
Amphiphilic Oligonucleotide Derivatives as a Tool to Study DNA Repair Proteins
 , , , , , ,
, , , , , , 
Abstract
1. Introduction
2. Results and Discussion
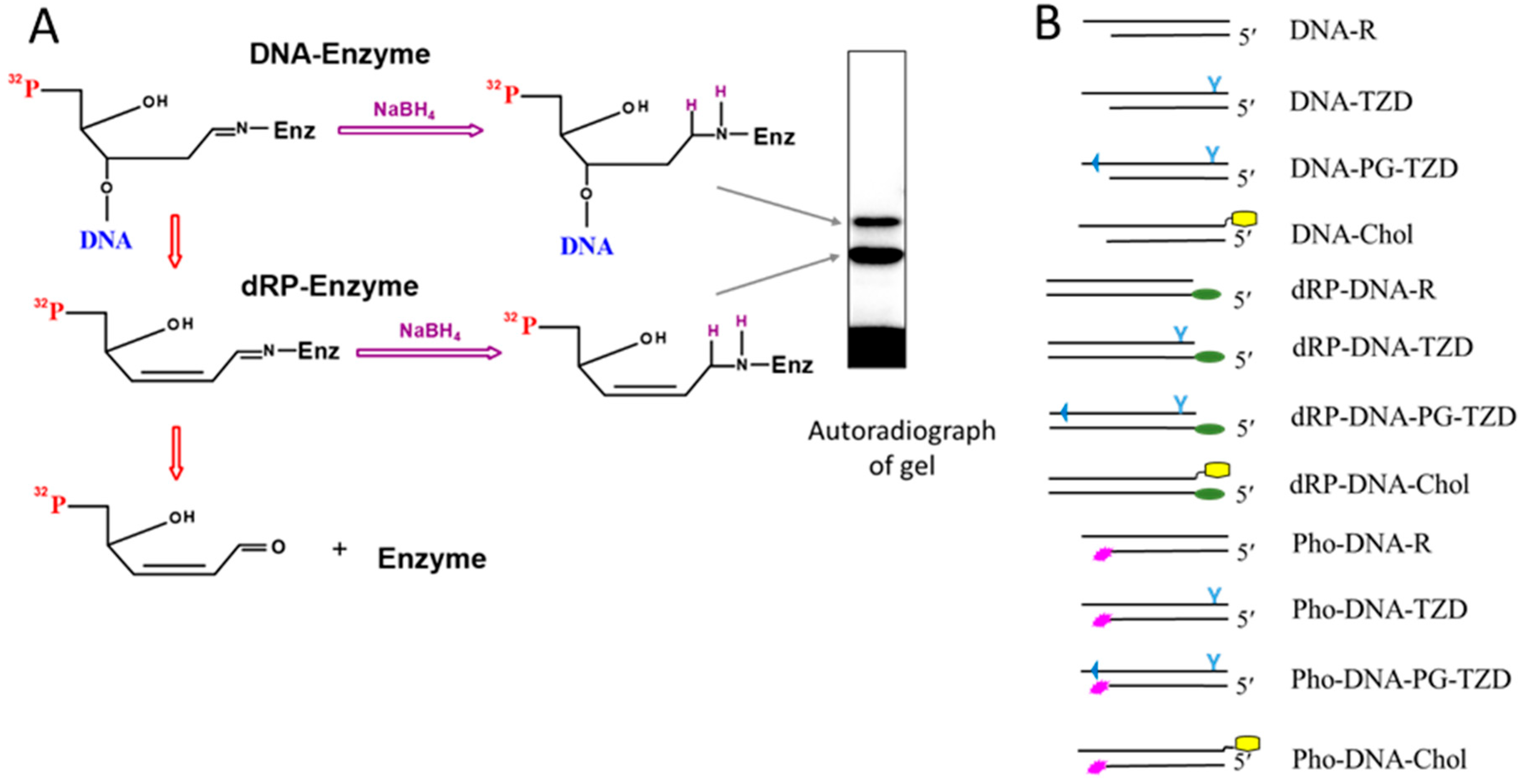
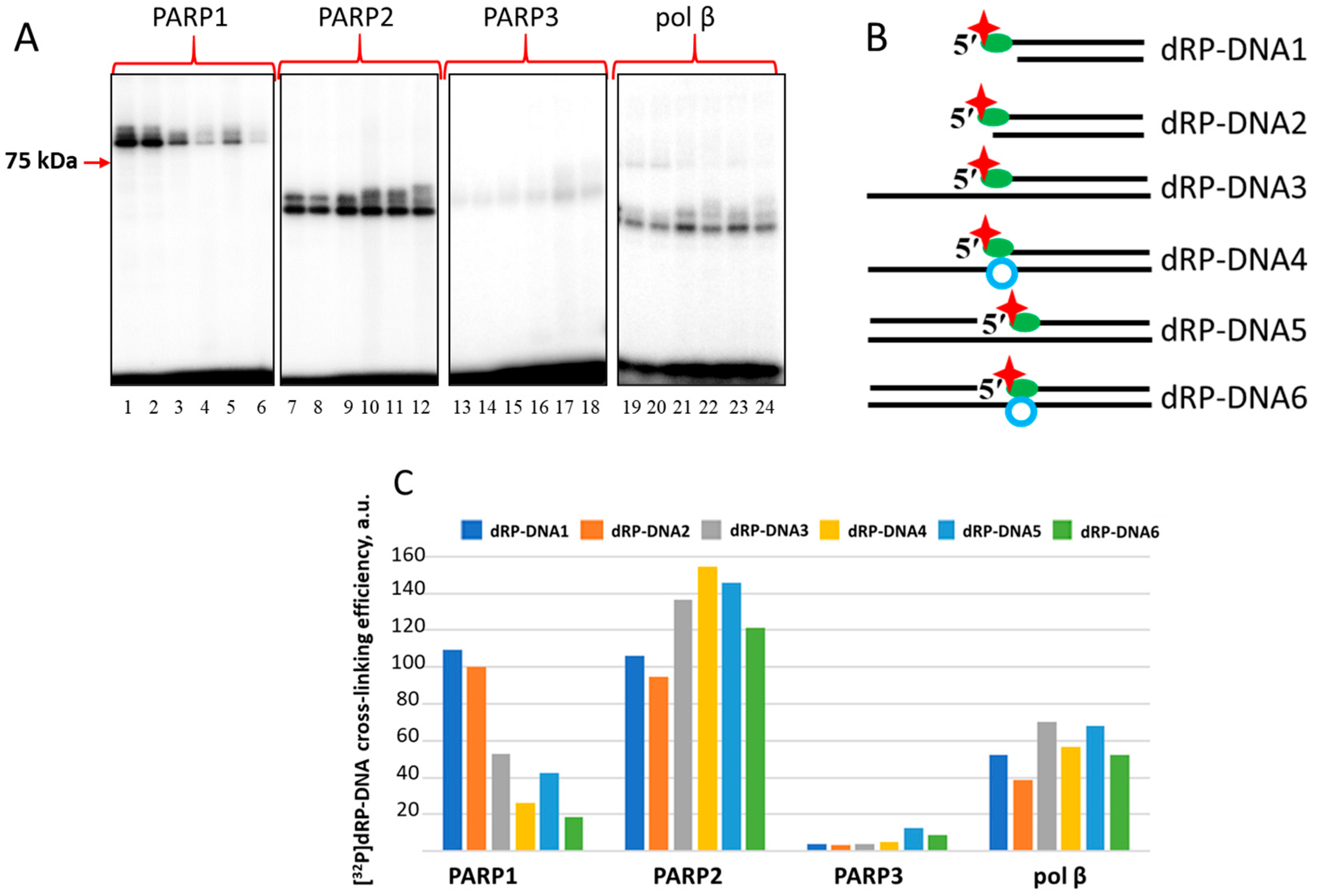
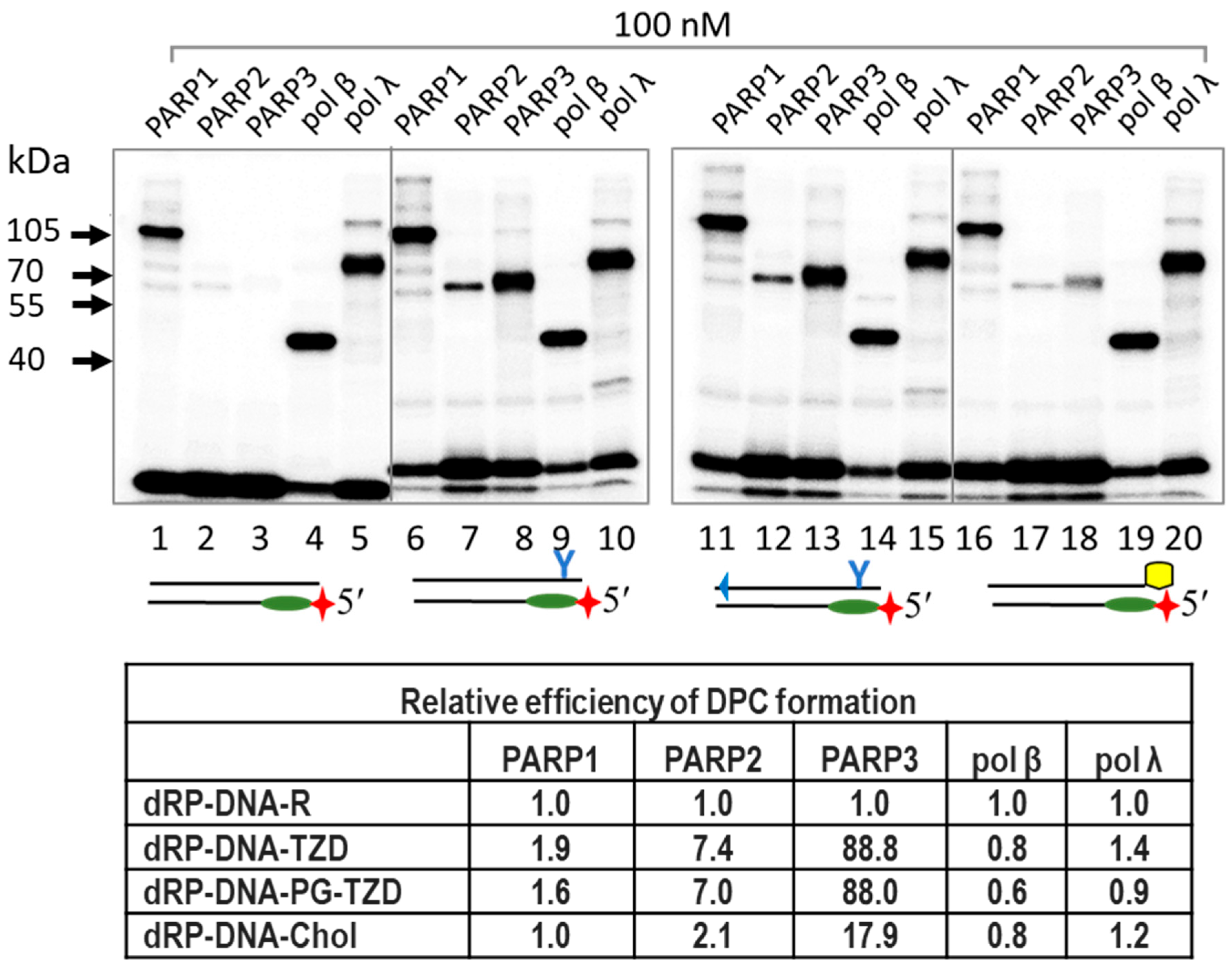
2.1. Borohydride Trapping of PARPs
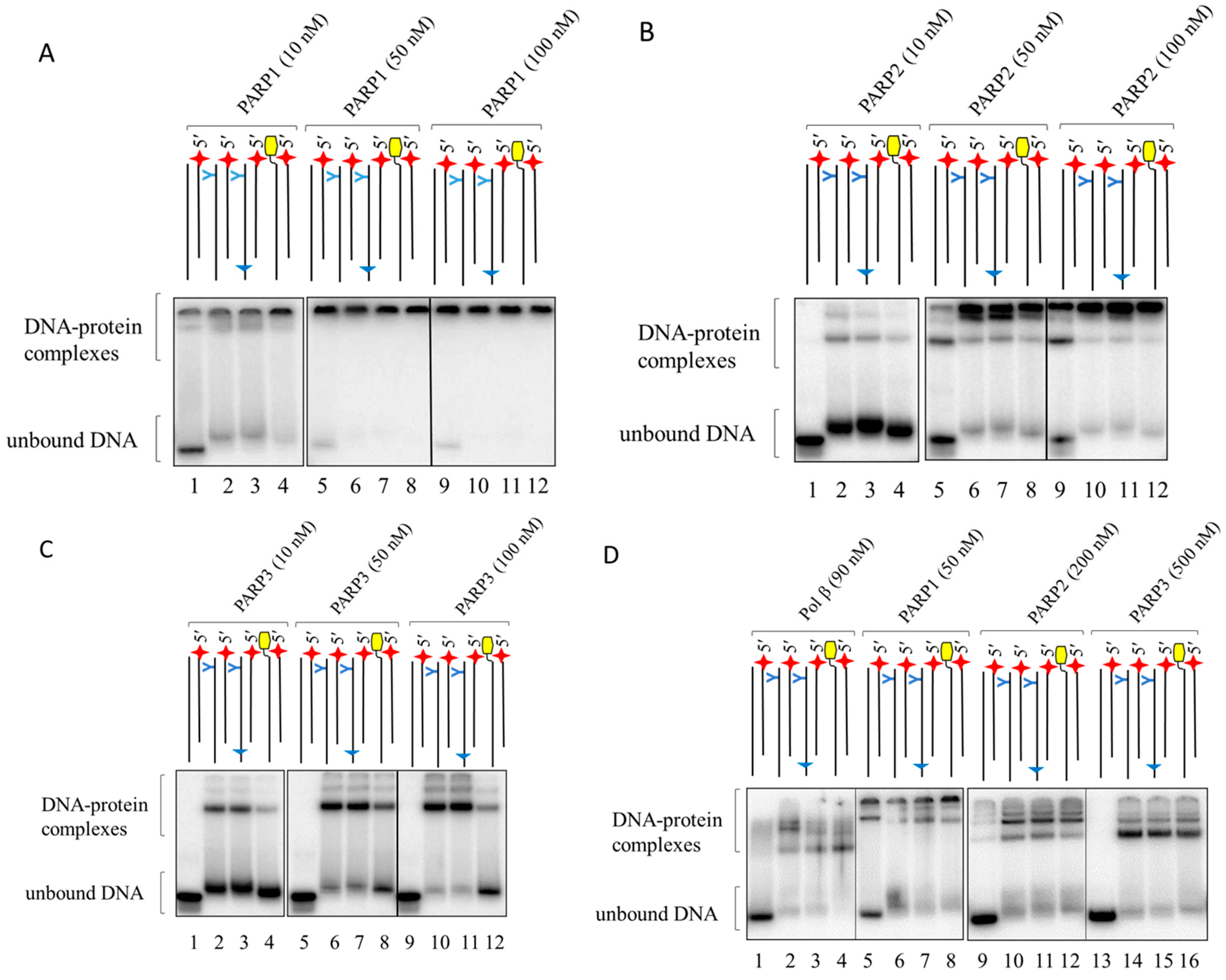
2.2. PARP Binding with LS-DNAs as Revealed by EMSA
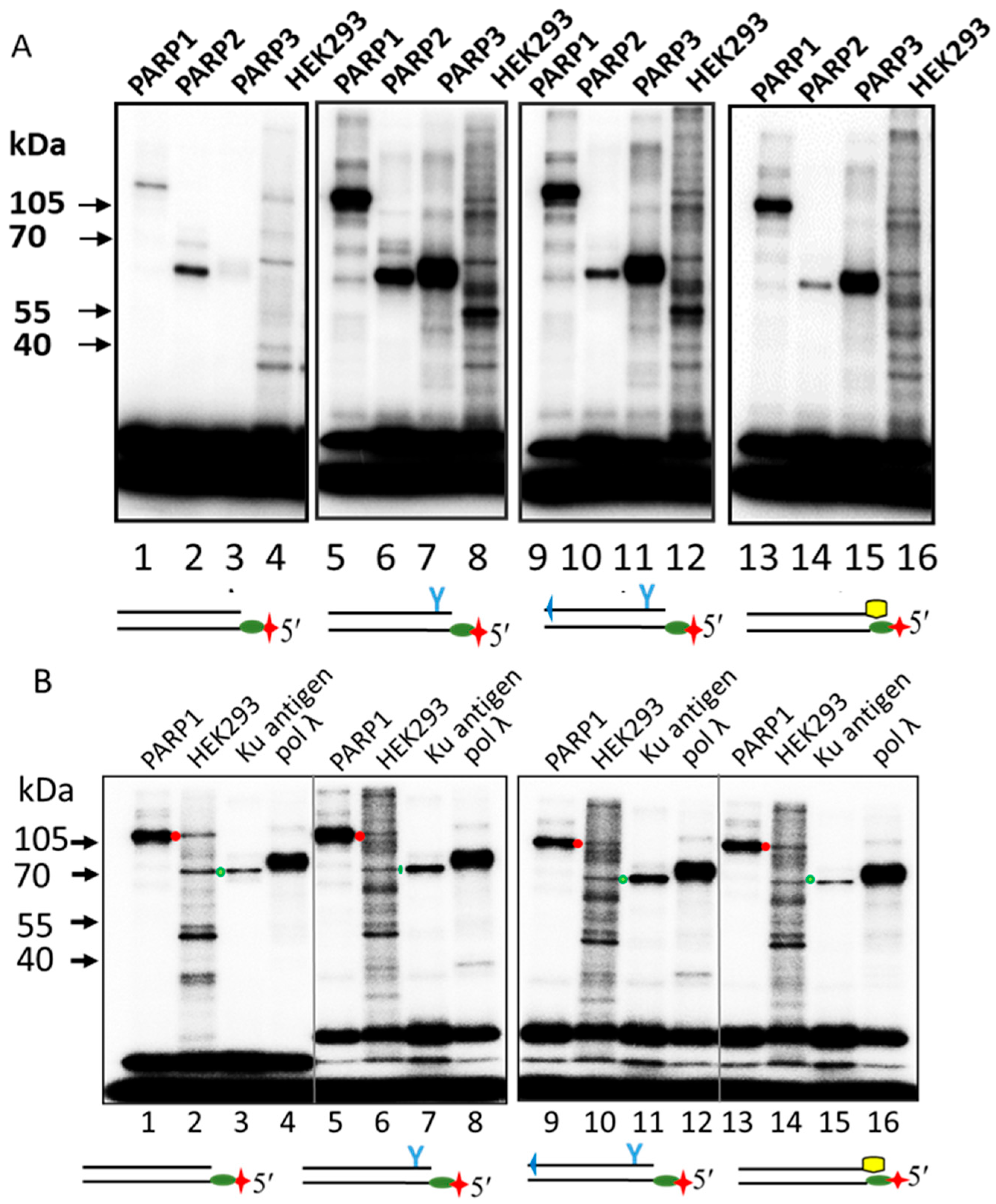
2.3. Probing of PARPs’ Interaction with Photoactivatable DNAs
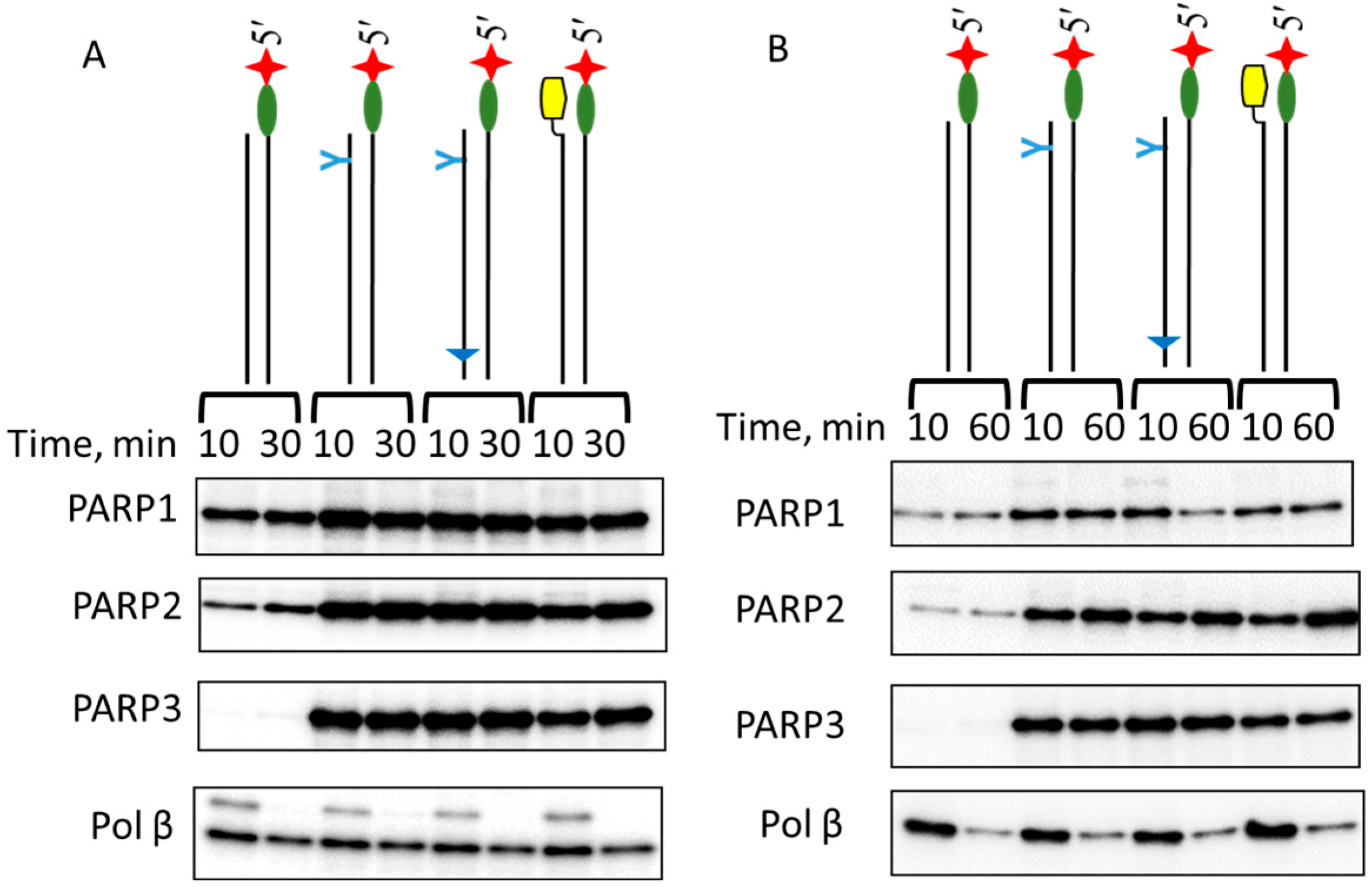
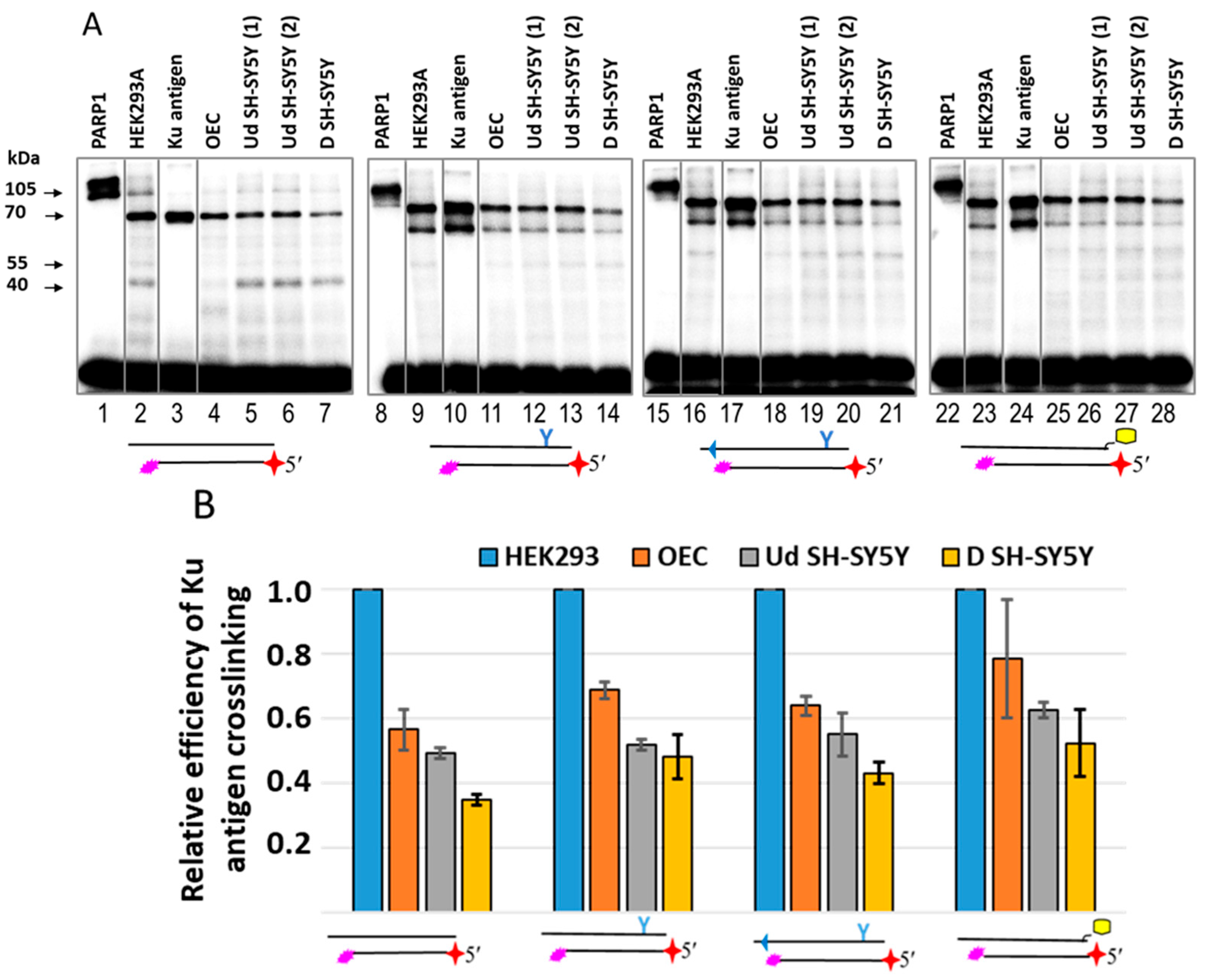
2.4. Probing of PARPs’ Interaction with 5′dRP DNAs
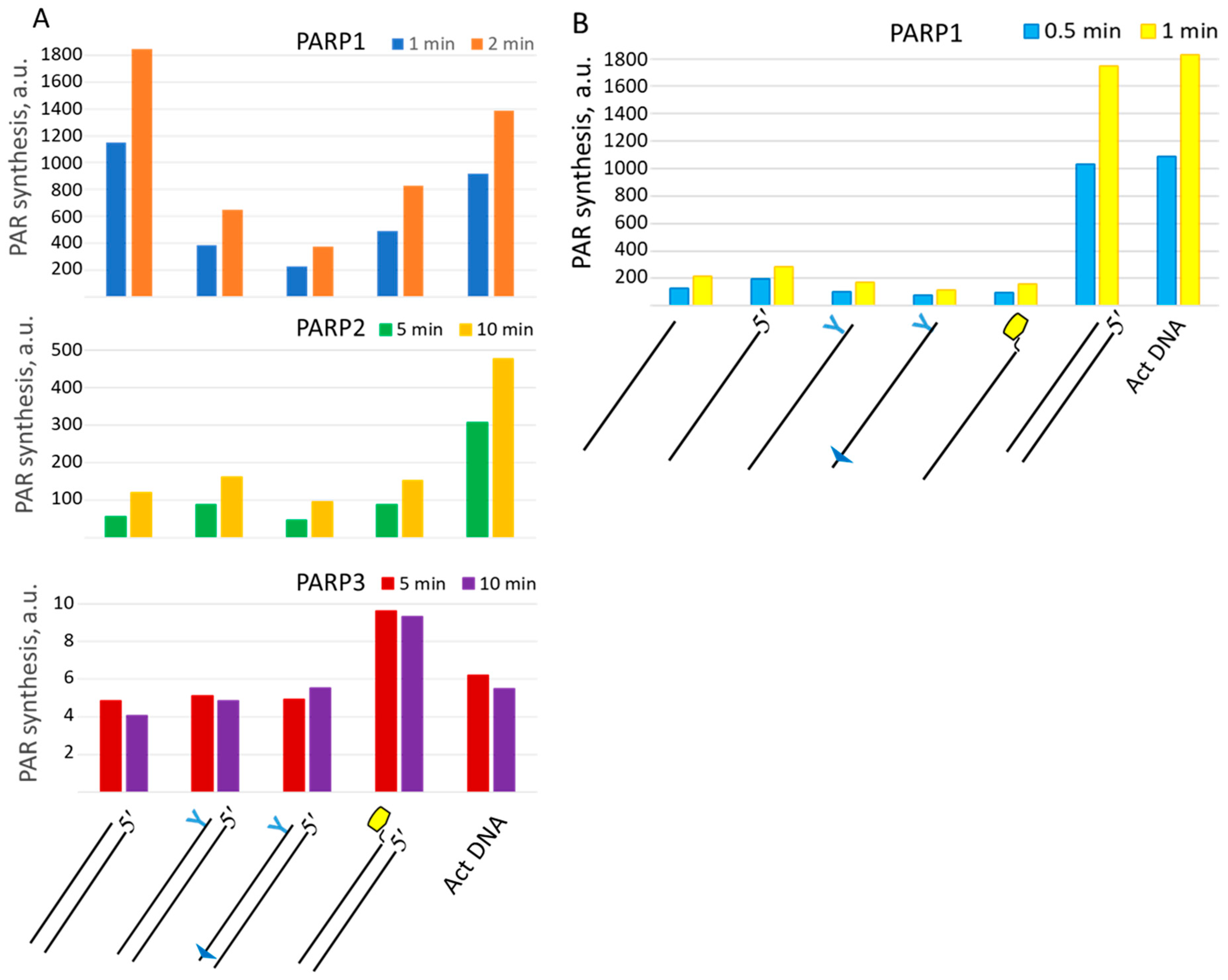
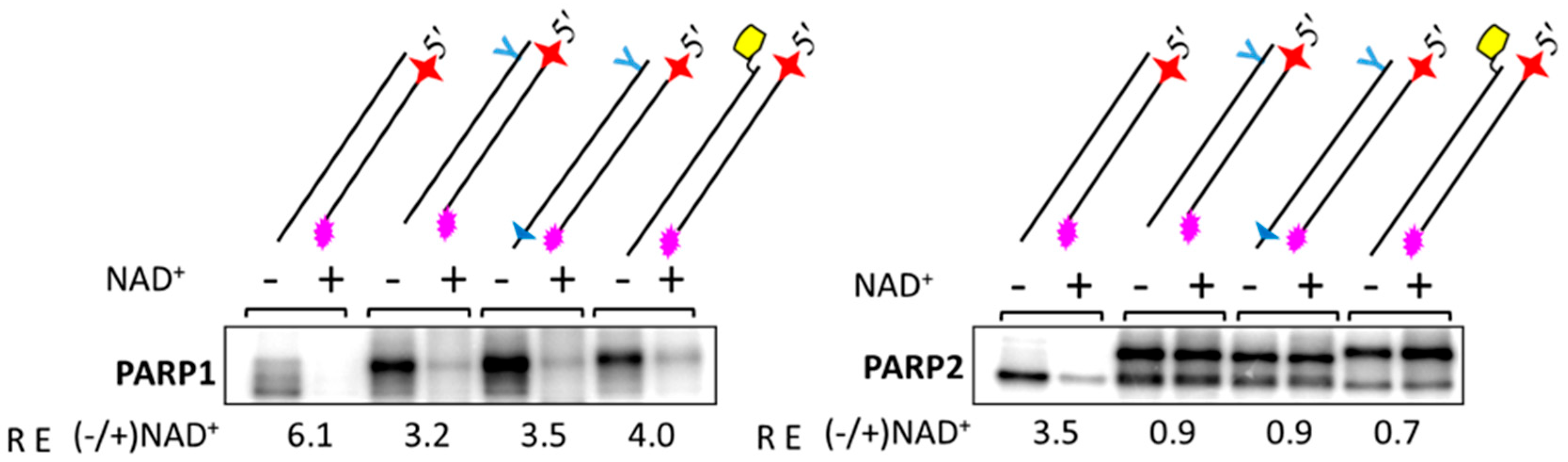
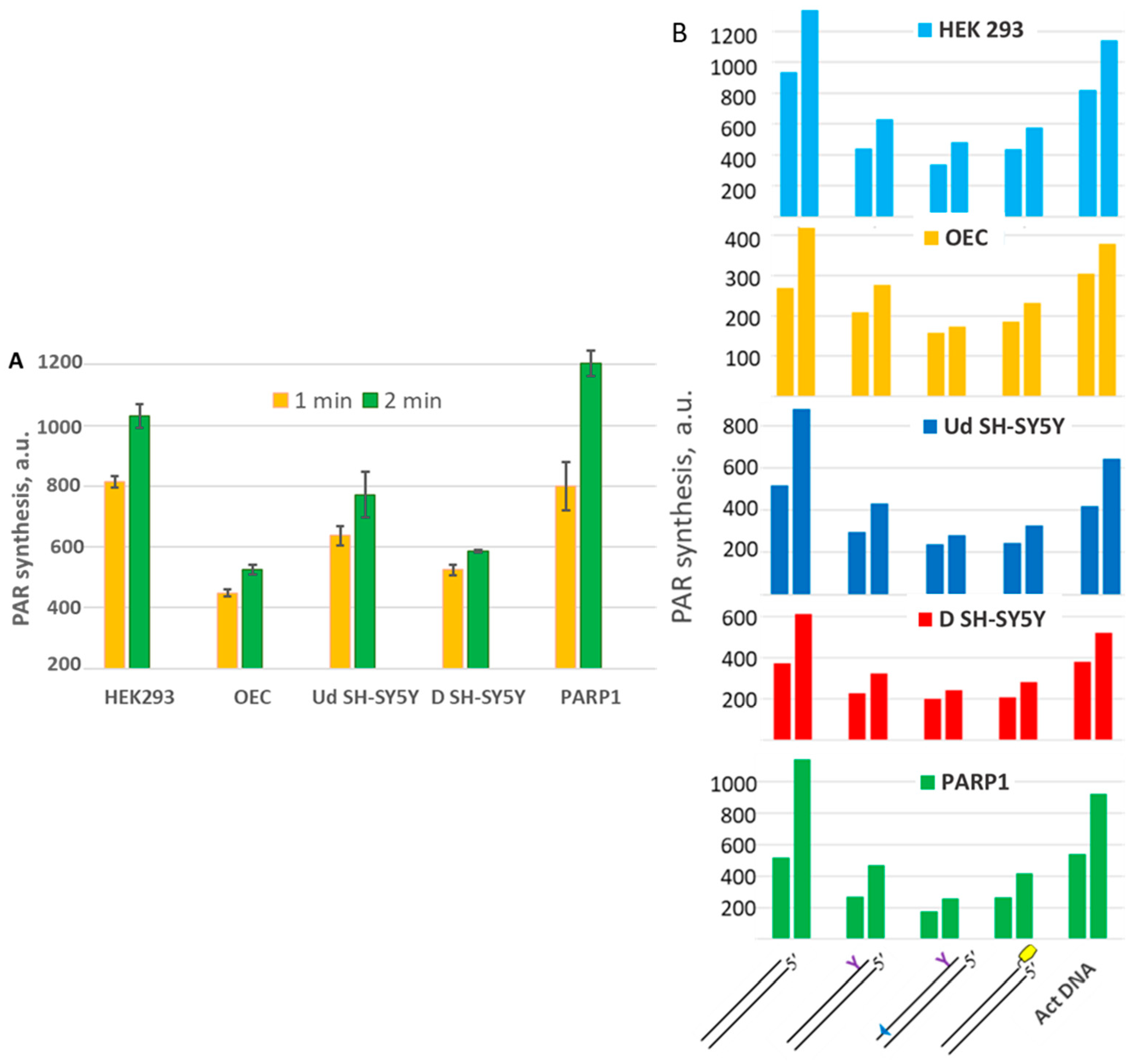
2.5. Probing of PARP Activation by LS DNAs
2.6. Probing of Chemically Reactive DNA Interaction with Proteins in WCEs of HEK293, SH-SY5Y, and Olfactory Epithelial Cells
2.7. Probing of Activation Properties of DNAs in WCEs
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis of [32P]NAD+
3.2.2. Preparation of DNA Substrates Containing 5′dRP Residue
3.2.3. Activating Properties of LS-DNA in the PAR Synthesis
3.2.4. Electrophoretic Mobility Shift Assay (EMSA)
3.2.5. Photoaffinity Modification of Proteins
3.2.6. Covalent Cross-Linking of Proteins to 5′dRP DNA
3.2.7. 5′dRP Lyase Activity Assay
3.2.8. Cell Growth and Differentiation
3.2.9. Oligonucleotide Synthesis
3.2.10. Oligonucleotide Purification and Identification
3.2.11. Quantification of the Results of Autoradiography
4. Conclusions
- (a)
- Increased efficiency in interacting with target proteins;
- (b)
- Poor PARP1 activation, which leads to its persistence on these DNAs rather than binding to damaged cellular DNA;
- (c)
- (d)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NHEJ | nonhomologous end joining |
| PAAGE | polyacrylamide gel electrophoresis |
| FAPdCMP | Exo-N-{2-[N-(4-azido-2,5-difluoro-3-chloropyridine-6-yl)-3-aminopropionyl]-aminoethyl}-2-deoxycytdine-5′-monophosphate |
| 5′dRP | 5′-deoxyribosephosphate |
| PARP1 | poly(ADP)ribose polymerase 1 |
| PARP2 | poly(ADP)ribose polymerase 2 |
| PARP3 | poly(ADP)ribose polymerase 3 |
| PAR | poly(ADP)ribose |
| PARylation | poly(ADP-ribosyl)ation |
| Pol β | DNA polymerase β |
| Pol λ | DNA polymerase λ |
| PG | phosphoryl guanidine |
| TZD | triazinylphosphoramidate group functionalised with two dodecyl substituents |
| OEC | human olfactory epithelium cells |
References
