 12820–12830. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Yuan, Z.; Hu, H.; Zhang, W.; Wang, G.; Wang, X. Genome-Wide Discovery of Circulating Cell-Free DNA Methylation Biomarkers for Colorectal Cancer Detection. Clin. Epigenet. 2023, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-J.; Shin, S.H.; Kim, H.H.; Min, N.Y.; Lim, Y.; Joo, T.; Lee, K.J.; Jeong, M.-S.; Kim, H.; Yun, S.; et al. Advances in Methylation Analysis of Liquid Biopsy in Early Cancer Detection of Colorectal and Lung Cancer. Sci. Rep. 2023, 13, 13502. [Google Scholar] [CrossRef] [PubMed]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised Colon Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, S.; Pan, P.; Xia, T.; Chang, X.; Yang, X.; Guo, L.; Meng, Q.; Yang, F.; Qian, W.; et al. Magnitude, Risk Factors, and Factors Associated with Adenoma Miss Rate of Tandem Colonoscopy: A Systematic Review and Meta-Analysis. Gastroenterology 2019, 156, 1661–1674.e11. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Wooldrage, K.; Robbins, E.C.; Kralj-Hans, I.; MacRae, E.; Piggott, C.; Stenson, I.; Prendergast, A.; Patel, B.; Pack, K.; et al. Faecal Immunochemical Tests (FIT) versus Colonoscopy for Surveillance after Screening and Polypectomy: A Diagnostic Accuracy and Cost-Effectiveness Study. Gut 2019, 68, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Lonardi, S.; Pietrantonio, F.; Tarazona Llavero, N.; Montagut Viladot, C.; Sartore Bianchi, A.; Zampino, M.G.; Elez Fernandez, M.E.; Santos Vivas, C.; Mandalà, M.; Tamberi, S.; et al. LBA28 The PEGASUS Trial: Post-Surgical Liquid Biopsy-Guided Treatment of Stage III and High-Risk Stage II Colon Cancer Patients. Ann. Oncol. 2023, 34, S1268–S1269. [Google Scholar] [CrossRef]
- Slater, S.; Bryant, A.; Aresu, M.; Begum, R.; Chen, H.-C.; Peckitt, C.; Lazaro-Alcausi, R.; Carter, P.; Anandappa, G.; Khakoo, S.; et al. Tissue-Free Liquid Biopsies Combining Genomic and Methylation Signals for Minimal Residual Disease Detection in Patients with Early Colorectal Cancer from the UK TRACC Part B Study. Clin. Cancer Res. 2024, 30, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A Colorectal Cancer Classification System That Associates Cellular Phenotype and Responses to Therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic Modulators, Modifiers and Mediators in Cancer Aetiology and Progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Panatta, E.; Butera, A.; Mammarella, E.; Pitolli, C.; Mauriello, A.; Leist, M.; Knight, R.A.; Melino, G.; Amelio, I. Metabolic Regulation by P53 Prevents R-Loop-Associated Genomic Instability. Cell Rep. 2022, 41, 111568. [Google Scholar] [CrossRef] [PubMed]
- Tew, B.Y.; Durand, J.K.; Bryant, K.L.; Hayes, T.K.; Peng, S.; Tran, N.L.; Gooden, G.C.; Buckley, D.N.; Der, C.J.; Baldwin, A.S.; et al. Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines. Sci. Rep. 2020, 10, 10149. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A. Epigenetic Plasticity, Selection, and Tumorigenesis. Biochem. Soc. Trans. 2020, 48, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A.; et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell 2018, 34, 982–995.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential Pre-Malignant Programs and Microenvironment Chart Distinct Paths to Malignancy in Human Colorectal Polyps. Cell 2021, 184, 6262–6280.e26. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, Y.R.; Krishna, V.; Powers, R.; Parmar, S.; Thursby, S.-J.; Gupta, E.; Kulak, O.; Gokare, P.; Reumers, J.; Van Wesenbeeck, L.; et al. Transcription Factor Expression Repertoire Basis for Epigenetic and Transcriptional Subtypes of Colorectal Cancers. Proc. Natl. Acad. Sci. USA 2023, 120, e2301536120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, B.; Shen, K.; Zhang, H.; Gong, Z.; Shi, H.; Jiang, Y. Identification of Transcription Factor/Gene Axis in Colon Cancer Using a Methylome Approach. Front. Genet. 2020, 11, 864. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-L.; Lin, X.; Yu, Y.-L.; Chen, L.; Hu, Q.-X.; Chen, M.; Cao, N.; Zhao, C.; Wang, C.-Y.; Huang, C.-W.; et al. Genome-Wide Profiling in Colorectal Cancer Identifies PHF19 and TBC1D16 as Oncogenic Super Enhancers. Nat. Commun. 2021, 12, 6470. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2023. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2024).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Catoni, M.; Tsang, J.M.; Greco, A.P.; Zabet, N.R. DMRcaller: A Versatile R/Bioconductor Package for Detection and Visualization of Differentially Methylated Regions in CpG and Non-CpG Contexts. Nucleic Acids Res. 2018, 46, e114. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J.; Cancer Genome Atlas Network. The Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New Functionalities in the TCGAbiolinks Package for the Study and Integration of Cancer Data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “Ggplot2”. 2024. Available online: https://rpkgs.datanovia.com/survminer/ (accessed on 1 September 2024).
