
Machine Learning-Based Prognostic Signature in Breast Cancer: Regulatory T Cells, Stemness, and Deep Learning for Synergistic Drug Discovery
 , , and
, , and
Abstract
1. Introduction
2. Results
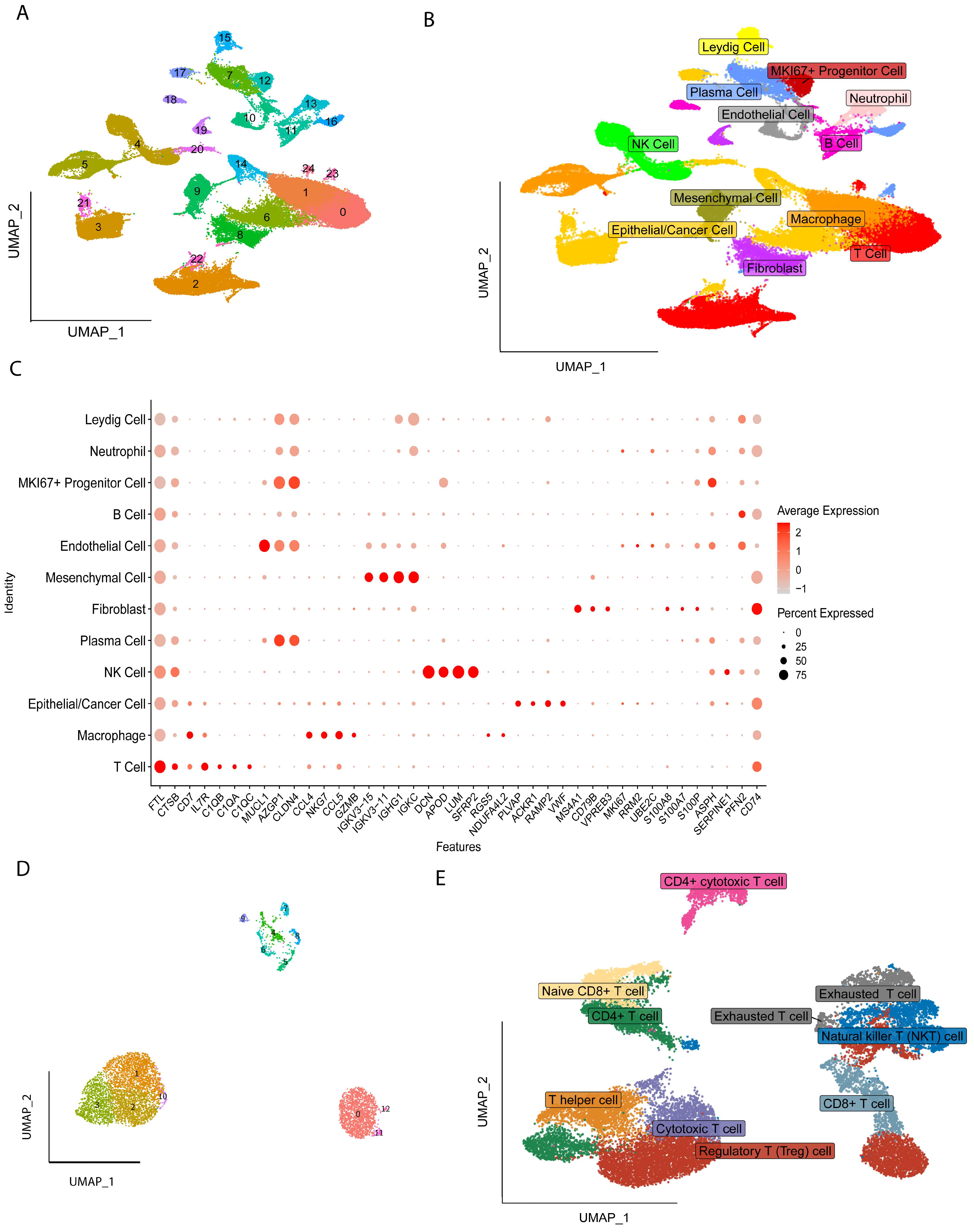
2.1. Identification of Cell Types
2.2. Different Differentiation Characteristics of Tregs
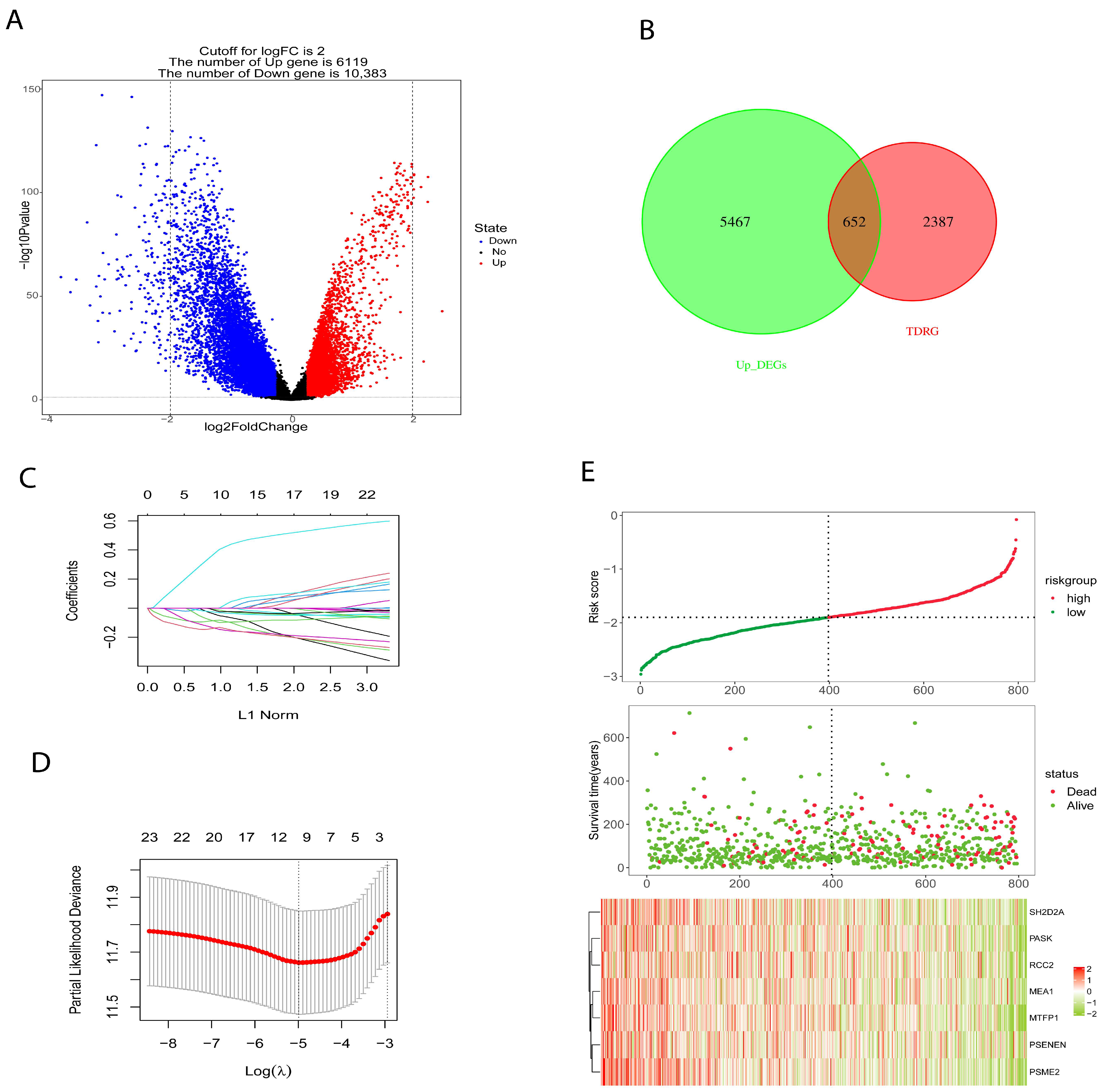
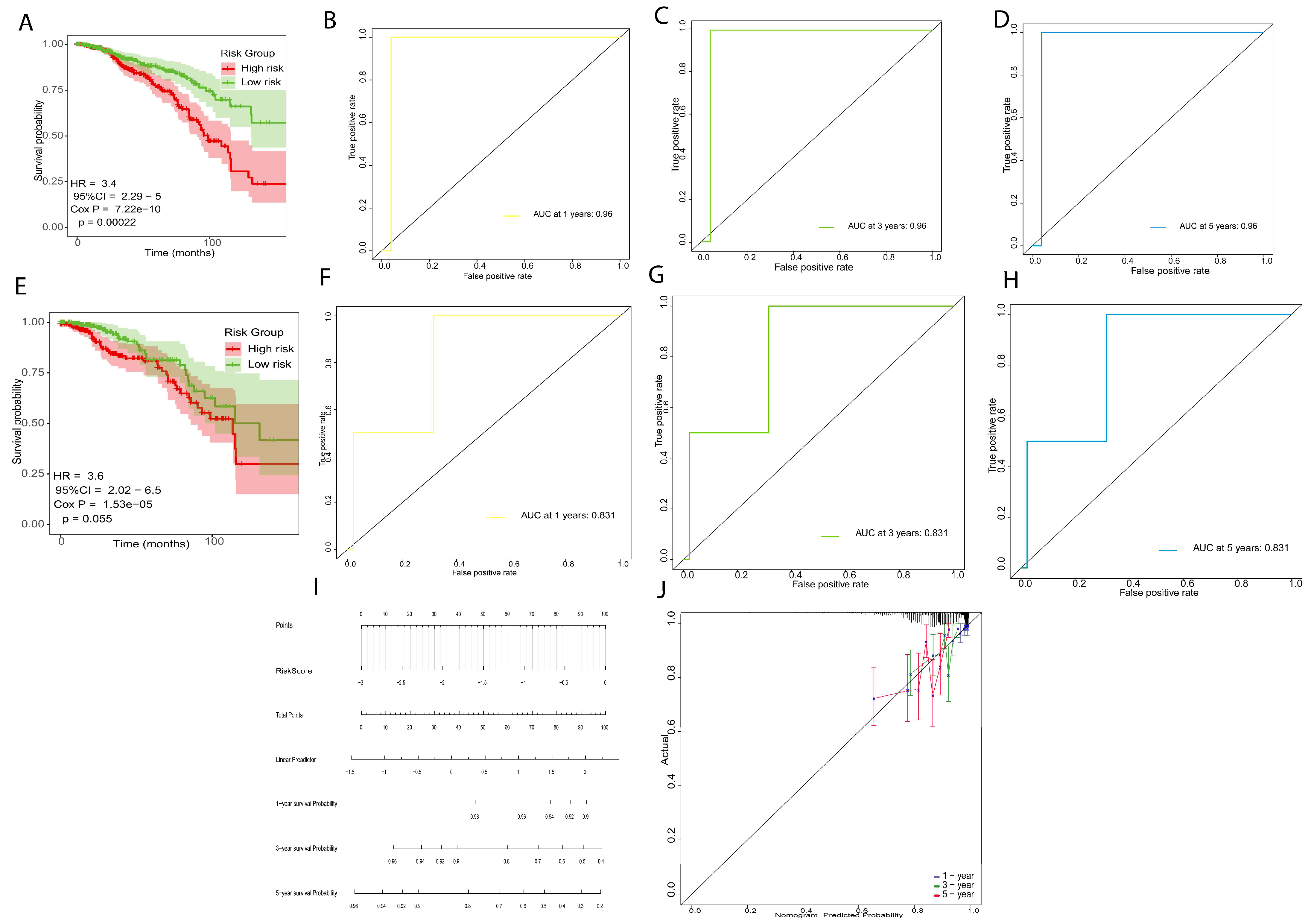
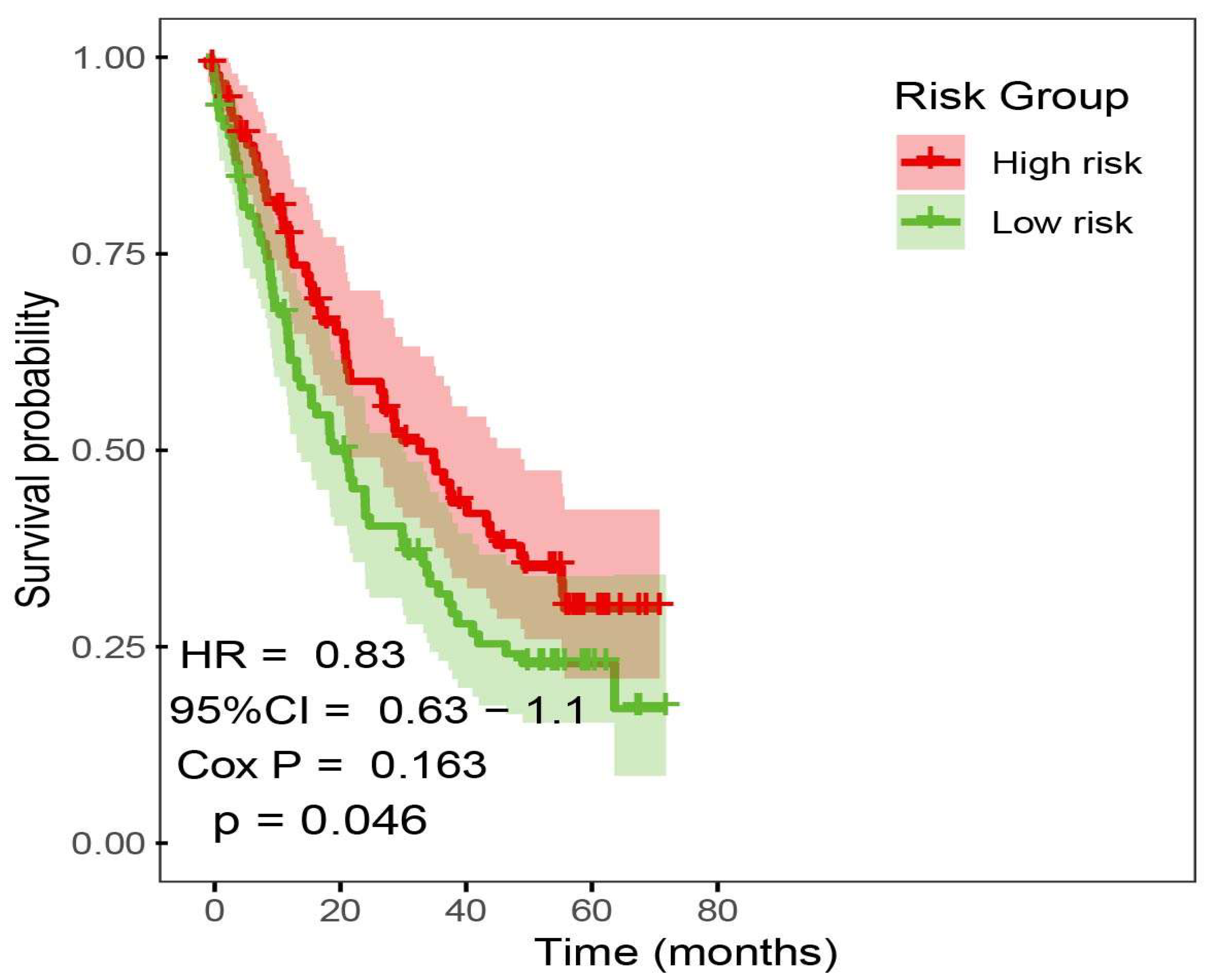
2.3. Development and Verification of Prognostic Risk Model
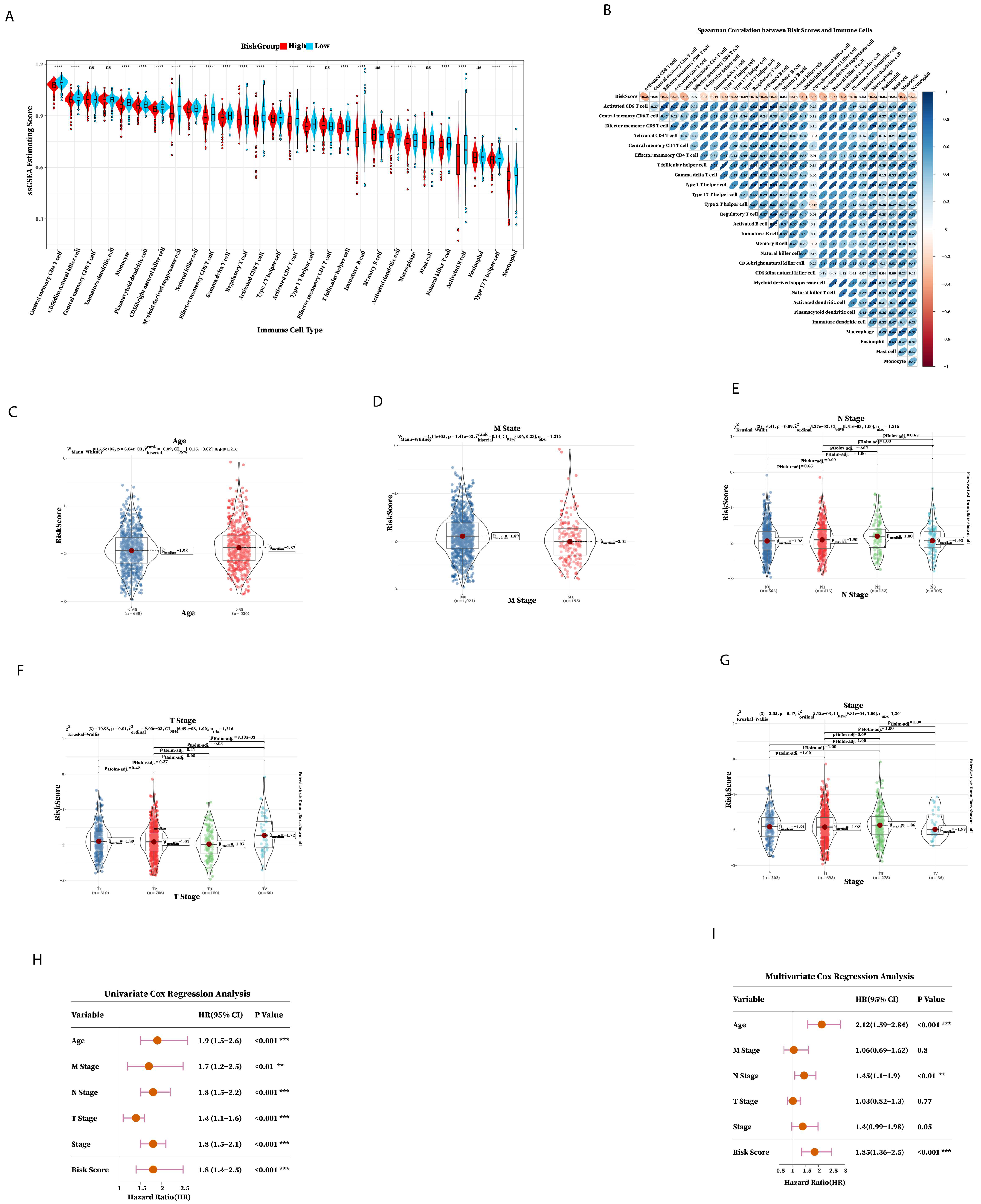
2.4. Immune Prediction and Clinical Application of Prognostic Risk Signature
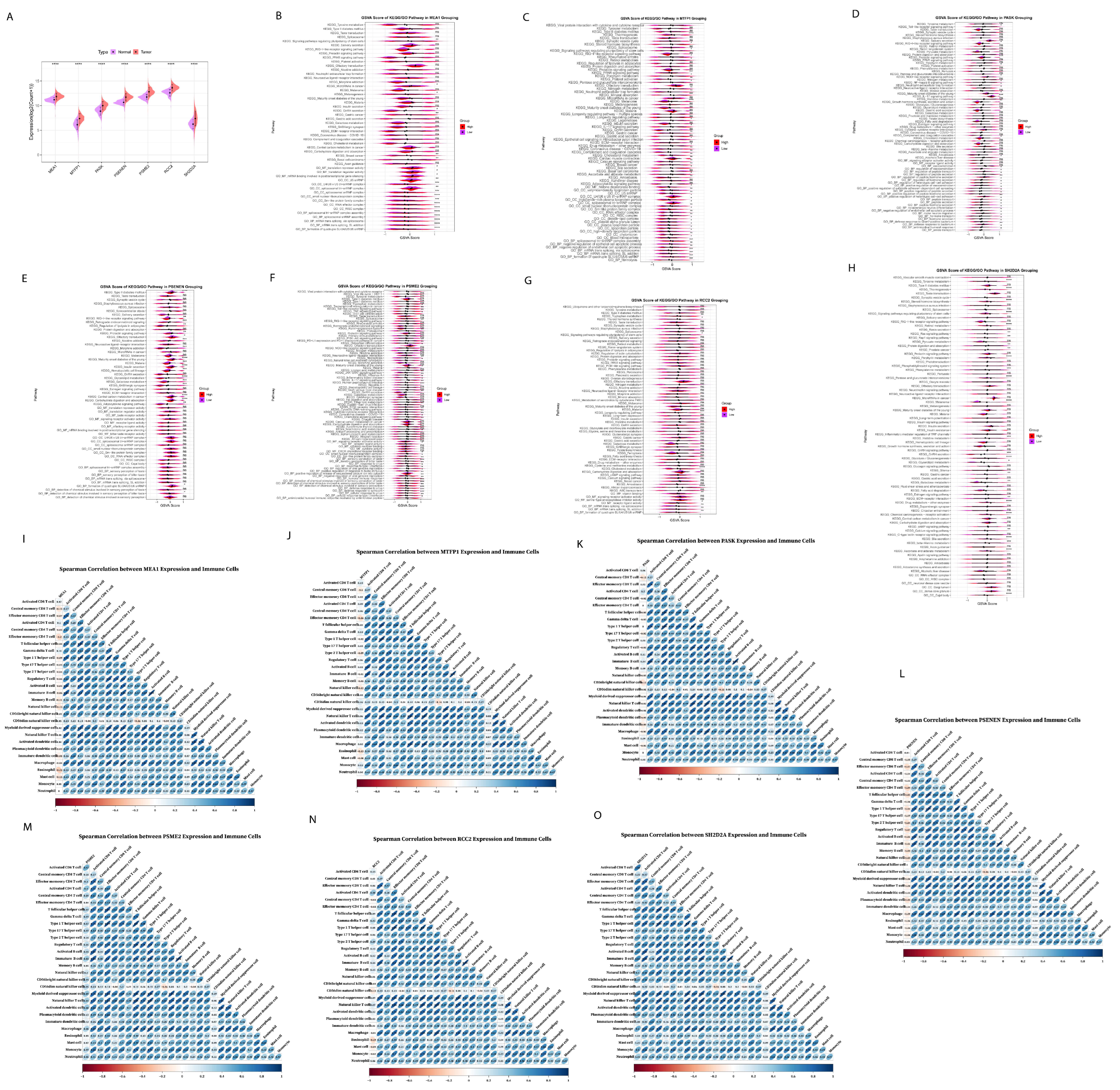
2.5. Functional Enrichment Analysis of Prognostic Risk Genes Signature
2.6. Immunotherapy Outcome Prediction by Prognostic Risk Signature
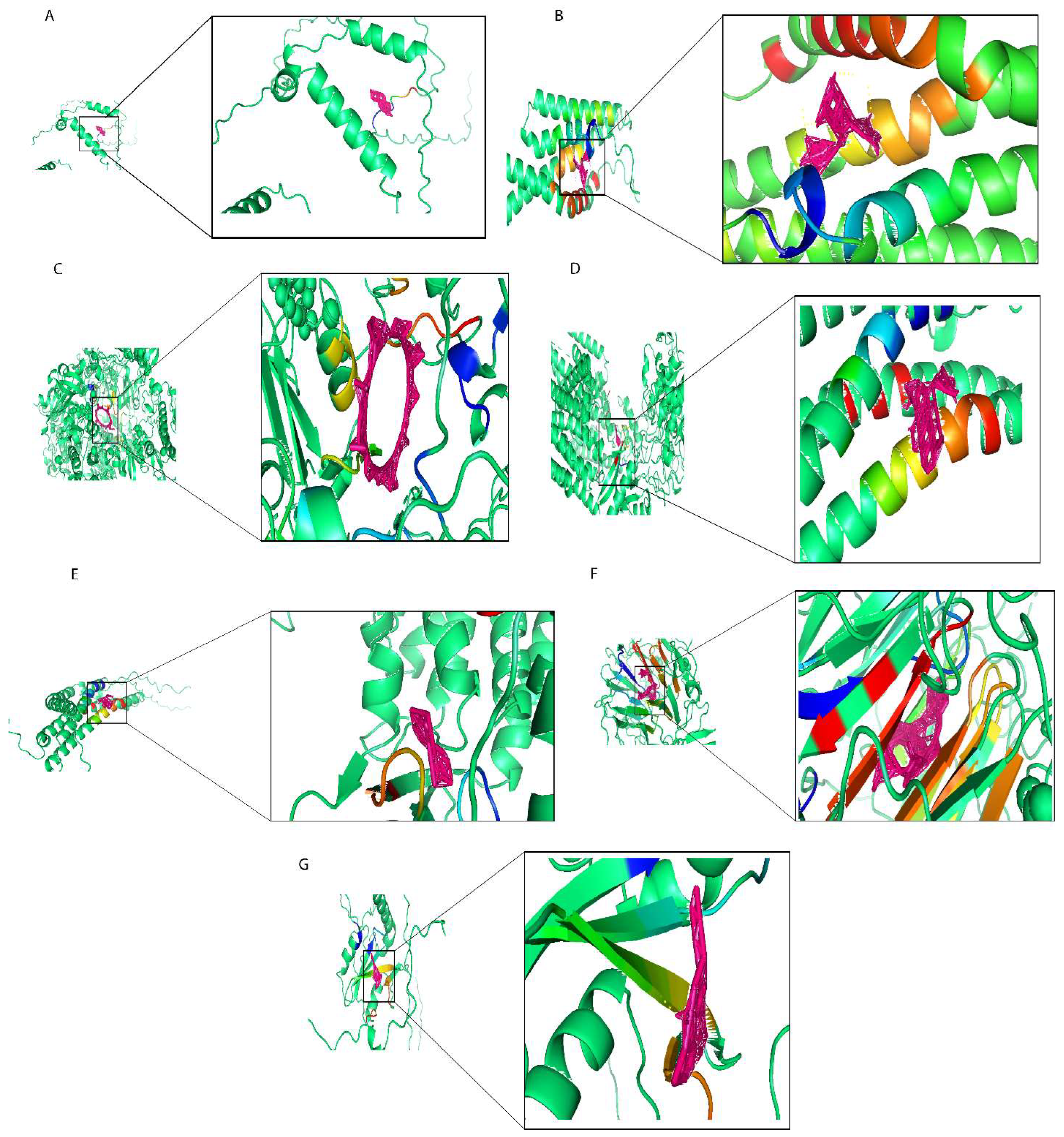
2.7. Molecular Docking of Prognostic Risk Signature Genes
2.8. Prediction of Synergistic Drug Combination by Deep Learning
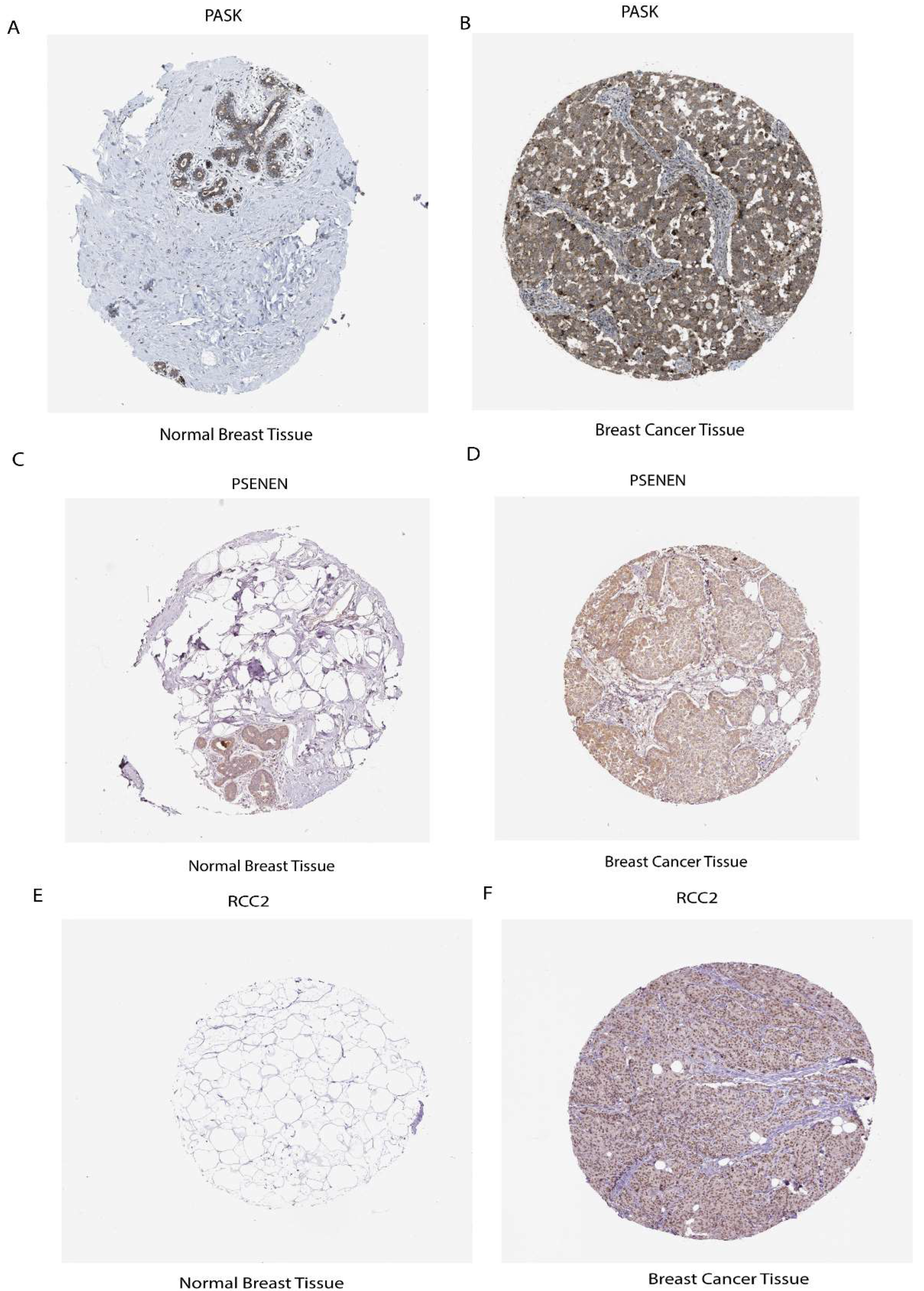
2.9. Elucidate the Expression Levels of mRNA and Protein of Prognostic Risk Signature Genes
3. Discussion
4. Materials and Methods
4.1. Data Sources Used for Analysis
4.2. Calculation of Stemness Index (mRNAsi)
4.3. Different Differentiation States of Tregs
4.4. Prognostic Risk Signature Construction and Validation
4.5. Immune Infiltration Analysis
4.6. Functional Enrichment Analysis of Prognostic Risk Model Genes
4.7. Predictive Model for ICI Response Datasets
4.8. Drugs Screening and Docking
4.9. Anticancer Drug Combination Prediction with Deep Learning
4.10. Detection of Prognostic Gene Expression
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gul, S.; Pang, J.; Yuan, H.; Chen, Y.; Yu, Q.; Wang, H.; Tang, W. Stemness signature and targeted therapeutic drugs identification for Triple Negative Breast Cancer. Sci. Data 2023, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, L.; Wei, D.Q. Editorial: Breast cancer resistance, biomarkers and therapeutics development in the era of artificial intelligence. Front. Mol. Biosci. 2022, 9, 2020–2022. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells: A. Immunology and Cell Biology 2018, 96, 21–33. [Google Scholar] [CrossRef]
