
Carbohydrate-Responsive Element-Binding Protein-Associated Metabolic Changes in Chemically Induced Hepatocarcinogenesis Mouse Model

Abstract
1. Introduction
2. Results
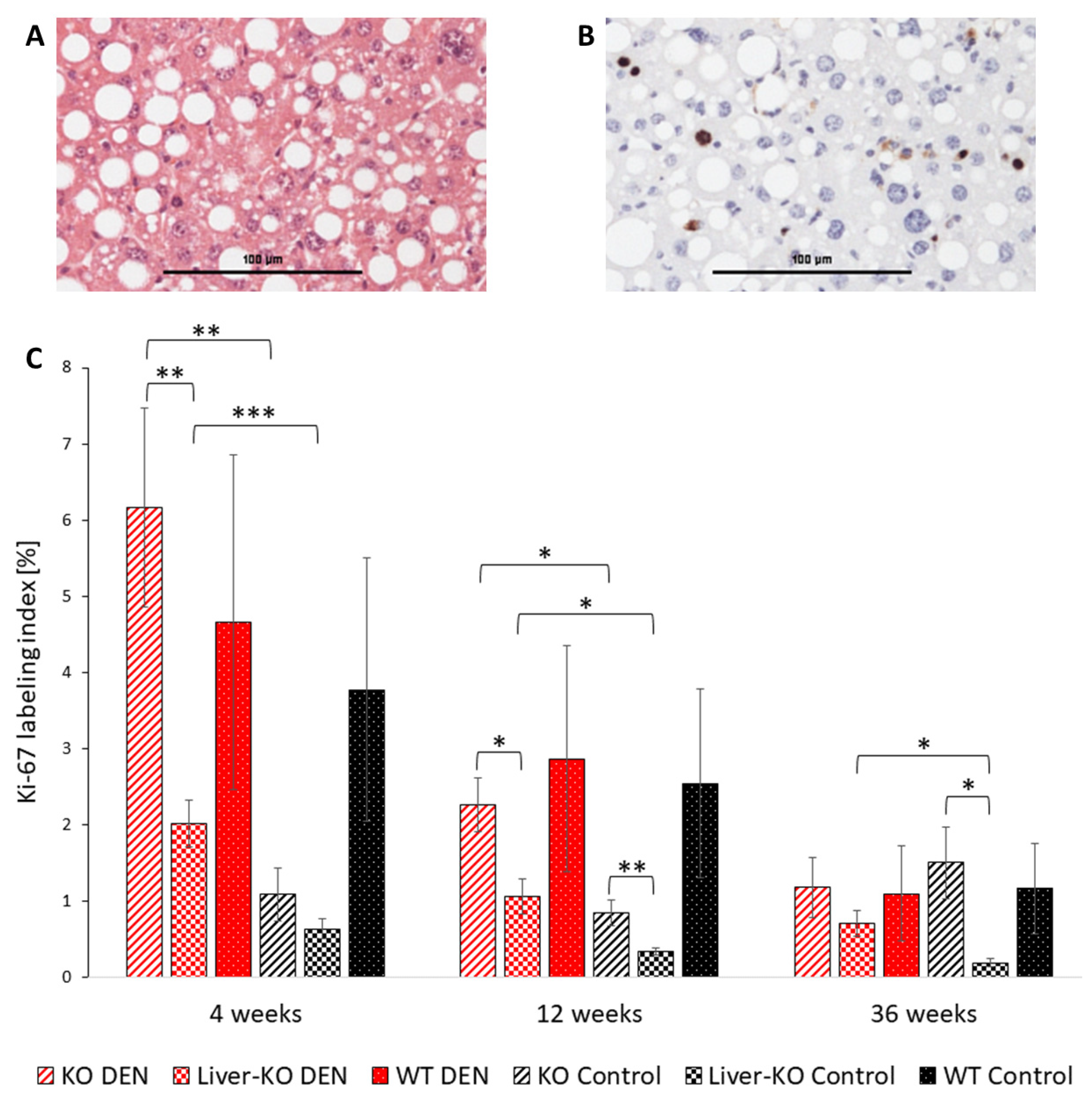
2.1. Proliferative Activity of Unaltered Liver Tissue
2.2. Glycogen Storage in Hepatocytes
2.3. Progression of Preneoplastic Foci to Liver Adenomas and Carcinomas
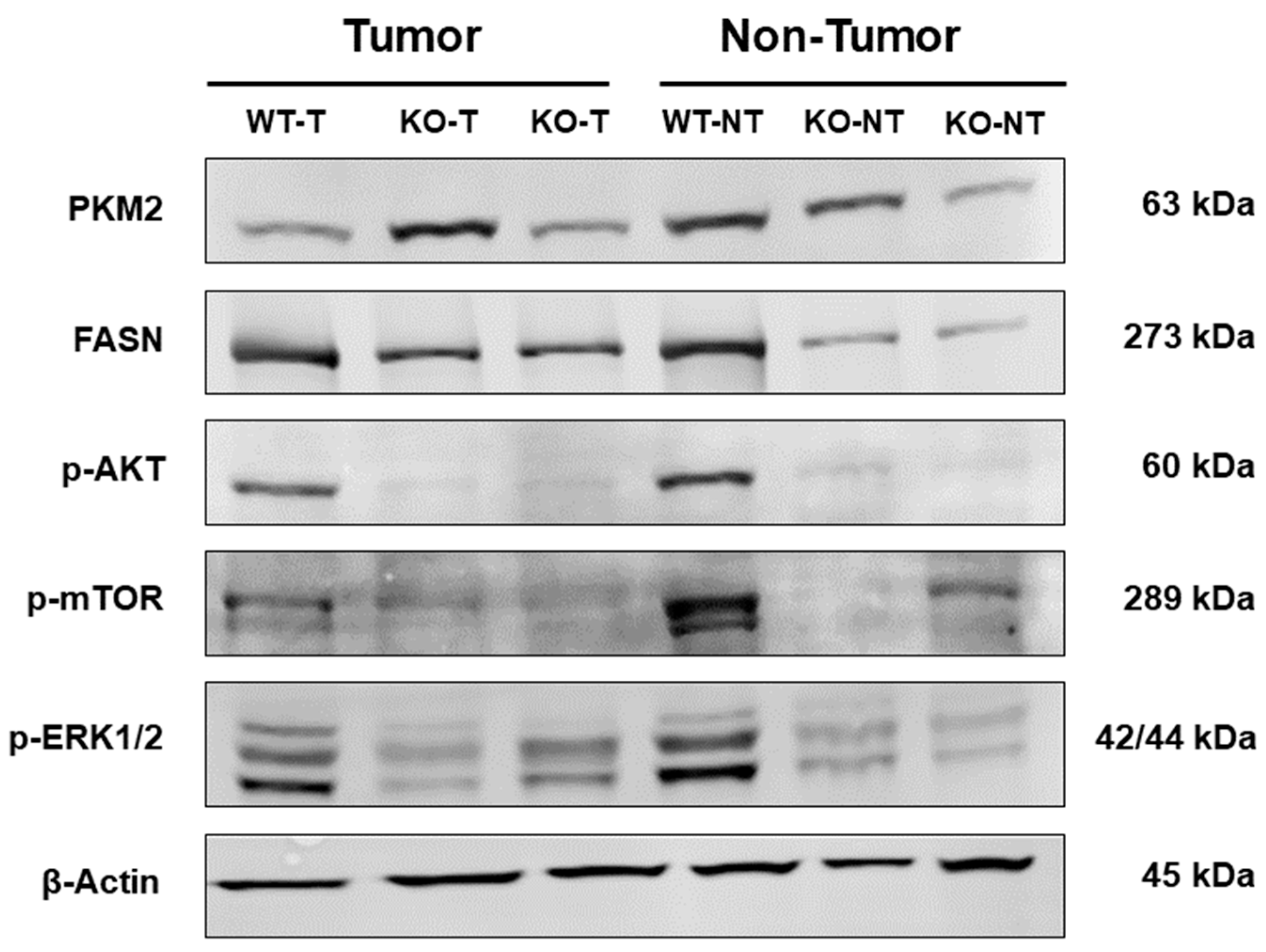
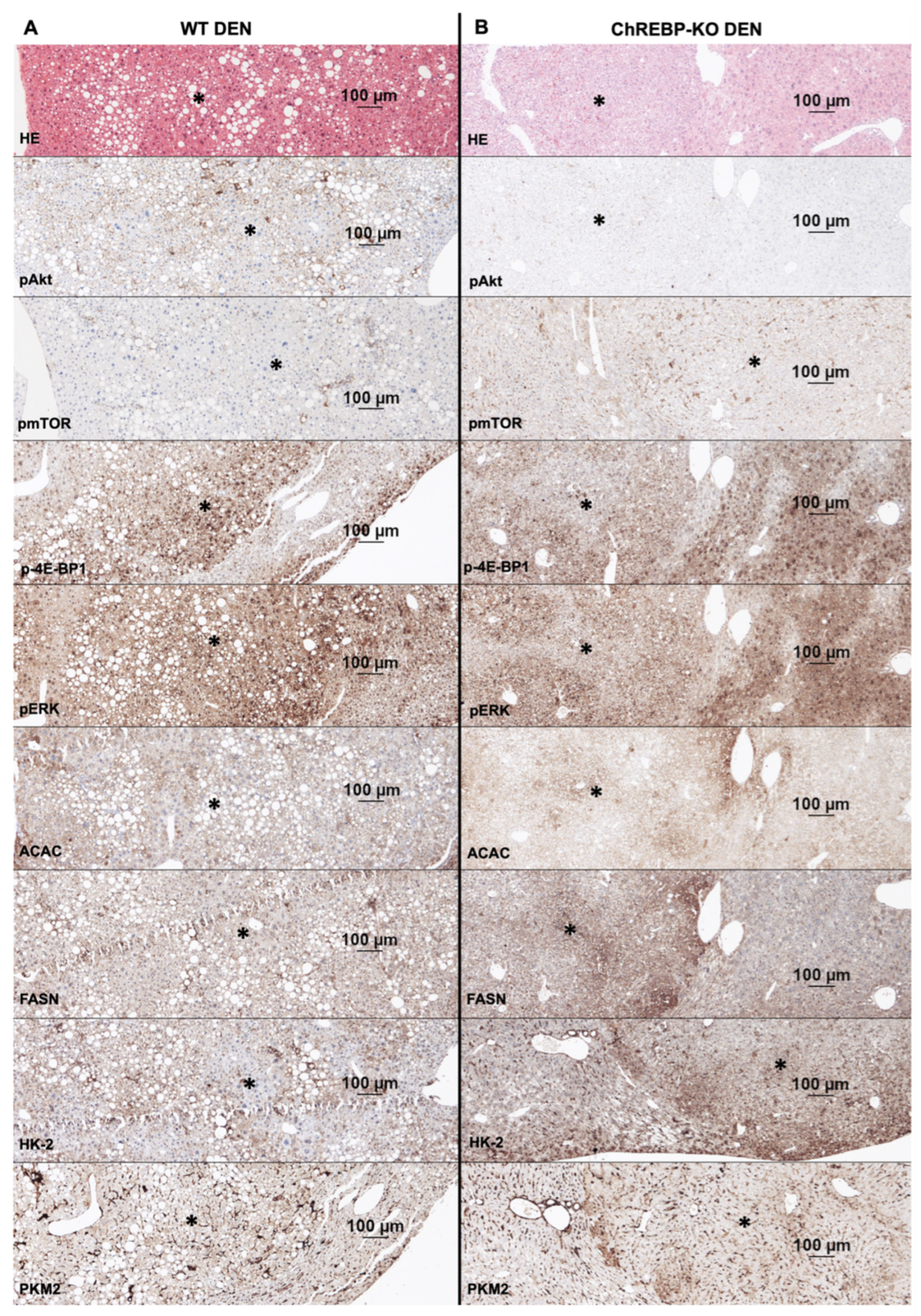
2.4. Altered Activation of Glycolysis, Lipogenesis, PI3K/AKT/mTOR, and Ras/MAPK Pathways in Liver Tumors
2.5. Protein Expression of Unaltered Liver Tissue
2.6. Body Weight Progression and Liver-to-Body-Weight Ratio
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Genotyping
4.3. Tissue Processing
4.4. Immunohistochemistry
4.5. Histological Evaluation
4.6. Protein Isolation
4.7. Western Blot
4.8. RNA Isolation and Quantitative PCR (qPCR)
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 3, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 6, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.W.; Lo, R.C.; Chan, L.K.; Ng, I.O. Molecular Pathogenesis of Hepatocellular Carcinoma. Liver Cancer 2016, 4, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.B.; Theve, E.J.; Feng, Y.; Fry, R.C.; Taghizadeh, K.; Clapp, K.M.; Boussahmain, C.; Cormier, K.S.; Fox, J.G. Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer Res. 2007, 24, 11536–11546. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.Y.; Lee, T.K.W. Preclinical mouse models of hepatocellular carcinoma: An overview and update. Exp. Cell Res. 2022, 2, 113042. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.P.; Colaço, A.A.; Oliveira, P.A. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumour Biol. 2017, 3, 1010428317695923. [Google Scholar] [CrossRef] [PubMed]