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Y.; Zhang, Y.; Hu, D.; Tang, L.; Zhou, B.; Yang, L. Landscape of small nucleic acid therapeutics: Moving from the bench to the clinic as next-generation medicines. Signal Transduct. Target Ther. 2025, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, S.; Milam, V.T. Modified nucleic acids: Expanding the capabilities of functional oligonucleotides. Molecules 2020, 25, 4659. [Google Scholar] [CrossRef] [PubMed]
- Berthault, N.; Bergam, P.; Pereira, F.; Girard, P.M.; Dutreix, M. Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA. Cells 2022, 11, 2149. [Google Scholar] [CrossRef] [PubMed]
- Sesink, A.; Becerra, M.; Ruan, J.L.; Leboucher, S.; Dubail, M.; Heinrich, S.; Jdey, W.; Petersson, K.; Fouillade, C.; Berthault, N.; et al. The AsiDNA™ decoy mimicking DSBs protects the normal tissue from radiation toxicity through a DNA-PK/p53/p21-dependent G1/S arrest. NAR Cancer 2024, 6, zcae011. [Google Scholar] [CrossRef] [PubMed]
- Kupryushkin, M.S.; Zharkov, T.D.; Ilina, E.S.; Markov, O.V.; Kochetkova, A.S.; Akhmetova, M.M.; Khodyreva, S.N. Triazinylamidophosphate Oligonucleotides: Synthesis and Study of Their Interaction with Cells and DNA-Binding Proteins. Russ. J. Bioorganic Chem. 2021, 47, 719–733. [Google Scholar] [CrossRef]
- Markov, O.V.; Filatov, A.V.; Kupryushkin, M.S.; Chernikov, I.V.; Patutina, O.A.; Strunov, A.A.; Chernolovskaya, E.L.; Vlassov, V.V.; Pyshnyi, D.V.; Zenkova, M.A. Transport Oligonucleotides—A Novel System for Intracellular Delivery of Antisense Therapeutics. Molecules 2020, 25, 3663. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Ilina, E.; Zharkov, T.; Grigorieva, E.; Chinak, O.; Kupryushkin, M.; Golyshev, V.; Mitin, D.; Chubarov, A.; Khodyreva, S.; et al. Self-Penetrating Oligonucleotide Derivatives: Features of Self-Assembly and Interactions with Serum and Intracellular Proteins. Pharmaceutics 2023, 15, 2779. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.A.; Dmitrienko, E.V. Amphiphilic Oligonucleotide Derivatives-Promising Tools for Therapeutics. Pharmaceutics 2024, 16, 1447. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Chassoux, D.; Berthault, N.; Agrario, C.; Sun, J.S.; Dutreix, M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS ONE 2009, 4, e6298. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; McClorey, G.; Shimizu, M.; Kothari, N.; Alam, R.; Iwamoto, N.; Kumarasamy, J.; Bommineni, G.R.; Bezigian, A.; Chivatakarn, O.; et al. Control of backbone chemistry and chirality boost oligonucleotide splice switching activity. Nucleic Acids Res. 2022, 50, 5443–5466. [Google Scholar] [CrossRef] [PubMed]
- Khodyreva, S.; Lavrik, O. Non-canonical interaction of DNA repair proteins with intact and cleaved AP sites. DNA Repair 2020, 90, 102847. [Google Scholar] [CrossRef] [PubMed]