- Zhao, R.; Cao, G.; Zhang, B.; Wei, L.; Zhang, X.; Jin, M.; He, B.; Zhang, B.; He, Z.; Bie, Q. TNF+ regulatory T cells regulate the stemness of gastric cancer cells through the IL13/STAT3 pathway. Front. Oncol. 2023, 13, 1162938. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, C.; Wang, B.; Zhang, H.; Qin, G.; Li, C.; Cao, L.; Gao, Q.; Ping, Y.; Zhang, K.; et al. Regulatory T cells promote glioma cell stemness through TGF-β-NF-κB-IL6-STAT3 signaling. Cancer Immunol. Immunother. 2021, 70, 2601–2616. [Google Scholar] [CrossRef]
- Xu, Y.; Mou, J.; Wang, Y.; Zhou, W.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Wang, M.; Wang, J. Regulatory T cells promote the stemness of leukemia stem cells through IL10 cytokine-related signaling pathway. Leukemia 2022, 36, 403–415. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, X.; Qi, P.; Ye, Y.; Shen, W.; Leng, L.; Wang, L.; Li, X.; Luo, X.; Chen, Y.; et al. Sox2 communicates with tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cells 2017, 35, 2351–2365. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, Q.; Pang, J.; Chen, Y.; Sheng, M.; Tang, W. The Value of the Stemness Index in Ovarian Cancer Prognosis. Genes 2022, 13, 993. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Zhu, D.; Wang, X.; Chen, J.; Zhang, Y.; Lian, Q.; Gu, B. Identification of prognostic stemness biomarkers in colon adenocarcinoma drug resistance. BMC Genom. Data 2022, 23, 51. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef]
- Chen, D.; Liu, J.; Zang, L.; Xiao, T.; Zhang, X.; Li, Z.; Zhu, H.; Gao, W.; Yu, X. Integrated Machine Learning and Bioinformatic Analyses Constructed a Novel Stemness-Related Classifier to Predict Prognosis and Immunotherapy Responses for Hepatocellular Carcinoma Patients. Int. J. Biol. Sci. 2022, 18, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.H.; Ha, D.; Lee, J.; Kim, I.; Park, M.; Im, S.H.; Shin, K.; Kim, S. Network-based machine learning approach to predict immunotherapy response in cancer patients. Nat. Commun. 