
AI-Driven Polypharmacology in Small-Molecule Drug Discovery

Abstract
1. Introduction
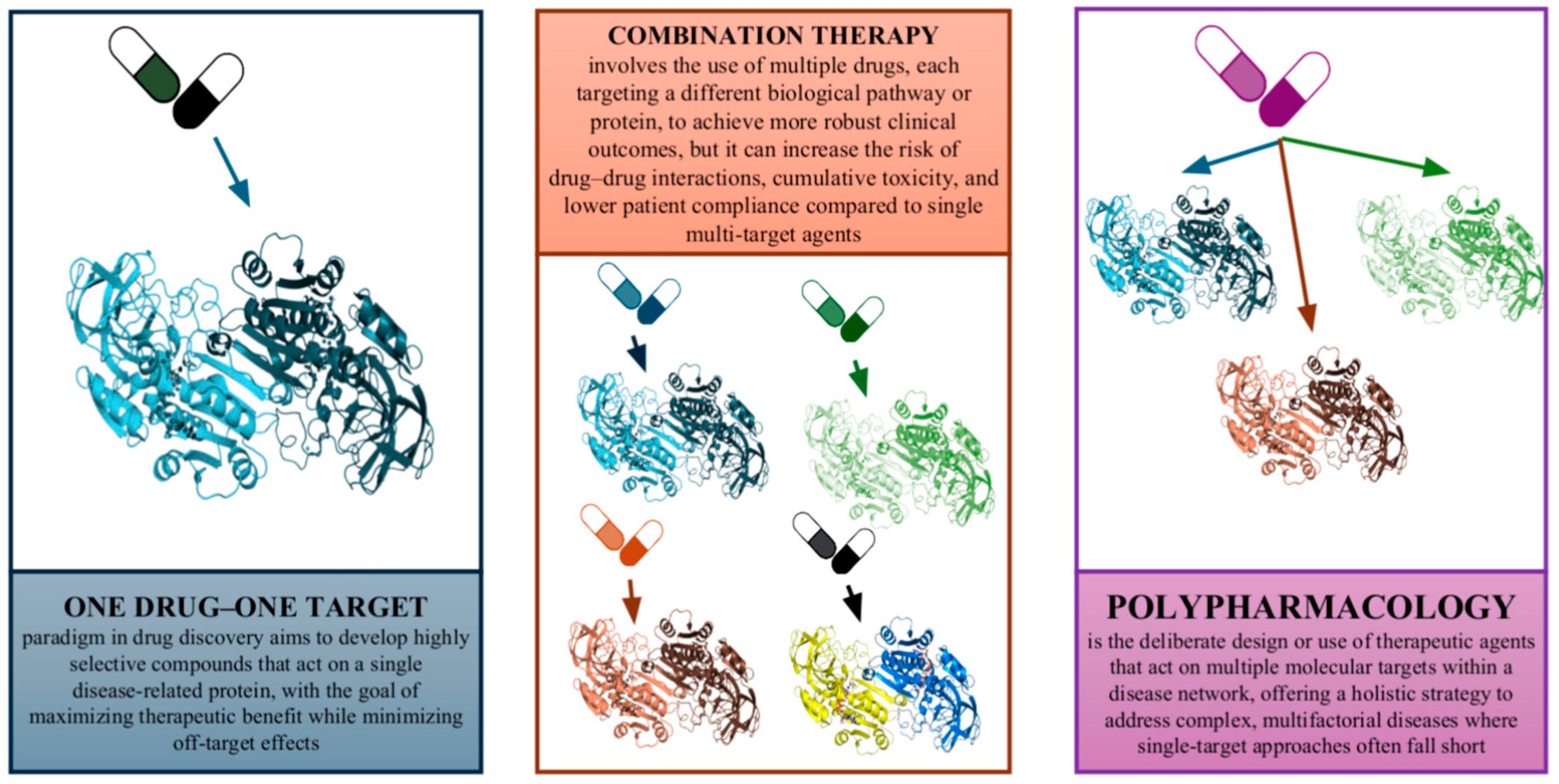
1.1. The One-Target–One-Drug Paradigm and Its Limitations
1.2. Rationale for Polypharmacology and Multi-Target Drug Design
1.3. Complex Diseases and the Need for Multi-Target Therapeutics
1.3.1. Cancer
1.3.2. Neurodegenerative Disorders
1.3.3. Metabolic and Endocrine Disorders
1.3.4. Infectious Diseases
2. Emerging Trends and Outlook for Polypharmacology in Drug Discovery
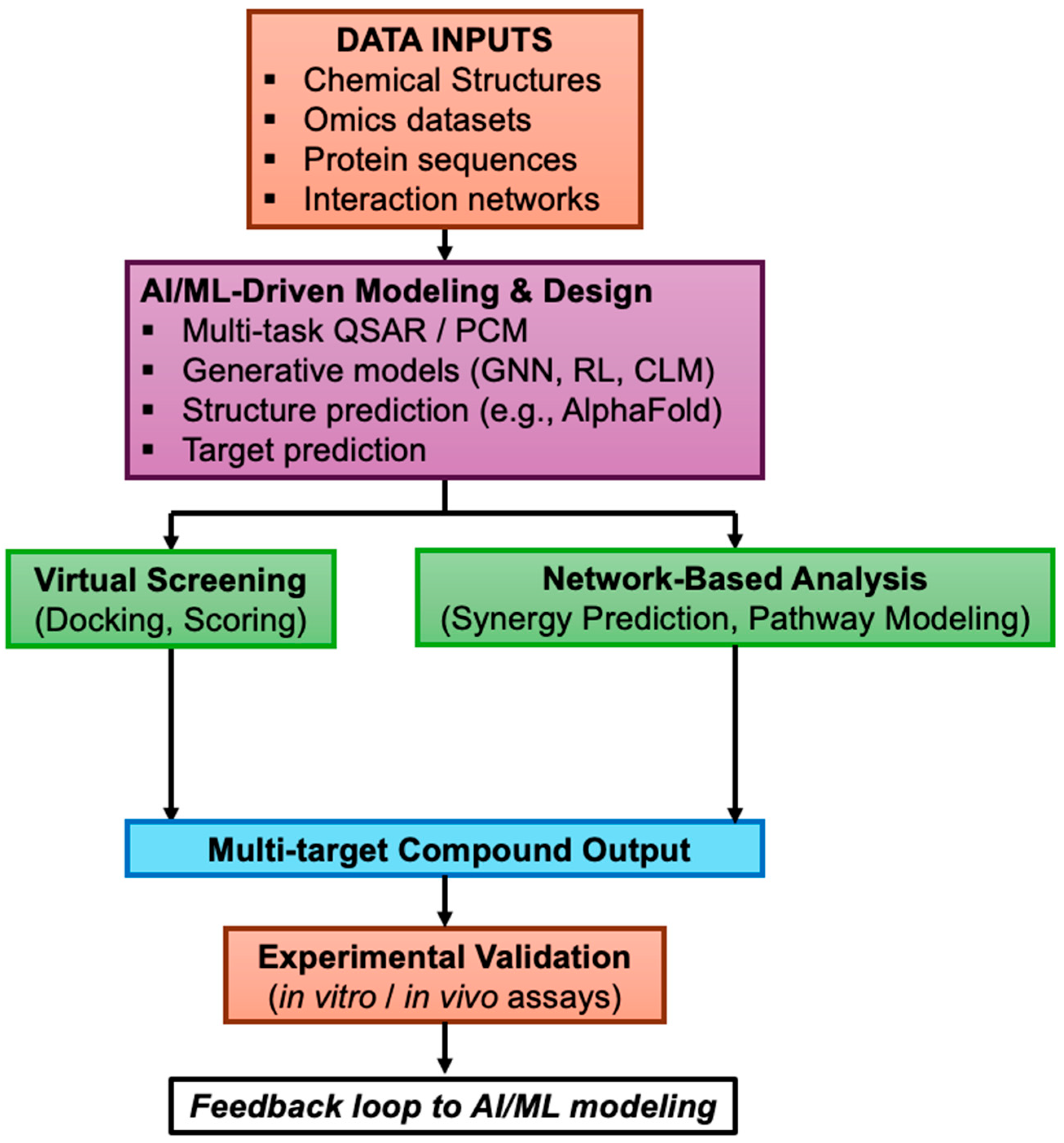
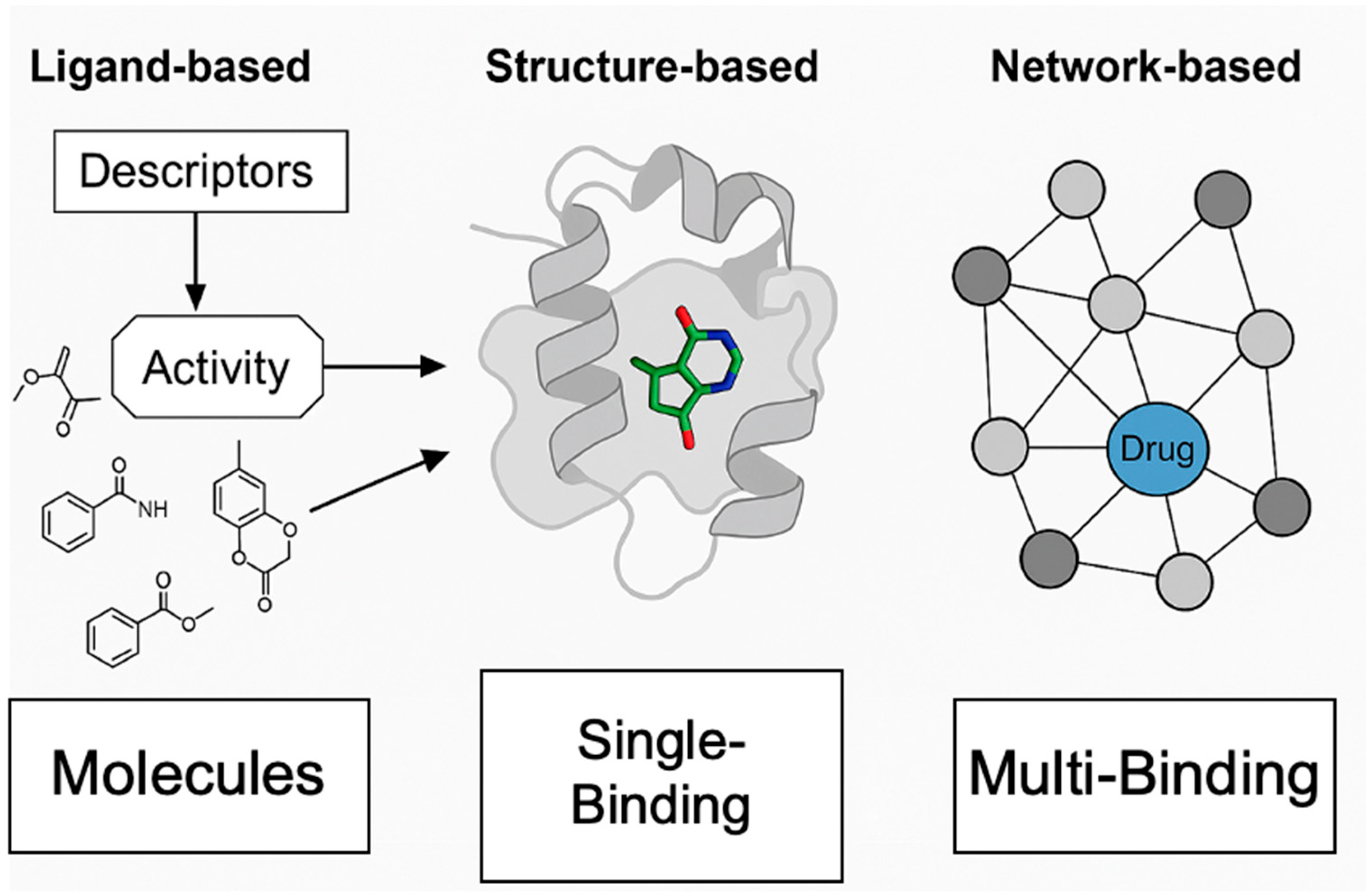
3. Computational Approaches for Polypharmacology in Small-Molecule Drug Design
3.1. Ligand-Based Multi-Target Modeling: Multi-Task QSAR and Proteochemometrics
3.2. Strengths and Limitations of Current Computational Techniques
4. Generative and AI Methods
4.1. Generative Deep Learning for Polypharmacology
4.2. Graph Neural Networks and Multi-Objective Reinforcement Learning
4.3. Emerging Tools and Case Studies
5. Systems Biology and Network Pharmacology in Polypharmacology
5.1. Omics-Driven Target Identification for Multi-Target Drug Design
5.2. Functional Genomic Screens and Network Pharmacology
5.3. Pathway Modeling and Simulation for Multi-Target Design
5.4. Risks and Limitations of Current AI Approaches
6. Conclusions
7. Future Directions
Funding
Conflicts of Interest
References
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic Shotguns versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network Pharmacology: The next Paradigm in Drug Discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery: Miniperspective. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A Network-Based Approach to Human Disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Paolini, G.V.; Shapland, R.H.B.; Van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global Mapping of Pharmacological Space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Mencher, S.K.; Wang, L.G. Promiscuous Drugs Compared to Selective Drugs (Promiscuity Can Be a Virtue). BMC Clin. Pharmacol. 