- Verna, L.; Whysner, J.; Williams, G.M. N-nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 12, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pérez, Y.; Carrasco-Legleu, C.; García-Cuellar, C.; Pérez-Carreón, J.; Hernández-García, S.; Salcido-Neyoy, M.; Alemán-Lazarini, L.; Villa-Treviño, S. Oxidative stress in carcinogenesis. Correlation between lipid peroxidation and induction of preneoplastic lesions in rat hepatocarcinogenesis. Cancer Lett. 2005, 217, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wang, Q.; Zhang, Q.; Lin, S.Y.; Zhu, Y.; Yang, X.; Guo, A.Y. Gene expression, regulation of DEN and HBx induced HCC mice models and comparisons of tumor, para-tumor and normal tissues. BMC Cancer 2017, 17, 862. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Wagner, E.F. Mouse models for liver cancer. Mol. Oncol. 2013, 7, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.Y.; Shim, J.J.; Shin, J.H.; Jeong, Y.S.; Kang, S.H.; Kim, S.B.; Eun, Y.G.; Lee, D.J.; Conner, E.A.; Factor, V.M.; et al. Integrated Genomic Comparison of Mouse Models Reveals Their Clinical Resemblance to Human Liver Cancer. Mol. Cancer Res. 2018, 11, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shay, C.; Saba, N.F.; Teng, Y. Cancer metabolism and carcinogenesis. Exp. Hematol. Oncol. 2024, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. Recent progress on the role of ChREBP in glucose and lipid metabolism. Endocr. J. 2013, 5, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 19, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Ribback, S.; Calvisi, D.F.; Cigliano, A.; Sailer, V.; Peters, M.; Rausch, J.; Heidecke, C.D.; Birth, M.; Dombrowski, F. Molecular and metabolic changes in human liver clear cell foci resemble the alterations occurring in rat hepatocarcinogenesis. J. Hepatol. 2013, 6, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Ribback, S.; Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Pes, G.M.; Porcu, A.; Pascale, R.M.; Li, L.; Qiao, Y.; et al. Oncogene-dependent addiction to carbohydrate-responsive element binding protein in hepatocellular carcinoma. Cell Cycle 2018, 12, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, V.; Mortoga, S.; Metzendorf, C.; Burkert, C.; Ehricke, K.; Knuth, E.; Zimmer, J.; Singer, S.; Nath, N.; Karim, M.; et al. Hormonally Induced Hepatocellular Carcinoma in Diabetic Wild Type and Carbohydrate Responsive Element Binding Protein Knockout Mice. Cells 2021, 10, 2787. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 7394, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wallace, M.; Sanchez-Gurmaches, J.; Hsiao, W.Y.; Li, H.; Lee, P.L.; Vernia, S.; Metallo, C.M.; Guertin, D.A. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun. 2016, 7, 11365. [Google Scholar] [CrossRef] [PubMed]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 2, E358–E364. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H.; Li, L.; Kuwahara-Otani, S.; Nakagawa, T.; Eguchi, H.; Yoshihara, D.; Shinohara, M.; Fujiwara, N.; Suzuki, K. A lack of ChREBP inhibits mitochondrial cristae formation in brown adipose tissue. Mol. Cell. Biochem. 2021, 10, 3577–3590. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Ma, X.; Su, K.; Qi, S.; Zhu, Y.; Lin, J.; Wang, C.; Yang, R.; Chen, X.; Wang, W.; et al. ChREBP-β regulates thermogenesis in brown adipose tissue. J. Endocrinol. 2020, 3, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Ribback, S.; Sonke, J.; Lohr, A.; Frohme, J.; Peters, K.; Holm, J.; Peters, M.; Cigliano, A.; Calvisi, D.F.; Dombrowski, F. Hepatocellular glycogenotic foci after combined intraportal pancreatic islet transplantation and knockout of the carbohydrate responsive element binding protein in diabetic mice. Oncotarget 2017, 61, 104315–104329. [Google Scholar] [CrossRef] [PubMed]
- Satomoto, K.; Aoki, M.; Wakita, A.; Yamagata, H.; Mitsumoto, T.; Okamoto, T.; Harada, R.; Hamada, S. Hepatocyte proliferation activity in untreated rats, measured by immunohistochemical detection of Ki-67: The effect of age on the repeated-dose liver micronucleus assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2023, 890, 503658. [Google