2022, 13, 3703. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Uhl, B.; Gires, O.; Reichel, C.A. A transcriptomic pan-cancer signature for survival prognostication and prediction of immunotherapy response based on endothelial senescence. J. Biomed. Sci. 2023, 30, 21. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xie, Y.; Xie, P.; Li, J.; Tong, Z.; Yang, Y. ZDHHC9: A promising therapeutic target for triple-negative breast cancer through immune modulation and immune checkpoint blockade resistance. Discov. Oncol. 2023, 14, 191. [Google Scholar] [CrossRef]
- Du, K.; Wei, S.; Wei, Z.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Sugarman, E.; Gabrilovich, D.I.; Sullivan, R.J.; et al. Pathway signatures derived from on-treatment tumor specimens predict response to anti-PD1 blockade in metastatic melanoma. Nat. Commun. 2021, 12, 6023. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef]
- Principe, D.R.; Chiec, L.; Mohindra, N.A.; Munshi, H.G. Regulatory T-Cells as an Emerging Barrier to Immune Checkpoint Inhibition in Lung Cancer. Front. Oncol. 2021, 11, 684098. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.X.; Chen, Y.X.; Wu, H.X.; Yin, L.; Zhao, Q.; Luo, H.Y.; Zeng, Z.L.; Qiu, M.Z.; Xu, R.H. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 2022, 14, 45. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ogawa, T.; Sano, R.; Takahara, T.; Inukai, D.; Akira, S.; Tsuchida, H.; Yoshikawa, K.; Ueda, R.; Tsuzuki, T. Immune-checkpoint molecules on regulatory T-cells as a potential therapeutic target in head and neck squamous cell cancers. Cancer Sci. 2020, 111, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Ohinata, Y.; Sutou, S.; Kondo, M.; Takahashi, T.; Mitsui, Y. Male-enhanced antigen-1 gene flanked by two overlapping genes is expressed in late spermatogenesis. Biol. Reprod. 