2005, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M. Polypharmacology: The Rise of Multitarget Drugs over Combination Therapies. Future Med. Chem. 2014, 6, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Ryszkiewicz, P.; Malinowska, B.; Schlicker, E. Polypharmacology: Promises and New Drugs in 2022. Pharmacol. Rep. 2023, 75, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Lisitsa, A.; Knight, R.; Melino, G.; Antonov, A. Polypharmacology of Approved Anticancer Drugs. Current Drug Target. 2017, 18, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the Cancer Kinome through Polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-Target Pharmacology: Possibilities and Limitations of the “Skeleton Key Approach” from a Medicinal Chemist Perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; De Sena Murteira Pinheiro, P.; Bolognesi, M.L. From Combinations to Multitarget-directed Ligands: A Continuum in Alzheimer’s Disease Polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fridkin, M.; Youdim, M. From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy. Pharmaceuticals 2014, 7, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Lillich, F.F.; Imig, J.D.; Proschak, E. Multi-Target Approaches in Metabolic Syndrome. Front. Pharmacol. 2021, 11, 554961. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lei, X.; Fu, S.; Liu, P.; Long, C.; Wang, Y.; Li, Z.; Xie, Q.; Chen, Q. Efficacy and Safety of Tirzepatide, Dual GLP-1/GIP Receptor Agonists, in the Management of Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Diabetol. Metab. Syndr. 2023, 15, 222. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, R.; Fong, L.W.; Tan, Z.; Huang, B.; Zhang, S. An Up-to-Date Overview of Computational Polypharmacology in Modern Drug Discovery. Expert Opin. Drug Discov. 2020, 15, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Leitão, M.M.; Gonçalves, A.S.C.; Borges, F.; Simões, M.; Borges, A. Polypharmacological Strategies for Infectious Bacteria. Pharmacol. Rev. 2025, 77, 100038. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Scotti, L.; Bezerra Mendonça Junior, F.J.; Muratov, E.; Scotti, M.T.; Speck-Planche, A. QSAR Modeling for Multi-Target Drug Discovery: Designing Simultaneous Inhibitors of Proteins in Diverse Pathogenic Parasites. Front. Chem. 2021, 9, 634663. [Google Scholar] [CrossRef] [PubMed]
