
NR4A1 Mediates Bronchopulmonary Dysplasia-Like Lung Injury Induced by Intrauterine Inflammation in Mouse Offspring

 and
and
Abstract
1. Introduction
2. Results
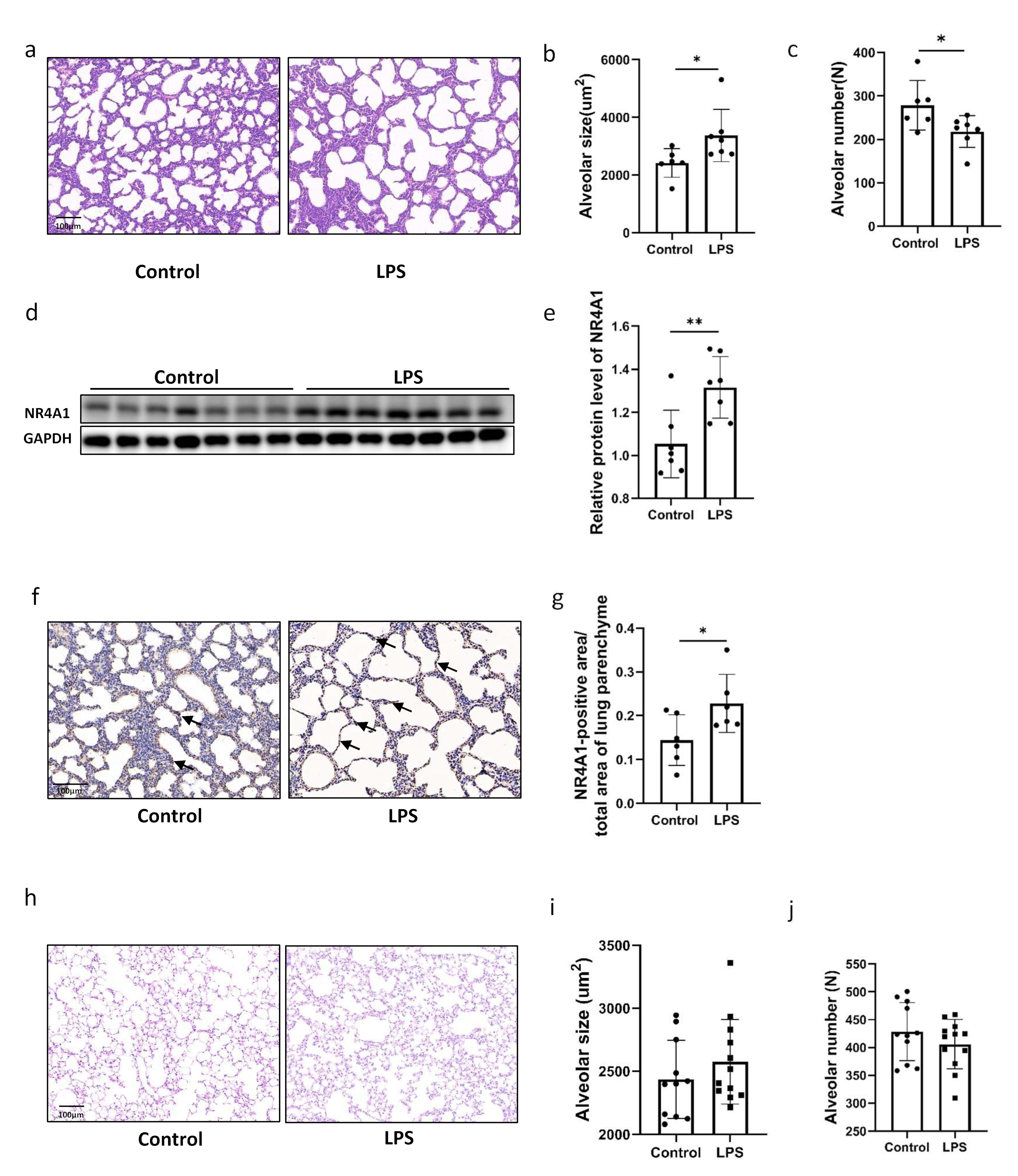
2.1. Intrauterine Inflammation Induces NR4A1 Expression and Causes Lung Injury in Offspring
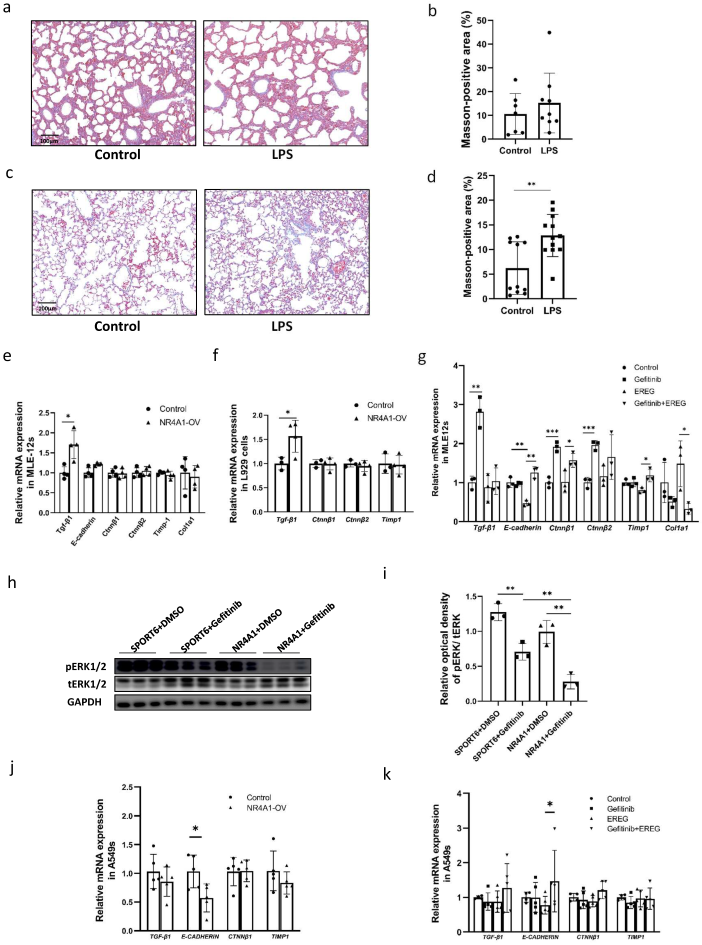
2.2. NR4A1 Involved in the Impaired the Lung Development Induced by Intrauterine Inflammation