2002, 67, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Sun, J.; Xing, Z.; Xie, F.; Yang, L.; Ding, W. MTFP1 overexpression promotes the growth of oral squamous cell carcinoma by inducing ROS production. Cell Biol. Int. 2020, 44, 821–829. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; Pérez-García, A.; Alvarez, E.; Sanz, C. PAS Kinase: A Nutrient and Energy Sensor “Master Key” in the Response to Fasting/Feeding Conditions. Front. Endocrinol. 2020, 11, 594053. [Google Scholar] [CrossRef]
- Hao, H.X.; Rutter, J. The role of PAS kinase in regulating energy metabolism. IUBMB Life 2008, 60, 204–209. [Google Scholar] [CrossRef]
- Yang, Z.; Wen, D.; Ye, Y.; Chen, K.; Qiu, Z.; Liu, X.; Li, X. Pan-cancer analysis highlights the role of PSENEN in the prognosis and immunology of cancer. J. Holist. Integr. Pharm. 2023, 4, 83–102. [Google Scholar] [CrossRef]
- Chen, K.; Liang, B.; Ma, W.; Wan, G.; Chen, B.; Lu, C.; Luo, Y.; Gu, X. Immunological and prognostic analysis of PSENEN in low-grade gliomas: An immune infiltration-related prognostic biomarker. Front. Mol. Neurosci. 2022, 15, 933855. [Google Scholar] [CrossRef]
- Wu, C.; Zhong, R.; Sun, X.; Shi, J. PSME2 identifies immune-hot tumors in breast cancer and associates with well therapeutic response to immunotherapy. Front. Genet. 2022, 13, 1071270. [Google Scholar] [CrossRef]
- Wang, X.; Wu, F.; Deng, Y.; Chai, J.; Zhang, Y.; He, G.; Li, X. Increased expression of PSME2 is associated with clear cell renal cell carcinoma invasion by regulating BNIP3-mediated autophagy. Int. J. Oncol. 2021, 59, 106. [Google Scholar] [CrossRef]
- Calderon-Aparicio, A.; Yamamoto, H.; de Vitto, H.; Zhang, T.; Wang, Q.; Bode, A.M.; Dong, Z. RCC2 promotes esophageal cancer growth by regulating activity and expression of the Sox2 transcription factor. Mol. Cancer Res. 2020, 18, 1660–1674. [Google Scholar] [CrossRef] [PubMed]