- Bongers, B.J.; IJzerman, A.P.; Van Westen, G.J.P. Proteochemometrics—Recent Developments in Bioactivity and Selectivity Modeling. Drug Discov. Today Technol. 2019, 32–33, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Di Stefano, M.; Martinelli, E.; Poli, G.; Tuccinardi, T. Recent Advances in In Silico Target Fishing. Molecules 2021, 26, 5124. [Google Scholar] [CrossRef] [PubMed]
- Isigkeit, L.; Hörmann, T.; Schallmayer, E.; Scholz, K.; Lillich, F.F.; Ehrler, J.H.M.; Hufnagel, B.; Büchner, J.; Marschner, J.A.; Pabel, J.; et al. Automated Design of Multi-Target Ligands by Generative Deep Learning. Nat. Commun. 2024, 15, 7946. [Google Scholar] [CrossRef] [PubMed]
- Munson, B.P.; Chen, M.; Bogosian, A.; Kreisberg, J.F.; Licon, K.; Abagyan, R.; Kuenzi, B.M.; Ideker, T. De Novo Generation of Multi-Target Compounds Using Deep Generative Chemistry. Nat. Commun. 2024, 15, 3636. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, K.; Van Vlijmen, H.W.T.; Emmerich, M.T.M.; IJzerman, A.P.; Van Westen, G.J.P. DrugEx v2: De Novo Design of Drug Molecules by Pareto-Based Multi-Objective Reinforcement Learning in Polypharmacology. J. Cheminform. 2021, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Mukaidaisi, M.; Vu, A.; Grantham, K.; Tchagang, A.; Li, Y. Multi-Objective Drug Design Based on Graph-Fragment Molecular Representation and Deep Evolutionary Learning. Front. Pharmacol. 2022, 13, 920747. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.L.; Singh, S.; Carpenter, A.E.; Way, G.P. Predicting Drug Polypharmacology from Cell Morphology Readouts Using Variational Autoencoder Latent Space Arithmetic. PLoS Comput. Biol. 2022, 18, e1009888. [Google Scholar] [CrossRef] [PubMed]