Scholar] [CrossRef] [PubMed]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 11, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Cigliano, A.; Simile, M.M.; Vidili, G.; Pes, G.M.; Dore, M.P.; Urigo, F.; Cossu, E.; Che, L.; Feo, C.; Steinmann, S.M.; et al. Fatty Acid Synthase Promotes Hepatocellular Carcinoma Growth via S-Phase Kinase-Associated Protein 2/p27KIP1 Regulation. Medicina 2024, 7, 1160. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Che, L.; Cigliano, A.; Wang, X.; Peitta, G.; Tao, J.; Zhong, S.; Ribback, S.; Evert, M.; Chen, X.; et al. Pivotal Role of Fatty Acid Synthase in c-MYC Driven Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 22, 8467. [Google Scholar] [CrossRef] [PubMed]
- Benichou, E.; Seffou, B.; Topçu, S.; Renoult, O.; Lenoir, V.; Planchais, J.; Bonner, C.; Postic, C.; Prip-Buus, C.; Pecqueur, C.; et al. The transcription factor ChREBP Orchestrates liver carcinogenesis by coordinating the PI3K/AKT signaling and cancer metabolism. Nat. Commun. 2024, 15, 1879. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, H.; Mei, Y.; Ding, X.; Yang, X.; Li, C.; Deng, M.; Gong, J. Clinicopathological and prognostic significance of PKM2 protein expression in cirrhotic hepatocellular carcinoma and non-cirrhotic hepatocellular carcinoma. Sci. Rep. 2017, 7, 15294. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 9, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Tian, C.; Jia, Y.; Cai, Y.; Yan, P. MLXIPL promotes the migration, invasion, and glycolysis of hepatocellular carcinoma cells by phosphorylation of mTOR. BMC Cancer 2023, 23, 176. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 5, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio-Protocol 2019, 24, e3465. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KO DEN | L-KO DEN | WT DEN | KO Control | L-KO Control | WT Control | |
|---|---|---|---|---|---|---|
| n | 22 | 22 | 23 | 25 | 24 | 25 |
| Preneoplastic foci (%) | 9.09 * # | 39.13 * § # | 40.91 § | 8.33 | 0 § | 8.00 § |