- Rendleman, J.; Shang, S.; Dominianni, C.; Shields, J.F.; Scanlon, P.; Adaniel, C.; Desrichard, A.; Ma, M.; Shapiro, R.; Berman, R.; et al. Melanoma risk loci as determinants of melanoma recurrence and survival. J. Transl. Med. 2013, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Wang, J.; Deng, Z.; Wang, M. RCC2 promotes proliferation and radio-resistance in glioblastoma via activating transcription of DNMT1. Biochem. Biophys. Res. Commun. 2019, 516, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Wu, N.; Guan, R.; Pang, L.; Li, X.; Li, S.; Tang, L.; Guo, Y.; Chen, J.; Sun, D.; et al. Overexpression of RCC2 enhances cell motility and promotes tumor metastasis in lung adenocarcinoma by inducing epithelial–mesenchymal transition. Clin. Cancer Res. 2017, 23, 5598–5610. [Google Scholar] [CrossRef]
- Gong, S.; Chen, Y.; Meng, F.; Zhang, Y.; Wu, H.; Li, C.; Zhang, G. RCC2, a regulator of the RalA signaling pathway, is identified as a novel therapeutic target in cisplatin-resistant ovarian cancer. FASEB J. 2019, 33, 5350–5365. [Google Scholar] [CrossRef]
- Matsuo, M.; Nakada, C.; Tsukamoto, Y.; Noguchi, T.; Uchida, T.; Hijiya, N.; Matsuura, K.; Moriyama, M. MiR-29c is downregulated in gastric carcinomas and regulates cell proliferation by targeting RCC2. Mol. Cancer 2013, 12, 15. [Google Scholar] [CrossRef]
- Kolltveit, K.M.; Granum, S.; Aasheim, H.C.; Forsbring, M.; Sundvold-Gjerstad, V.; Dai, K.Z.; Molberg, Ø.; Schjetne, K.W.; Bogen, B.; Shapiro, V.S.; et al. Expression of SH2D2A in T-cells is regulated both at the transcriptional and translational level. Mol. Immunol. 2008, 45, 2380–2390. [Google Scholar] [CrossRef]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef]
- Sun, Z.; Li, X.; Massena, S.; Kutschera, S.; Padhan, N.; Gualandi, L.; Sundvold-Gjerstad, V.; Gustafsson, K.; Choy, W.W.; Zang, G.; et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAD. J. Exp. Med. 2012, 209, 1363–1377. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, D.; Chen, J.; Jiang, Q.; Song, S.; Mi, Y.; Wang, T.; Ye, Q. Interaction between BEND5 and RBPJ suppresses breast cancer growth and metastasis via inhibiting Notch signaling. Int. J. Biol. Sci. 2022, 18, 4233–4244. [Google Scholar] [CrossRef]
