
Targeting Bcl-xL with Navitoclax Effectively Eliminates Senescent Tumor Cells That Appear Following CEP-1347-Induced Differentiation of Glioma Stem Cells

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
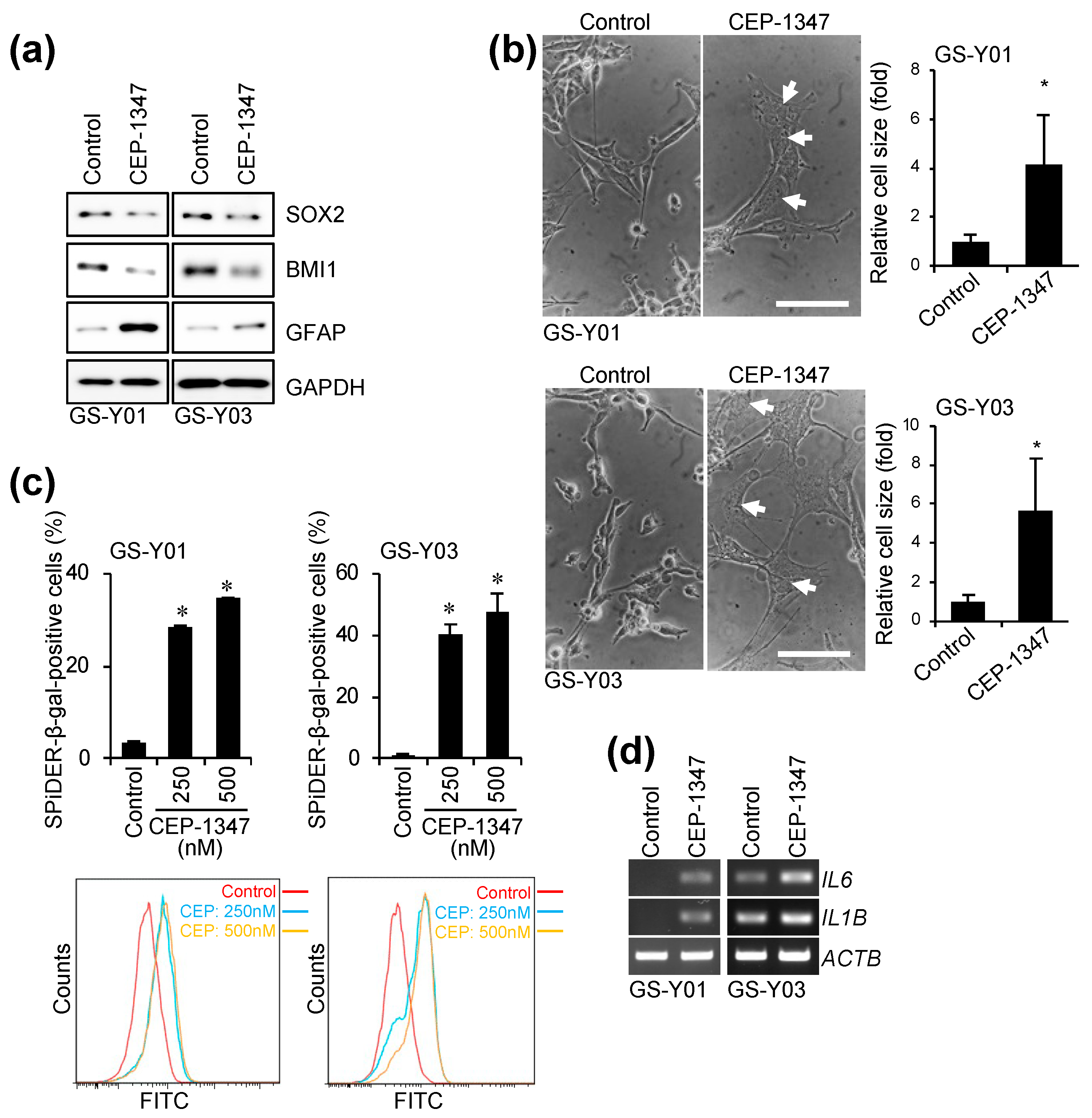
2.1. CEP-1347 Induces a Senescent-like Phenotype in GSCs
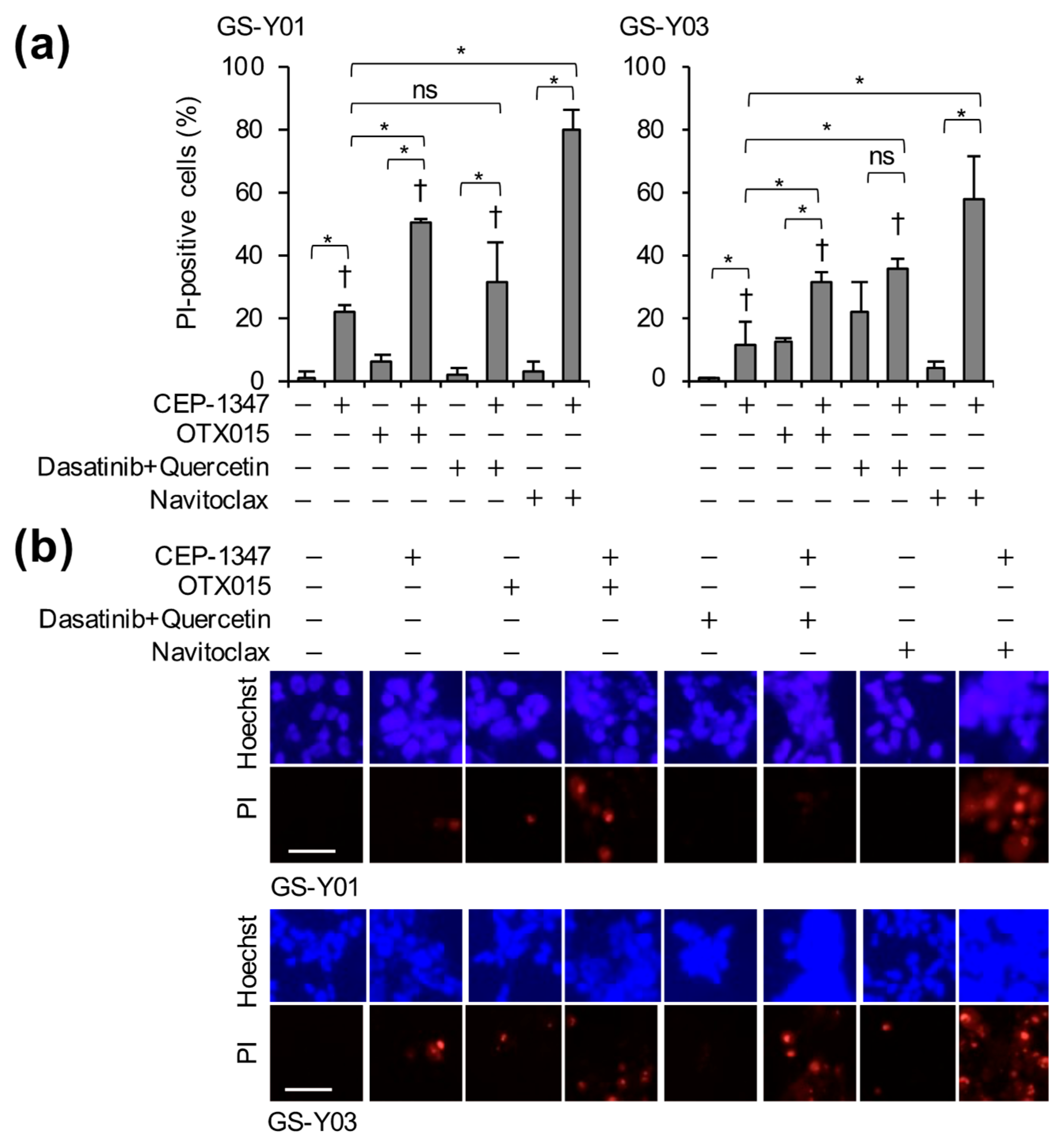
2.2. The Combination of CEP-1347 and Senolytics Potently Induces Cell Death in GSCs
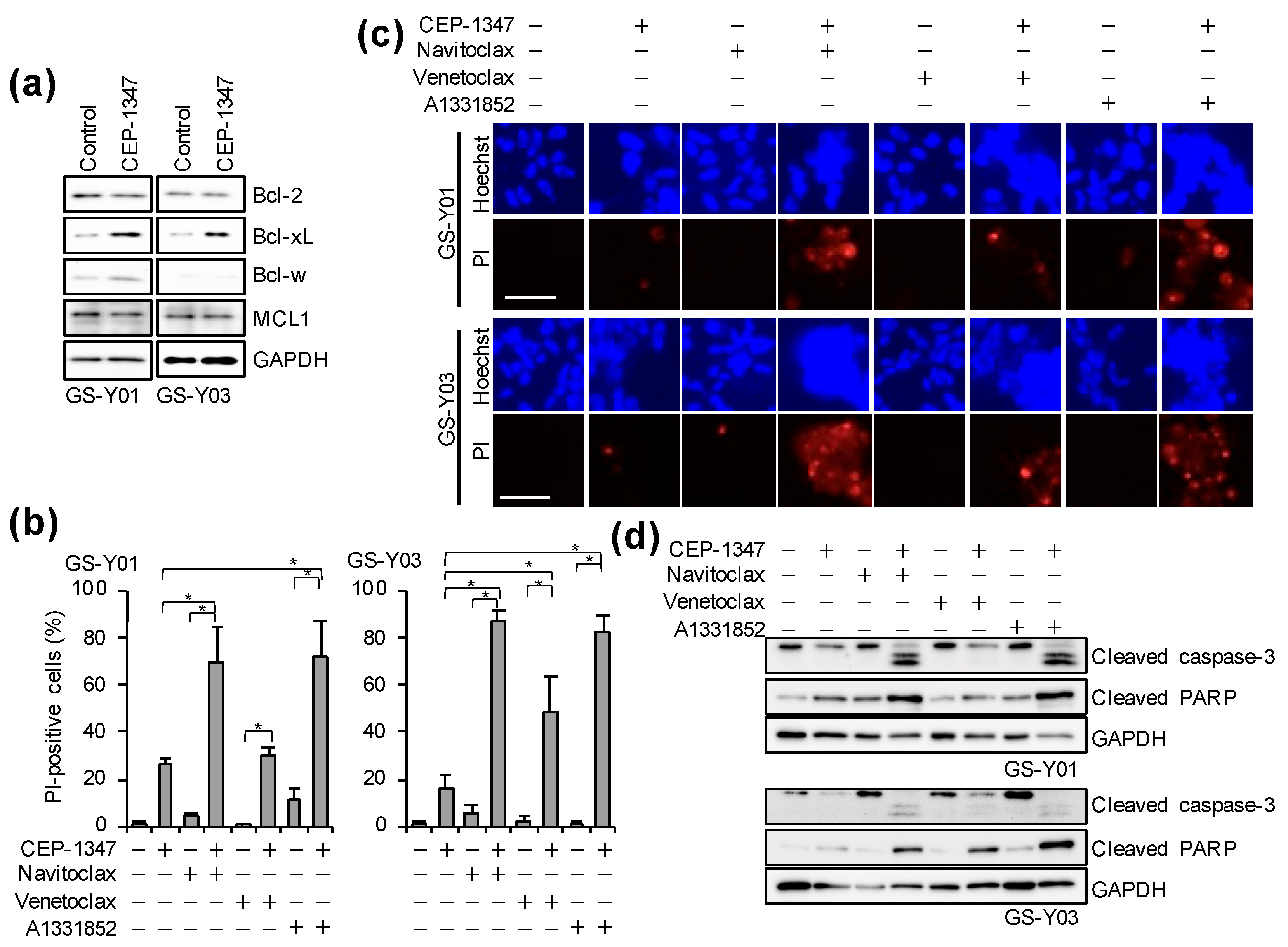
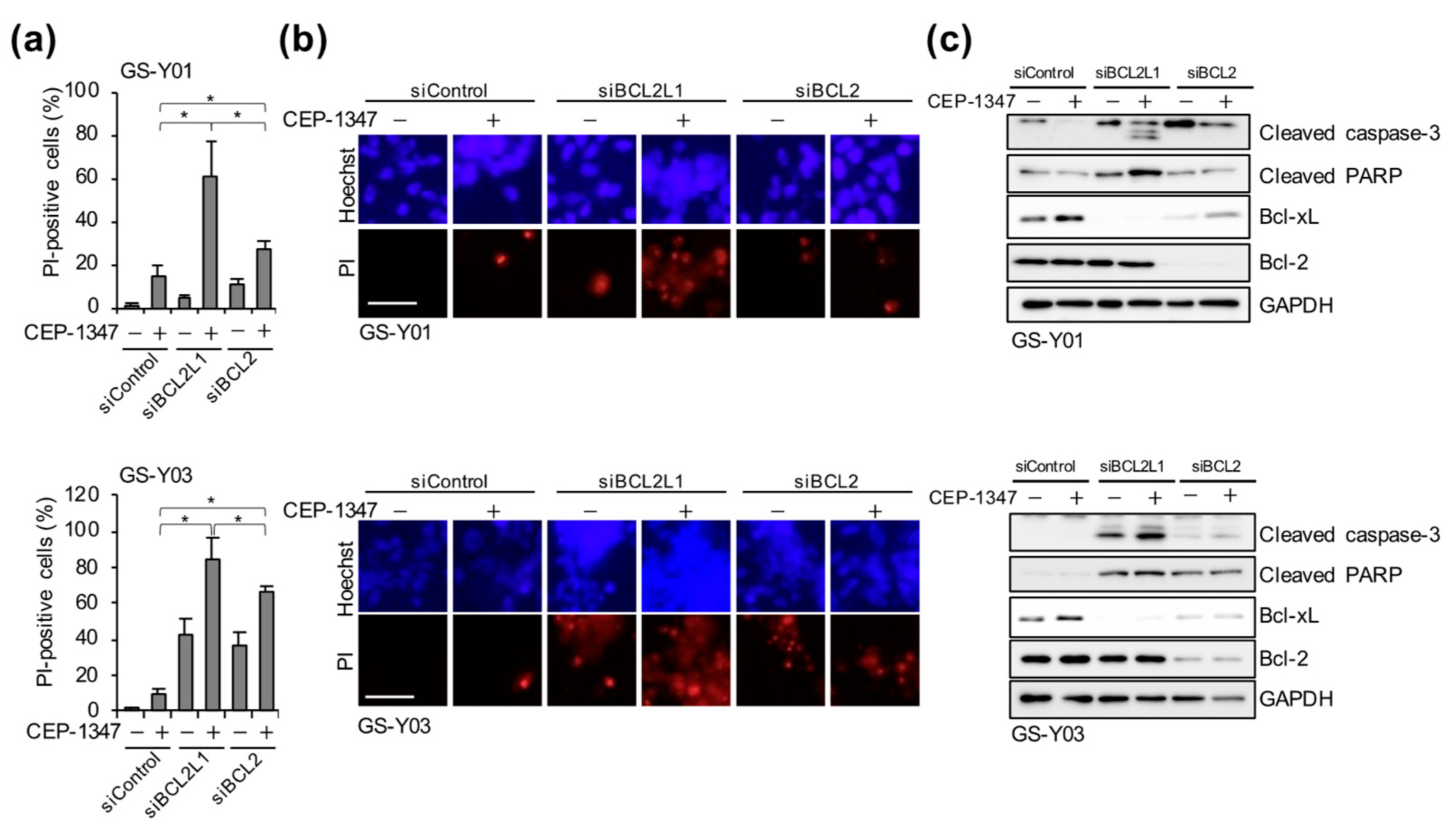
2.3. The Combination of CEP-1347 and Bcl-xL Inhibition Potently Induces Apoptosis in GSCs
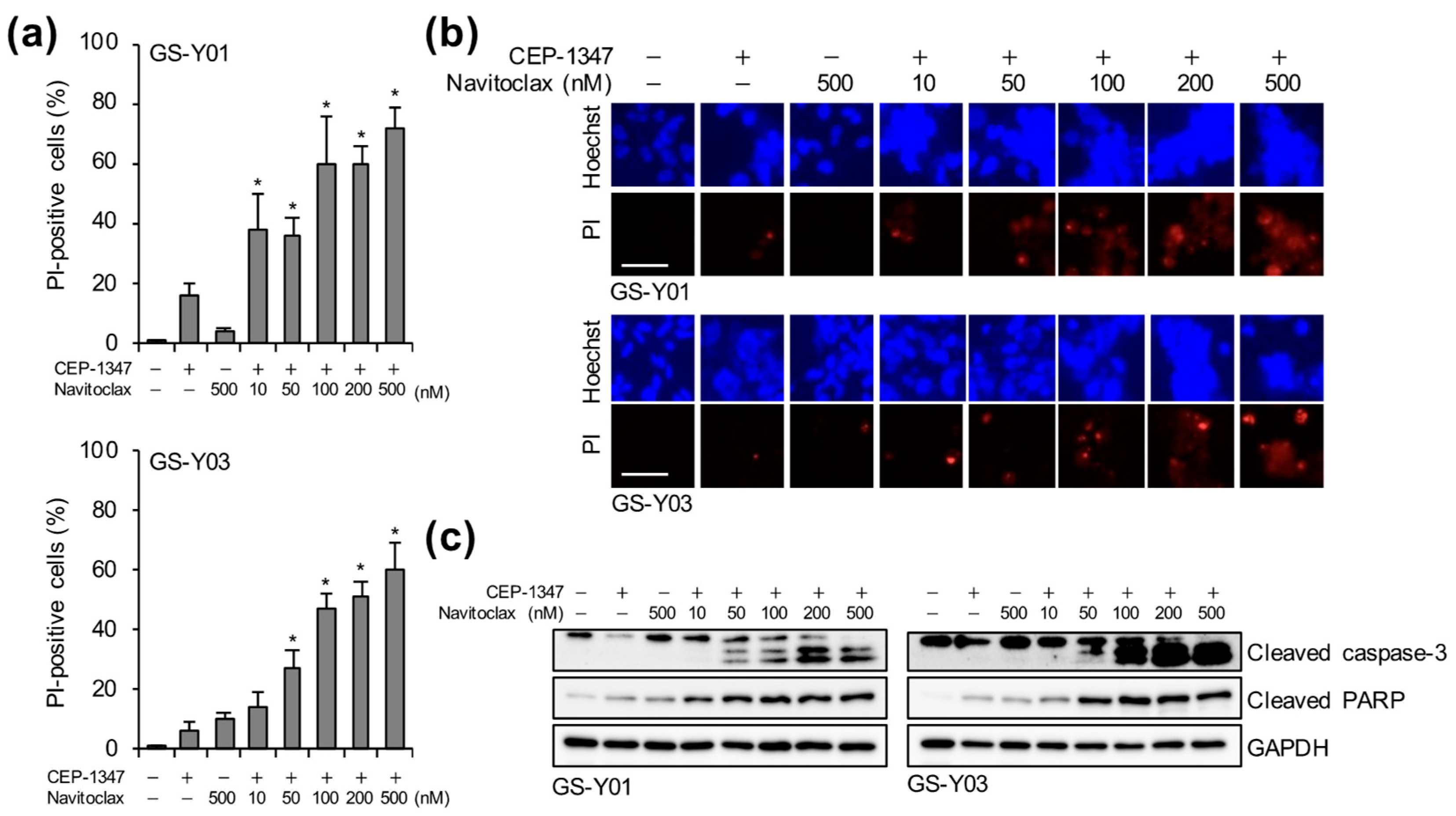
2.4. Navitoclax Exhibits Potent GSC-Killing Activity in Combination with CEP-1347 at Clinically Achievable Concentrations for the Brain
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Propidium Iodide (PI) Uptake Assay
4.4. Western Blot Analysis
4.5. Reverse Transcription (RT)-PCR Analysis
4.6. Fluorescence-Activated Cell Sorting (FACS) Analysis
4.7. Gene Silencing by siRNA
4.8. Data Reproducibility and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef] [PubMed]