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and Integration of Resources for the Estimation of Human Transcription Factor Activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional Characterization of Somatic Mutations in Cancer Using Network-Based Inference of Protein Activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- del Toro, N.; Shrivastava, A.; Ragueneau, E.; Meldal, B.; Combe, C.; Barrera, E.; Perfetto, L.; How, K.; Ratan, P.; Shirodkar, G.; et al. The IntAct Database: Efficient Access to Fine-Grained Molecular Interaction Data. Nucleic Acids Res. 2022, 50, D648–D653. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the Molecular Interaction Database: 2012 Update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Lesurf, R.; Cotto, K.C.; Wang, G.; Griffith, M.; Kasaian, K.; Jones, S.J.M.; Montgomery, S.B.; Griffith, O.L. ORegAnno 3.0: A Community-Driven Resource for Curated Regulatory Annotation. Nucleic Acids Res. 2016, 44, D126–D132. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, M.; Krallinger, M.; Leitner, F.; Kuiper, M.; Valencia, A.; Laegreid, A. ExTRI: Extraction of Transcription Regulation Interactions from Literature. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2022, 1865, 194778. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to Characterize Multiomics Data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG Island Methylator Phenotype Underlies Sporadic Microsatellite Instability and Is Tightly Associated with BRAF Mutation in Colorectal Cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of Markers for CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer by a Large Population-Based Sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | n | Age ± SD | Sex Distribution | Age Range |
|---|---|---|---|---|
| HGD | 9 | 67 ± 8 | F 11%, M 88.9% | 53–77 |
| LGD | 6 | 72.8 ± 9 | F 66.7%, M 33.7% | 59–82 |
| Diagnosis | n | Age ± SD | Sex Distribution | Age Range |
|---|---|---|---|---|
| HGD | 4 | 63 ± 10.3 | F 25%, M 75% | 51–73 |
| LGD | 26 | 62.2 ± 6.5 | F 53.8%, M 46.2% | 51–74 |
| Control | 28 | 62.3 ± 12.2 | F 53.6%, M 46.2% | 30–84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higareda-Almaraz, J.C.; Mancuso, F.M.; Canal-Noguer, P.; Kruusmaa, K.; Bertossi, A. Multiomics Signature Reveals Network Regulatory Mechanisms in a CRC Continuum. Int. J. Mol. Sci. 2025, 26, 7077. https://doi.org/10.3390/ijms26157077
Higareda-Almaraz JC, Mancuso FM, Canal-Noguer P, Kruusmaa K, Bertossi A. Multiomics Signature Reveals Network Regulatory Mechanisms in a CRC Continuum. International Journal of Molecular Sciences. 2025; 26(15):7077. https://doi.org/10.3390/ijms26157077
Chicago/Turabian StyleHigareda-Almaraz, Juan Carlos, Francesco Mattia Mancuso, Pol Canal-Noguer, Kristi Kruusmaa, and Arianna Bertossi. 2025. "Multiomics Signature Reveals Network Regulatory Mechanisms in a CRC Continuum" International Journal of Molecular Sciences 26, no. 15: 7077. https://doi.org/10.3390/ijms26157077
APA StyleHigareda-Almaraz, J. C., Mancuso, F. M., Canal-Noguer, P., Kruusmaa, K., & Bertossi, A. (2025). Multiomics Signature Reveals Network Regulatory Mechanisms in a CRC Continuum. International Journal of Molecular Sciences, 26(15), 7077. https://doi.org/10.3390/ijms26157077