- Rs, J. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions—Evidence for Estrogen as an Immunomodulator. Front. Cell. Infect. Microbiol. 2021, 11, 649815. [Google Scholar] [CrossRef]
- Rani, A.; Saini, V.; Patra, P.; Prashar, T.; Pandey, R.K.; Mishra, A.; Jha, H.C. Epigallocatechin Gallate: A Multifaceted Molecule for Neurological Disorders and Neurotropic Viral Infections. ACS Chem. Neurosci. 2023, 14, 2968–2980. [Google Scholar] [CrossRef] [PubMed]
- Du, G.J.; Zhang, Z.; Wen, X.D.; Yu, C.; Calway, T.; Yuan, C.S.; Wang, C.Z. Epigallocatechin gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 2012, 4, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; He, B.; Lai, L.; Chen, Q.; Liu, Y.; Guo, Q.; Wang, Q. Cyclosporine A inhibits breast cancer cell growth by downregulating the expression of pyruvate kinase subtype M2. Int. J. Mol. Med. 2012, 30, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Kashiwagi, E.; Ide, H.; Li, Y.; Zheng, Y.; Miyamoto, Y.; Netto, G.J.; Ishiguro, H.; Miyamoto, H. Cyclosporine A and tacrolimus inhibit bladder cancer growth through down-regulation of NFATc1. Oncotarget 2015, 6, 1582–1593. [Google Scholar] [CrossRef]
- Codini, M.; Cataldi, S.; Ambesi-Impiombato, F.S.; Lazzarini, A.; Floridi, A.; Lazzarini, R.; Curcio, F.; Beccari, T.; Albi, E. Gentamicin arrests cancer cell growth: The intriguing involvement of nuclear sphingomyelin metabolism. Int. J. Mol. Sci. 2015, 16, 2307–2319. [Google Scholar] [CrossRef]
- Albi, E.; Cataldi, S.; Ceccarini, M.R.; Conte, C.; Ferri, I.; Fettucciari, K.; Patria, F.F.; Beccari, T.; Codini, M. Gentamicin targets acid sphingomyelinase in cancer: The case of the human gastric cancer NCI-N87 cells. Int. J. Mol. Sci. 2019, 20, 4375. [Google Scholar] [CrossRef]
- Yun, U.J.; Lee, J.H.; Shim, J.; Yoon, K.; Goh, S.H.; Yi, E.H.; Ye, S.K.; Lee, J.S.; Lee, H.; Park, J.; et al. Anti-cancer effect of doxorubicin is mediated by downregulation of HMG-Co A reductase via inhibition of EGFR/Src pathway. Lab. Investig. 2019, 99, 1157–1172. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, Y.J.; Song, X.-H. Ivermectin inhibits tumor metastasis by regulating the Wnt/β catenin/integrin β1/FAK signaling pathway. Am. J. Cancer Res. 2022, 12, 4502–4512. [Google Scholar]