- Dezhurov, S.V.; Khodyreva, S.N.; Plekhanova, E.S.; Lavrik, O.I. A new highly efficient photoreactive analogue of dCTP. Synthesis, characterization, and application in photoaffinity modification of DNA binding proteins. Bioconjug. Chem. 2005, 16, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Piersen, C.E.; McCullough, A.K.; Lloyd, R.S. AP lyases and dRPases: Commonality of mechanism. Mutat. Res. 2000, 459, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pinz, K.G.; Bogenhagen, D.F. Characterization of a catalytically slow AP lyase activity in DNA polymerase gamma and other family A DNA polymerases. J. Biol. Chem. 2000, 275, 12509–12514. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Reed, A.J.; Zahurancik, W.J.; Daskalova, S.M.; Hecht, S.M.; Suo, Z. Interlocking activities of DNA polymerase β in the base excision repair pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2118940119. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.S.; Lavrik, O.I.; Khodyreva, S.N. Ku antigen interacts with abasic sites. Biochim. Biophys. Acta 2008, 1784, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Prakash, L.; Prakash, S. A mechanism for the exclusion of low-fidelity human Y-family DNA polymerases from base excision repair. Genes Dev. 2003, 17, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Poltoratsky, V.; Hou, E.W.; Wilson, S.H. Rev1 is a base excision repair enzyme with 5′-deoxyribose phosphate lyase activity. Nucleic Acids Res. 2016, 44, 10824–10833. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Wong, R.S.; McKnight, J.N.; Bowman, G.D.; Greenberg, M.M. Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Natl. Acad. Sci. USA 2010, 107, 22475–22480. [Google Scholar] [CrossRef] [PubMed]
- Khodyreva, S.N.; Prasad, R.; Ilina, E.S.; Sukhanova, M.V.; Kutuzov, M.M.; Liu, Y.; Hou, E.W.; Wilson, S.H.; Lavrik, O.I. Apurinic/apyrimidinic (AP) site recognition by the 50-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. USA 2010, 107, 22090–22095. [Google Scholar] [CrossRef] [PubMed]
- Yudkina, A.V.; Zharkov, D.O. The hidden elephant: Modified abasic sites and their consequences. DNA Repair 2025, 148, 103823. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. DNA polymerase gamma in mitochondrial DNA replication and repair. Sci. World J. 2003, 3, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hussen, A.S.; Freudenthal, B.D.; Suo, Z.; Zhao, L. Mitochondrial transcription factor A (TFAM) has 5′-deoxyribose phosphate lyase activity in vitro. DNA Repair 2024, 137, 103666. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Longley, M.J.; Sharief, F.S.; Hou, E.W.; Copeland, W.C.; Wilson, S.H. Human DNA polymerase theta possesses 5′-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009, 37, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5ʹ-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.; Roberts, S.A.; Oh, S.; Hendrickson, E.A.; Ramsden, D.A. Specificity of the dRP/AP lyase of Ku promotes nonhomologous end joining (NHEJ) fidelity at damaged ends. J. Biol. Chem. 2012, 287, 13686–13693. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.T.; Carvajal-Garcia, J.; Hallett, R.A.; Waters, C.A.; Roberts, S.A.; Strom, C.; Kuhlman, B.; Ramsden, D.A. Requirements for 5′dRP/AP lyase activity in Ku. Nucleic Acids Res. 2014, 42, 11136–11143. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2022, 289, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Conceição, C.J.F.; Moe, E.; Ribeiro, P.A.; Raposo, M. PARP1: A comprehensive review of its mechanisms, therapeutic implications and emerging cancer treatments. Biochim. Biophys. Acta Rev. Cancer 2025, 1880, 189282. [Google Scholar] [CrossRef] [PubMed]
- Feltes, B.C.; Alvares, L.O. PARP1 in the intersection of different DNA repair pathways, memory formation, and sleep pressure in neurons. J. Neurochem. 2024, 168, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Seif El Dahan, M.; Iehl, F.; Fernandez-Varela, P.; Le, D.M.H.; Ropars, V.; Charbonnier, J.B. The Multifaceted Roles of Ku70/80. Int. J. Mol. Sci. 2021, 22, 4134. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Roy, S.; Kamyab, J.; Danzter, F.; Franco, S. Common and unique genetic interactions of the poly(ADP-ribose) polymerases PARP1 and PARP2 with DNA double-strand break repair pathways. DNA Repair 2016, 45, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.A.W.; Molloy, J.E. Single-molecule force spectroscopy reveals binding and bridging dynamics of PARP1 and PARP2 at DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2023, 120, e2214209120. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Luedeman, M.E.; Stroik, S.; Feng, W.; Luthman, A.J.; Gupta, G.P.; Ramsden, D.A. Poly(ADP) ribose polymerase promotes DNA polymerase theta-mediated end joining by activation of end resection. Nat. Commun. 2022, 13, 4547. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Boehler, C.; Guirouilh Barbat, J.; Bonnet, M.E.; Illuzzi, G.; Ronde, P.; Gauthier, L.R.; Magroun, N.; Rajendran, A.; Lopez, B.S.; et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014, 42, 5616–5632. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Fisher, A.E.; Robert, I.; Zuma, M.C.; Rouleau, M.; Ju, L.; Poirier, G.; Reina-San-Martin, B.; Caldecott, K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell 2011, 411, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Khodyreva, S.N.; Ilina, E.S.; Sukhanova, M.V.; Amé, J.C.; Lavrik, O.I. Interaction of PARP-2 with AP site containing DNA. Biochimie 2015, 112, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Allinson, S.L.; Dianova, I.I.; Dianov, G.L. DNA polymerase beta is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. EMBO J. 2001, 20, 6919–6926. [Google Scholar] [CrossRef] [PubMed]
- Laverty, D.J.; Greenberg, M.M. Expanded Substrate Scope of DNA Polymerase θ and DNA Polymerase β: Lyase Activity on 5′-Overhangs and Clustered Lesions. Biochemistry 2018, 57, 6119–6127. [Google Scholar] [CrossRef] [PubMed]
- Vekariya, U.; Toma, M.; Nieborowska-Skorska, M.; Le, B.V.; Caron, M.C.; Kukuyan, A.M.; Sullivan-Reed, K.; Podszywalow-Bartnicka, P.; Chitrala, K.N.; Atkins, J.; et al. DNA polymerase θ protects leukemia cells from metabolically induced DNA damage. Blood 2023, 141, 2372–2389. [Google Scholar] [PubMed]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Zahn, K.E.; Jensen, R.B. Polymerase θ Coordinates Multiple Intrinsic Enzymatic Activities during DNA Repair. Genes 2021, 12, 1310. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 108, 53193. [Google Scholar]
- Gordon, J.; Amini, S. General Overview of Neuronal Cell Culture. In Neuronal Cell Culture. Methods in Molecular Biology; Amini, S., White, M.K., Eds.; Humana: New York, NY, USA, 2021; Volume 2311. [Google Scholar]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Lavrik, O.I. Protein-Protein Interactions in DNA Base Excision Repair. Biochemistry 2018, 83, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Vasil’eva, I.; Moor, N.; Anarbaev, R.; Kutuzov, M.; Lavrik, O. Functional Roles of PARP2 in Assembling Protein-Protein Complexes Involved in Base Excision DNA Repair. Int. J. Mol. Sci. 2021, 22, 4679. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- García-Díaz, M.; Bebenek, K.; Kunkel, T.A.; Blanco, L. Identification of an intrinsic 5’-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: A possible role in base excision repair. J. Biol. Chem. 2001, 276, 34659–34663. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Amin, H.; Zahid, S.; Barboule, N.; Hall, C.; Matharu, G.; Hardwick, S.W.; Chauvat, J.; Britton, S.; Chirgadze, D.Y.; et al. Structural and functional insights into the interaction between Ku70/80 and Pol X family polymerases in NHEJ. Nat. Commun. 2025, 16, 4208. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. Pol X DNA polymerases contribute to NHEJ flexibility. Nat. Struct. Mol. Biol. 2023, 30, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Maldonado, D.; Drogalis Beckham, L.; Wood, R.D.; Doublié, S. When DNA Polymerases Multitask: Functions Beyond Nucleotidyl Transfer. Front. Mol. Biosci. 2022, 8, 815845. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat. Res. 2010, 685, 80–99. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Khodyreva, S.N.; Amé, J.C.; Ilina, E.S.; Sukhanova, M.V.; Schreiber, V.; Lavrik, O.I. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie 2013, 95, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.S.; Khodyreva, S.N.; Lavrik, O.I. Unusual interaction of human apurinic/apyrimidinic endonuclease 1 (APE1) with abasic sites via the Schiff-base-dependent mechanism. Biochimie 2018, 150, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.S.; Khodyreva, S.N.; Berezhnoy, A.E.; Larin, S.S.; Lavrik, O.I. Tracking Ku antigen levels in cell extracts with DNA containing abasic sites. Mutat Res. 2010, 685, 90–96. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, I.; Pelletier, J.D.; Lagueux, J.; D’Amours, D.; Chaudhry, M.A.; Weinfeld, M.; Lees-Miller, S.P.; Poirier, G.G. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1999, 1430, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Lilyestrom, W.; Woerd, M.J.v.d.; Clark, N.; Luger, K. Structural and biophysical studies of human PARP-1 in complex with damaged DNA. J. Mol. Biol. 2010, 395, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, T.; Wennink, T.; Lebbink, J.H.G.; Keijzers, G.; Florea, B.I.; Verkaik, N.S.; van Gent, D.C. C-Terminal Extensions of Ku70 and Ku80 Differentially Influence DNA End Binding Properties. Int. J. Mol. Sci. 2020, 21, 6725. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Tucker, P.W.; Capra, J.D. Production and characterization of recombinant human Ku antigen. Nucleic Acids Res. 1994, 22, 3918–3924. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed]
- Golovin, A.; Dzarieva, F.; Rubetskaya, K.; Shamadykova, D.; Usachev, D.; Pavlova, G.; Kopylov, A. In Silico Born Designed Anti-EGFR Aptamer Gol1 Has Anti-Proliferative Potential for Patient Glioblastoma Cells. Int. J. Mol. Sci. 2025, 26, 1072. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase. Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.C.; Kalisch, T.; Dantzer, F.; Schreiber, V. Purification of Recombinant Poly(ADP-Ribose) Polymerases. Methods Mol. Biol. 2011, 780, 135–152. [Google Scholar] [PubMed]
- Belousova, E.A.; Ishchenko, A.A.; Lavrik, O.I. DNA is a New Target of PARP3. Sci. Rep. 2018, 8, 4176. [Google Scholar] [CrossRef] [PubMed]