- Ai, C.; Yang, H.; Liu, X.; Dong, R.; Ding, Y.; Guo, F. MTMol-GPT: De Novo Multi-Target Molecular Generation with Transformer-Based Generative Adversarial Imitation Learning. PLoS Comput. Biol. 2024, 20, e1012229. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.M.; Zhu, H.; Lu, Z.; Wang, K.; Tang, J.; Li, M. DeepDTAGen: A Multitask Deep Learning Framework for Drug-Target Affinity Prediction and Target-Aware Drugs Generation. Nat. Commun. 2025, 16, 5021. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Jeon, M.; Allaway, R.J.; Kang, J.; Park, D.; Lee, J.; Jeon, H.; Ko, M.; Jiang, H.; Zheng, M.; et al. Crowdsourced Identification of Multi-Target Kinase Inhibitors for RET- and TAU-Based Disease: The Multi-Targeting Drug DREAM Challenge. PLoS Comput. Biol. 2021, 17, e1009302. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.K.; Loscalzo, J. Developing New Drug Treatments in the Era of Network Medicine. Clin. Pharmacol. Ther. 2013, 93, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Aittokallio, T. Improving the Efficacy-Safety Balance of Polypharmacology in Multi-Target Drug Discovery. Expert. Opin. Drug Discov. 2018, 13, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-Omics Analysis for Drug-Target Identification. Biomolecules 2024, 14, 692. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Park, B.-S.; Heo, S.; Jeon, H.; Kim, J.; Kim, D.; Kook Lee, S.; Jung, S.-Y.; Kong, S.-Y.; Lu, T. Combinatorial CRISPR Screen Reveals FYN and KDM4 as Targets for Synergistic Drug Combination for Treating Triple Negative Breast Cancer. eLife 2025, 13, RP93921. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.; Segura-Cabrera, A.; Pacini, C.; Picco, G.; Behan, F.M.; Jaaks, P.; Coker, E.A.; Van Der Meer, D.; Barthorpe, A.; Lightfoot, H.; et al. Drug Mechanism-of-action Discovery Through the Integration of Pharmacological and CRISPR Screens. Mol. Syst. Biol. 2020, 16, e9405. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gautam, P.; Gupta, A.; He, L.; Akimov, Y.; Wang, W.; Szwajda, A.; Jaiswal, A.; Turei, D.; Yadav, B.; et al. Network Pharmacology Modeling Identifies Synergistic Aurora B and ZAK Interaction in Triple-Negative Breast Cancer. npj Syst. Biol. Appl. 2019, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.; Hwang, Y.; Lee, S.; Lee, D. Rule-Based Multi-Scale Simulation for Drug Effect Pathway Analysis. BMC Med. Inform. Decis. Mak. 2013, 13, S4. [Google Scholar] [CrossRef] [PubMed]
- Hasselgren, C.; Oprea, T.I. Artificial Intelligence for Drug Discovery: Are We There Yet? Annu. Rev. Pharmacol. Toxicol. 2024, 64, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Aittokallio, T. Network Pharmacology Strategies Toward Multi-Target Anticancer Therapies: From Computational Models to Experimental Design Principles. Curr. Pharm. design. 2019, 32, 89–98. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Method | Principle/Approach | Key Strengths | Limitations | Typical Application Scenarios |
|---|---|---|---|---|
| Ligand-based | Uses known ligands’ chemical features to predict new actives (QSAR, PCM) | Fast; no target structure needed; leverages existing bioactivity data | Limited by data quality/coverage; struggles with novel targets | Virtual screening, off-target prediction, drug repurposing [7,8] |
| Structure-based | Uses 3D structures of protein targets to dock and score ligands | Provides structural insights; suitable for novel chemotypes | Requires accurate protein structures; docking/scoring errors | Lead optimization, binding mode analysis, novel target screening [8,9] |
| Network-based | Integrates biological networks to identify target combinations | Captures system-level effects; can suggest synergistic targets | Networks often incomplete; translation to chemistry is nontrivial | Target prioritization, multi-target design, systems pharmacology [7,8,9] |
| Screening Mode | Example Methods/Tools | Example Application/Case Study | Reference(s) |
|---|---|---|---|
| Ligand-based | Multi-task QSAR, Proteochemometrics, Similarity Ensemble Approach | Classification of antiparasitic inhibitors, target fishing | [7,8,23,24,25] |