- Badowski Paa Kwesi Yanful, M. Therapeutics and Clinical Risk Management Dovepress Dronabinol oral solution in the management of anorexia and weight loss in AiDS and cancer. Ther. Clin. Risk Manag. 2018, 14, 643–651. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef]
- Quinn, B.A.; Dash, R.; Sarkar, S.; Azab, B.; Bhoopathi, P.; Das, S.K.; Emdad, L.; Wei, J.; Pellecchia, M.; Sarkar, D.; et al. Pancreatic Cancer Combination Therapy Using a BH3 Mimetic and a Synthetic Tetracycline. Cancer Res. 2015, 23, 5–15. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Homet, B.; Hu-lieskovan, S.; Berent-maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transriptomic features of anti-PD1 response. Cell 2017, 165, 35–44. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Angelo, M.S.; Forman, J.; Ross-macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Amon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e15. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Preuer, K.; Lewis, R.P.I.; Hochreiter, S.; Bender, A.; Bulusu, K.C.; Klambauer, G. DeepSynergy: Predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 2018, 34, 1538–1546. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tissue | Synergy | Drug1 | Drug2 |
|---|---|---|---|---|
| T47D | BREAST | 18.295 | Ethinyl Estradiol | Doxorubicin |
| T47D | BREAST | 17.79508 | Gentamicin | Doxorubicin |
| T47D | BREAST | 16.03166 | Dronabinol | Doxorubicin |
| T47D | BREAST | 14.89733 | Cyclosporine | Doxorubicin |
| T47D | BREAST | 14.25005 | Epigallocatechin gallate | Doxorubicin |
| T47D | BREAST | 12.93522 | Ivermectin | Doxorubicin |
| T47D | BREAST | 9.996137 | Epigallocatechin gallate | Gentamicin |
| T47D | BREAST | 9.803739 | Gentamicin | Ethinyl Estradiol |
| T47D | BREAST | 9.124499 | Gentamicin | Dronabinol |
| OCUBM | BREAST | 7.538448 | Ivermectin | Ethinyl Estradiol |