- Ramar, V.; Guo, S.; Hudson, B.; Liu, M. Progress in Glioma Stem Cell Research. Cancers 2023, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, F.; Magni, F.; Leonardi, A.; Crescenzi, E. Therapy-Induced Senescence: Novel Approaches for Markers Identification. Int. J. Mol. Sci. 2024, 25, 8448. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Dorsey, J.F.; Fan, Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol. Ther. 2022, 231, 107985. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, L. Senescence Promotes the Recovery of Stemness among Cancer Cells via Reprograming. Biomolecules 2024, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Group, P.S. The safety and tolerability of a mixed lineage kinase inhibitor (CEP-1347) in PD. Neurology 2004, 62, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T. Attacking cancer’s Achilles heel: Antagonism of anti-apoptotic BCL-2 family members. FEBS J. 2016, 283, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Basu, A. The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy. Pharmacol. Ther. 2022, 230, 107943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. Febs J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef] [PubMed]
- Leten, C.; Struys, T.; Dresselaers, T.; Himmelreich, U. In vivo and ex vivo assessment of the blood brain barrier integrity in different glioblastoma animal models. J. Neurooncol. 2014, 119, 297–306. [Google Scholar] [CrossRef] [PubMed]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.F.; Voorbach, M.J.; Smith, A.; Reuter, D.R.; Zhuang, Y.; Wang, J.Q.; Wooten, D.W.; Asque, E.; Hu, M.; Hoft, C.; et al. Navitoclax safety, tolerability, and effect on biomarkers of senescence and neurodegeneration in aged nonhuman primates. Heliyon 2024, 10, e36483. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, D.; Gelfo, V.; Pagano, F.; Ferlizza, E.; Sgarzi, M.; Mazzeschi, M.; Morselli, A.; Miano, C.; D’Uva, G.; Lauriola, M. Senescence-associated reprogramming induced by interleukin-1 impairs response to EGFR neutralization. Cell. Mol. Biol. Lett. 2022, 27, 20. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, M.; Dalmo, E.; Segerman, A.; Rendo, V.; Westermark, B. p21-Dependent Senescence Induction by BMP4 Renders Glioblastoma Cells Vulnerable to Senolytics. Int. J. Mol. Sci. 2025, 26, 3974. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Okada, M.; Suzuki, S.; Sanomachi, T.; Seino, S.; Yamamoto, M.; Yamashita, H.; Kitanaka, C. Inhibition of Retinoblastoma Cell Growth by CEP1347 Through Activation of the P53 Pathway. Anticancer Res. 2020, 40, 4961–4968. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, Y.; Nakagawa-Saito, Y.; Togashi, K.; Suzuki, S.; Sugai, A.; Matsuda, K.I.; Sonoda, Y.; Kitanaka, C.; Okada, M. CEP-1347 Targets MDM4 Protein Expression to Activate p53 and Inhibit the Growth of Glioma Cells. Anticancer Res. 2022, 42, 4727–4733. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, Y.; Suzuki, S.; Nakagawa-Saito, Y.; Togashi, K.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; Okada, M. The Novel MDM4 Inhibitor CEP-1347 Activates the p53 Pathway and Blocks Malignant Meningioma Growth In Vitro and In Vivo. Biomedicines 2023, 11, 1967. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, V.S.; Loweth, C.J.; Ravula, S.B.; Wiemann, T.; Nguyen, T.; Xu, Y.; Todd, D.E.; Sheppard, D.; Pollack, S.; Polesskaya, O.; et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3. J. Med. Chem. 2013, 56, 8032–8048. [Google Scholar] [CrossRef] [PubMed]
- Huck, J.J.; Zhang, M.; McDonald, A.; Bowman, D.; Hoar, K.M.; Stringer, B.; Ecsedy, J.; Manfredi, M.G.; Hyer, M.L. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol. Cancer Res. 2010, 8, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Kang, C.H.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. JNK inhibitor SP600125 promotes the formation of polymerized tubulin, leading to G2/M phase arrest, endoreduplication, and delayed apoptosis. Exp. Mol. Med. 2009, 41, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial with Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients with Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Miyazaki, M.; Okamoto, I.; Ito, Y.; Ueda, K.; Seriu, T.; Nakagawa, K.; Hatake, K. Phase I study