- Biade, S.; Sobol, R.W.; Wilson, S.H.; Matsumoto, Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 1998, 273, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.A. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, G.V.; Zverkov, Y.B.; Shram, S.I.; Lazurkina, T.Y.; Myasoedov, N.F. Chemical and enzymatic synthesis of tritium labelled coenzymes. J. Label. Comp. Radiopharm. 2003, 46, 465–473. [Google Scholar] [CrossRef]
- Popova, D.; Karlsson, J.; Jacobsson, S.O.P. Comparison of neurons derived from mouse P19, rat PC12 and human SH-SY5Y cells in the assessment of chemical- and toxin-induced neurotoxicity. BMC Pharmacol. Toxicol. 2017, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 6, pdb-prot5439. [Google Scholar] [CrossRef] [PubMed]
- Yasumitsu, H.; Ozeki, Y.; Kawsar, S.M.; Fujii, Y.; Sakagami, M.; Matuo, Y.; Toda, T.; Katsuno, H. RAMA stain: A fast, sensitive and less protein-modifying CBB R250 stain. Electrophoresis 2010, 31, 1913–1917. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligos | Sequence | DNA Name |
|---|---|---|
| O-R | 5′-TCCTGACATACTTGATACTTAGACATTCTT-3′ | |
| O-TZD | 5′-TCCTGACATACTTGATACTTAGACATTCT*T-3′ | |
| O-PG-TZD | 5′-TxCxCTGACATACTTGATACTTAGACATTCT*T-3′ | |
| O-Chol | 5′-TCCTGACATACTTGATACTTAGACATTCTT-[Chol]-3′ | |
| U-O1 | 5′-UAAGAATGTCTAAGTATCAAGTATGTCAGGA-3′ | |
| dRP-O1 | 5′-dRP-AAGAATGTCTAAGTATCAAGTATGTCAGGA-3′ | |
| O1-Pho | 5′-AAGAATGTCTAAGTATCAAGTATGT-3′ | |
| O1-Pho-FAPdCMP | 5′-AAGAATGTCTAAGTATCAAGTATGTCφ-3′ | |
| O-R/O-R1 | 5′-TCCTGACATACTTGATACTTAGACATTCTT-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-5′ | DNA-R |
| O-R/O-TZD | 5′-TCCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-5′ | DNA-TZD |
| O-R/O-PG-TZD | 5′-TxCxCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-5′ | DNA-PG-TZD |
| O-R/O-Chol | 5′-TCCTGACATACTTGATACTTAGACATTCTT-[Chol]-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-5′ | DNA-Chol |
| O-R/dRP-O1 | 5′-TCCTGACATACTTGATACTTAGACATTCTT-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-dRP-5′ | dRP-DNA-R |
| O-TZD/dRP-O1 | 5′-TCCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-dRP-5′ | dRP-DNA-TZD |
| O-PG-TZD/dRP-O1 | 5′-TxCxCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-dRP-5′ | dRP-DNA-PG-TZD |
| O-Chol/dRP-O1 | 5′-TCCTGACATACTTGATACTTAGACATTCTT-[Chol]-3′ 3′-AGGTCAACTGTATGAACTATGAATCTGTAAGAA-dRP-5′ | dRP-DNA-Chol |
| O-R/O1-Pho-FAPdCMP | 5′-TCCTGACATACTTGATACTTAGACATTCTT-3′ 3′-CφAACTGTATGAACTATGAATCTGTAAGAA-5′ | Pho-DNA-R |
| O-TZD/O1-Pho-FAPdCMP | 5′-TCCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-CφAACTGTATGAACTATGAATCTGTAAGAA-5′ | Pho-DNA-TZD |
| O-PG-TZD/O1-Pho-FAPdCMP | 5′-TxCxCTGACATACTTGATACTTAGACATTCT*T-3′ 3′-CφAACTGTATGAACTATGAATCTGTAAGAA-5′ | Pho-DNA-PG-TZD |