| OCUBM | BREAST | 7.024859 | Ethinyl Estradiol | Dronabinol |
| T47D | BREAST | 6.901407 | Gentamicin | Ivermectin |
| OCUBM | BREAST | 6.568412 | Gentamicin | Ethinyl Estradiol |
| OCUBM | BREAST | 6.47709 | Ethinyl Estradiol | Cyclosporine |
| T47D | BREAST | 5.857921 | Epigallocatechin gallate | Ethinyl Estradiol |
| OCUBM | BREAST | 4.984258 | Gentamicin | Ivermectin |
| KPL1 | BREAST | 4.943874 | Ethinyl Estradiol | Cyclosporine |
| ZR751 | BREAST | 4.943094 | Ethinyl Estradiol | Cyclosporine |
| KPL1 | BREAST | 4.416937 | Ivermectin | Ethinyl Estradiol |
| ZR751 | BREAST | 4.41583 | Ivermectin | Ethinyl Estradiol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gul, S.; Pang, J.; Chen, Y.; Qi, Q.; Tang, Y.; Sun, Y.; Wang, H.; Tang, W.; Zhou, X. Machine Learning-Based Prognostic Signature in Breast Cancer: Regulatory T Cells, Stemness, and Deep Learning for Synergistic Drug Discovery. Int. J. Mol. Sci. 2025, 26, 6995. https://doi.org/10.3390/ijms26146995
Gul S, Pang J, Chen Y, Qi Q, Tang Y, Sun Y, Wang H, Tang W, Zhou X. Machine Learning-Based Prognostic Signature in Breast Cancer: Regulatory T Cells, Stemness, and Deep Learning for Synergistic Drug Discovery. International Journal of Molecular Sciences. 2025; 26(14):6995. https://doi.org/10.3390/ijms26146995
Chicago/Turabian StyleGul, Samina, Jianyu Pang, Yongzhi Chen, Qi Qi, Yuheng Tang, Yingjie Sun, Hui Wang, Wenru Tang, and Xuhong Zhou. 2025. "Machine Learning-Based Prognostic Signature in Breast Cancer: Regulatory T Cells, Stemness, and Deep Learning for Synergistic Drug Discovery" International Journal of Molecular Sciences 26, no. 14: 6995. https://doi.org/10.3390/ijms26146995
APA StyleGul, S., Pang, J., Chen, Y., Qi, Q., Tang, Y., Sun, Y., Wang, H., Tang, W., & Zhou, X. (2025). Machine Learning-Based Prognostic Signature in Breast Cancer: Regulatory T Cells, Stemness, and Deep Learning for Synergistic Drug Discovery. International Journal of Molecular Sciences, 26(14), 6995. https://doi.org/10.3390/ijms26146995