of dasatinib (BMS-354825) in Japanese patients with solid tumors. Cancer Sci. 2011, 102, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Southon, A.G.; Saleh, E.; Brinkmann, K.; Ke, F.; Iliopoulos, M.; Cross, R.S.; Jenkins, M.R.; Nhu, D.; Wang, Z.; et al. BH3 mimetic drugs cooperate with Temozolomide, JQ1 and inducers of ferroptosis in killing glioblastoma multiforme cells. Cell Death Differ. 2022, 29, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Matsuda, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takenouchi, S.; Ito, Y.; Nakamura, K.; Nakagawa-Saito, Y.; Mitobe, Y.; Togashi, K.; Suzuki, S.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; et al. Targeting Bcl-xL with Navitoclax Effectively Eliminates Senescent Tumor Cells That Appear Following CEP-1347-Induced Differentiation of Glioma Stem Cells. Int. J. Mol. Sci. 2025, 26, 6984. https://doi.org/10.3390/ijms26146984
Takenouchi S, Ito Y, Nakamura K, Nakagawa-Saito Y, Mitobe Y, Togashi K, Suzuki S, Sugai A, Sonoda Y, Kitanaka C, et al. Targeting Bcl-xL with Navitoclax Effectively Eliminates Senescent Tumor Cells That Appear Following CEP-1347-Induced Differentiation of Glioma Stem Cells. International Journal of Molecular Sciences. 2025; 26(14):6984. https://doi.org/10.3390/ijms26146984
Chicago/Turabian StyleTakenouchi, Senri, Yasufumi Ito, Kazuki Nakamura, Yurika Nakagawa-Saito, Yuta Mitobe, Keita Togashi, Shuhei Suzuki, Asuka Sugai, Yukihiko Sonoda, Chifumi Kitanaka, and et al. 2025. "Targeting Bcl-xL with Navitoclax Effectively Eliminates Senescent Tumor Cells That Appear Following CEP-1347-Induced Differentiation of Glioma Stem Cells" International Journal of Molecular Sciences 26, no. 14: 6984. https://doi.org/10.3390/ijms26146984
APA StyleTakenouchi, S., Ito, Y., Nakamura, K., Nakagawa-Saito, Y., Mitobe, Y., Togashi, K., Suzuki, S., Sugai, A., Sonoda, Y., Kitanaka, C., & Okada, M. (2025). Targeting Bcl-xL with Navitoclax Effectively Eliminates Senescent Tumor Cells That Appear Following CEP-1347-Induced Differentiation of Glioma Stem Cells. International Journal of Molecular Sciences, 26(14), 6984. https://doi.org/10.3390/ijms26146984