| O-Chol/O1-Pho-FAPdCMP | 5′-TCCTGACATACTTGATACTTAGACATTCTT-[Chol]-3 3′-CφAACTGTATGAACTATGAATCTGTAAGAA-5′ | Pho-DNA-Chol |
| ds, 16/15 | 5′-dRPCCCGGCTTAGTCGCC-3′ 3′-GGCCGAATCAGCGG-5′ | dRP-DNA1 |
| ds, 16/16 | 5′-dRPCCCGGCTTAGTCGCC-3′ 3′-GGGCCGAATCAGCGG-5′ | dRP-DNA2 |
| ext/G, 16/32 | 5′-dRPCCCGGCTTAGTCGCC-3′ 3′-CCCTCCGGGACCGCAAGGGGCCGAATCAGCGG-5′ | dRP-DNA3 |
| ext/AP site, 16/32 | 5′-dRPCCCGGCTTAGTCGCC-3′ 3′-CCCTCCGGGACCGCAAOGGGCCGAATCAGCGG-5′ | dRP-DNA4 |
| nick/G, 16/32 | 5′-GGGAGGCCCTGGCGTT dRPCCCGGCTTAGTCGCC-3′ 3′-CCCTCCGGGACCGCAA GGGGCCGAATCAGCGG-5′ | dRP-DNA5 |
| nick/AP site, 16/32 | 5′-GGGAGGCCCTGGCGTT dRPCCCGGCTTAGTCGCC-3′ 3′-CCCTCCGGGACCGCAAOGGGCCGAATCAGCGG-5′ | dRP-DNA6 |
| x = PG | 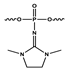 Phosphoryl guanidine modification | |
| * = TZD |  Triazinyl phosphoramidate modification with two dodecyl residues | |
| Chol = Cholesterol | 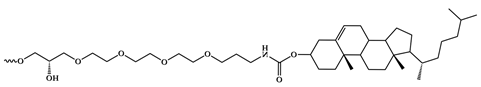 Cholesterol modification | |
| Cφ = FAPdCMP | Exo-N-{2-[N-(4-azido-2,5-difluoro-3-chloropyridine-6-yl)-3-aminopropionyl]aminoethyl}-2′-deoxycytidine-5′-monophosphate | |
| 5′-dRP | 5′-deoxyribosephosphate | |
| O | Apurinic/apyrimidinic site |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodyreva, S.N.; Yamskikh, A.A.; Ilina, E.S.; Kutuzov, M.M.; Belousova, E.A.; Kupryushkin, M.S.; Zharkov, T.D.; Koval, O.A.; Zvereva, S.P.; Lavrik, O.I. Amphiphilic Oligonucleotide Derivatives as a Tool to Study DNA Repair Proteins. Int. J. Mol. Sci. 2025, 26, 7078. https://doi.org/10.3390/ijms26157078
Khodyreva SN, Yamskikh AA, Ilina ES, Kutuzov MM, Belousova EA, Kupryushkin MS, Zharkov TD, Koval OA, Zvereva SP, Lavrik OI. Amphiphilic Oligonucleotide Derivatives as a Tool to Study DNA Repair Proteins. International Journal of Molecular Sciences. 2025; 26(15):7078. https://doi.org/10.3390/ijms26157078
Chicago/Turabian StyleKhodyreva, Svetlana N., Alexandra A. Yamskikh, Ekaterina S. Ilina, Mikhail M. Kutuzov, Ekaterina A. Belousova, Maxim S. Kupryushkin, Timofey D. Zharkov, Olga A. Koval, Sofia P. Zvereva, and Olga I. Lavrik. 2025. "Amphiphilic Oligonucleotide Derivatives as a Tool to Study DNA Repair Proteins" International Journal of Molecular Sciences 26, no. 15: 7078. https://doi.org/10.3390/ijms26157078
APA StyleKhodyreva, S. N., Yamskikh, A. A., Ilina, E. S., Kutuzov, M. M., Belousova, E. A., Kupryushkin, M. S., Zharkov, T. D., Koval, O. A., Zvereva, S. P., & Lavrik, O. I. (2025). Amphiphilic Oligonucleotide Derivatives as a Tool to Study DNA Repair Proteins. International Journal of Molecular Sciences, 26(15), 7078. https://doi.org/10.3390/ijms26157078