| HCA (%) | 4.55 # | 8.7 | 27.27 # § | 8.33 | 0 | 4.00 § |
| HCC (%) | 13.64 | 0 | 9.09 | 8.33 | 0 | 12 |
| Tumors (HCA + HCC) (%) | 18.18 | 8.7 | 27.27 § | 8.33 | 0 | 12 |
| 4 Weeks | 12 Weeks | 36 Weeks | ||||
|---|---|---|---|---|---|---|
| DEN | Control | DEN | Control | DEN | Control | |
| C57Bl/6J WT | n = 16 | n = 24 | n = 8 | n = 24 | n = 22 | n = 25 |
| ChREBP-KO | n = 17 | n = 20 | n = 8 | n = 25 | n = 22 | n = 24 |
| Liver-ChREBP-KO | n = 17 | n = 25 | n = 8 | n = 25 | n = 23 | n = 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engeler, M.; Karim, M.; Gischke, M.; Willer, F.; Leiner, H.; Prey, J.; Ziegler, P.F.; Dombrowski, F.; Ribback, S. Carbohydrate-Responsive Element-Binding Protein-Associated Metabolic Changes in Chemically Induced Hepatocarcinogenesis Mouse Model. Int. J. Mol. Sci. 2025, 26, 6932. https://doi.org/10.3390/ijms26146932
Engeler M, Karim M, Gischke M, Willer F, Leiner H, Prey J, Ziegler PF, Dombrowski F, Ribback S. Carbohydrate-Responsive Element-Binding Protein-Associated Metabolic Changes in Chemically Induced Hepatocarcinogenesis Mouse Model. International Journal of Molecular Sciences. 2025; 26(14):6932. https://doi.org/10.3390/ijms26146932
Chicago/Turabian StyleEngeler, Maren, Majedul Karim, Marcel Gischke, Franziska Willer, Helen Leiner, Jessica Prey, Paul Friedrich Ziegler, Frank Dombrowski, and Silvia Ribback. 2025. "Carbohydrate-Responsive Element-Binding Protein-Associated Metabolic Changes in Chemically Induced Hepatocarcinogenesis Mouse Model" International Journal of Molecular Sciences 26, no. 14: 6932. https://doi.org/10.3390/ijms26146932
APA StyleEngeler, M., Karim, M., Gischke, M., Willer, F., Leiner, H., Prey, J., Ziegler, P. F., Dombrowski, F., & Ribback, S. (2025). Carbohydrate-Responsive Element-Binding Protein-Associated Metabolic Changes in Chemically Induced Hepatocarcinogenesis Mouse Model. International Journal of Molecular Sciences, 26(14), 6932. https://doi.org/10.3390/ijms26146932