| Structure-based | Molecular docking, Homology modeling, Structure-based virtual screening | Multi-target kinase inhibitor design, binding mode prediction | [8,9,26] |
| Network-based | Network pharmacology models, Omics integration, CRISPR screening | Synergistic target identification, pathway simulation | [13,14,15,16] |
| Tool/Platform | Main Functionality | Method/Algorithm | Application Domain(s) | Notable Features | Reference |
|---|---|---|---|---|---|
| POLYGON | Generative design of multi-target ligands | VAE + RL | Oncology, kinase inhibitors | Dual-target optimization, experimental validation | [11,27] |
| MTMol-GPT | Multi-target molecule generation | Transformer + Imitation Learning | Kinases, CNS | Conditional generation, novelty | [11,31] |
| DrugEx v2 | Multi-objective molecule design | RNN + Pareto RL | GPCRs, ADMET | Pareto optimization, anti-target avoidance | [11,28] |
| DeepDTAGen | Affinity prediction and target-aware generation | Multitask Deep Learning | Multiple protein classes | Integrated affinity and generation | [11,33] |
| Chemprop * | Target activity prediction | Message Passing Neural Net | Polypharmacology, ADMET | User-friendly, open source | - |
| DeepChem * | General molecular ML platform | Multiple (GNNs, DL, etc.) | Broad: prediction, generation | Extensive library, tutorials | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelsayed, M. AI-Driven Polypharmacology in Small-Molecule Drug Discovery. Int. J. Mol. Sci. 2025, 26, 6996. https://doi.org/10.3390/ijms26146996
Abdelsayed M. AI-Driven Polypharmacology in Small-Molecule Drug Discovery. International Journal of Molecular Sciences. 2025; 26(14):6996. https://doi.org/10.3390/ijms26146996
Chicago/Turabian StyleAbdelsayed, Mena. 2025. "AI-Driven Polypharmacology in Small-Molecule Drug Discovery" International Journal of Molecular Sciences 26, no. 14: 6996. https://doi.org/10.3390/ijms26146996
APA StyleAbdelsayed, M. (2025). AI-Driven Polypharmacology in Small-Molecule Drug Discovery. International Journal of Molecular Sciences, 26(14), 6996. https://doi.org/10.3390/ijms26146996