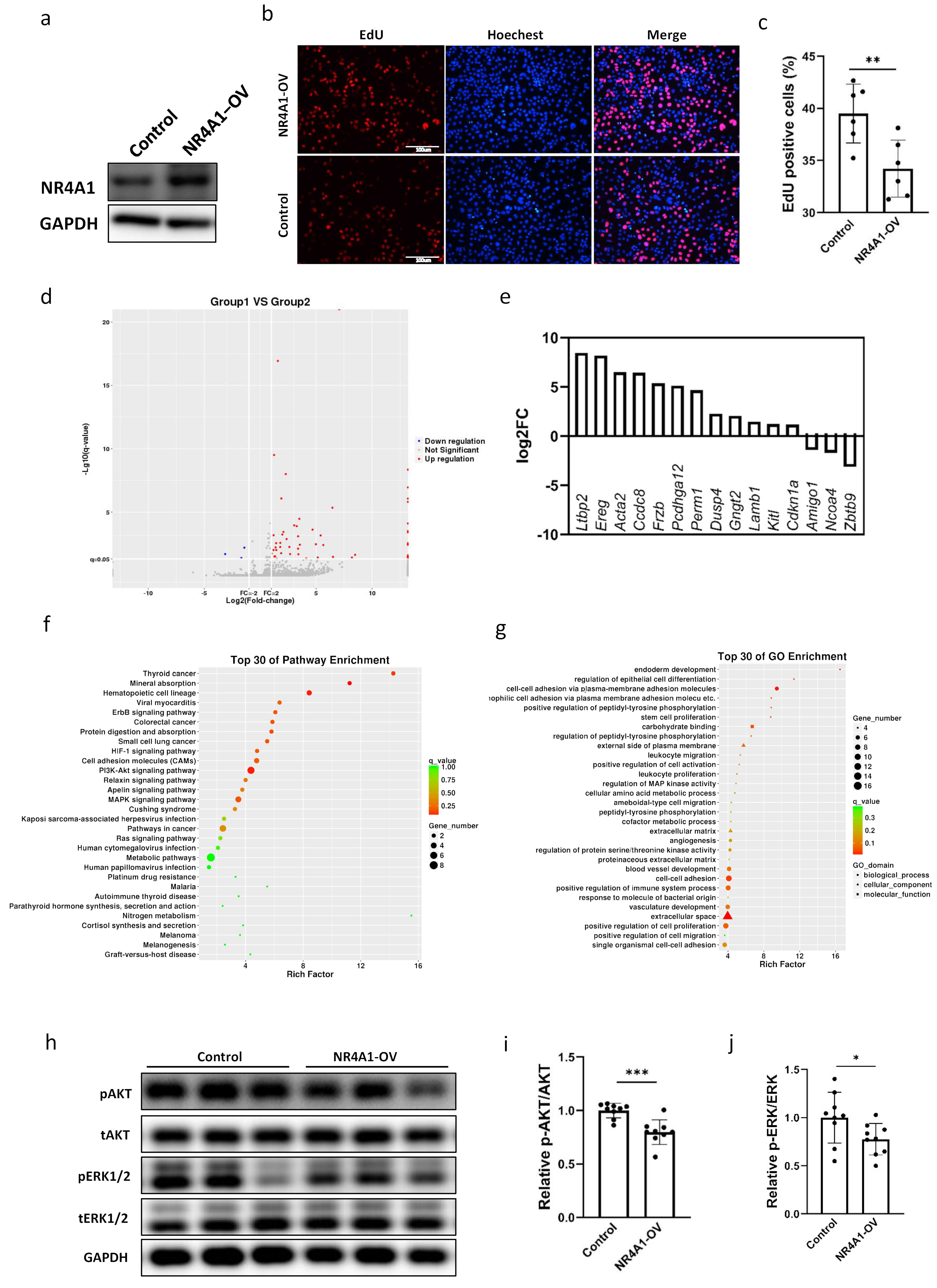
2.3. NR4A1 Promoted the Proliferation and Altered the Transcriptome of Lung Epithelial Cells
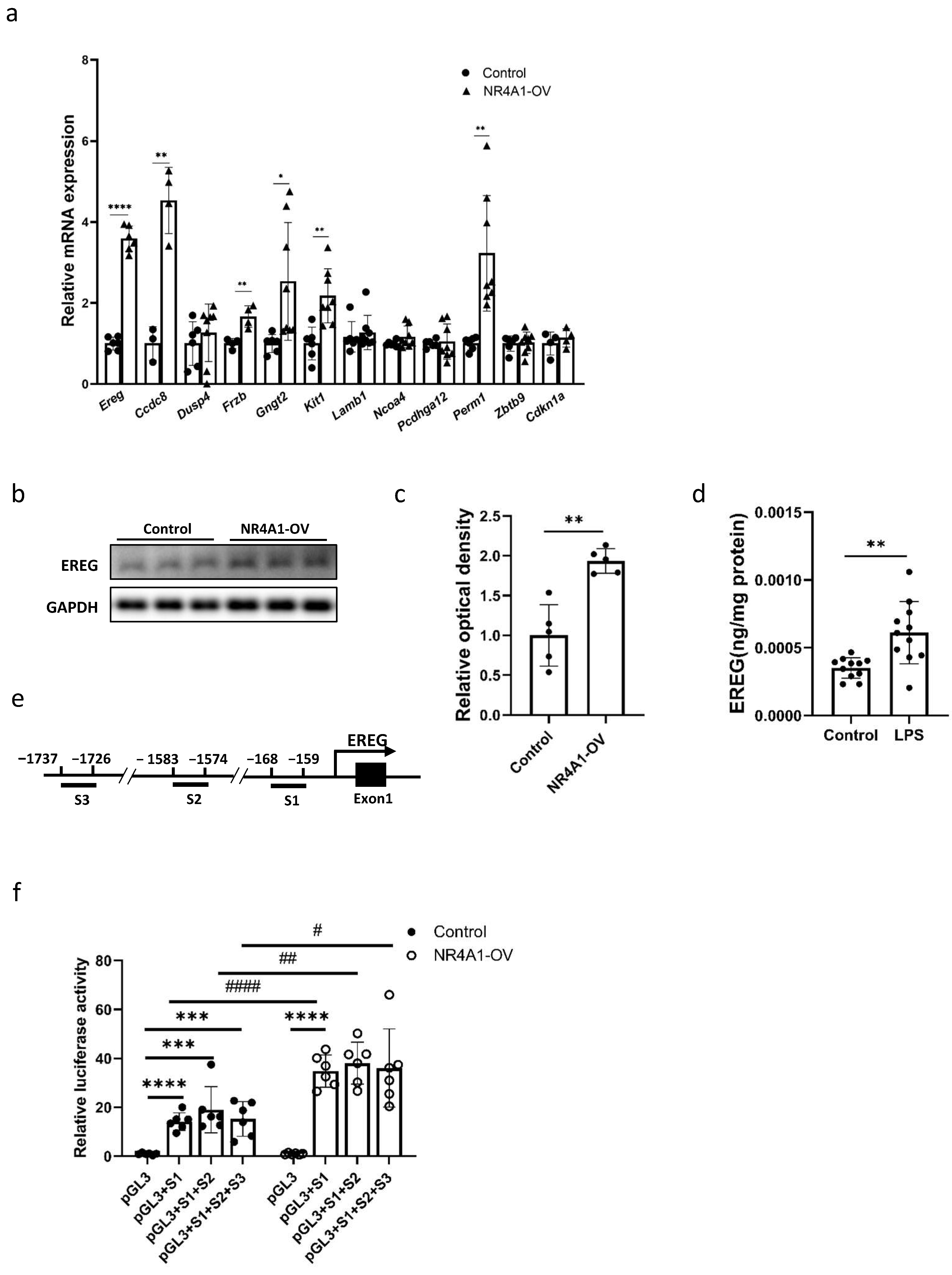
2.4. EREG Is a Downstream Target of NR4A1
3. Discussion
4. Materials and Methods
4.1. Mouse Model of Intrauterine Inflammation
4.2. Histological Staining Procedures
4.3. Morphometric Analysis of Alveolar Structure
4.4. Cell Culture and Treatments
4.5. Plasmid Construction and Transfection
4.6. RNA Sequencing and Data Analysis
4.7. Immunohistochemistry
4.8. RT-qPCR
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Western Blot
4.11. Cell Proliferation Assay
4.12. Cell Cycle Assay
4.13. Luciferase Assay
4.14. RNA Interference
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BPD | Bronchopulmonary dysplasia |
| DEGs | differentially expressed genes |
| EdU | 5-ethynyl-2′-deoxyuridine |
| EGFR | Epidermal growth factor receptor |
| EREG | Epiregulin |
| EMT | Epithelial–mesenchymal transition |
| MLE | Mouse lung epithelial |
| IUI | Intrauterine inflammation |
| NR4A1 | Nuclear receptor subfamily 4, group A, member 1 |
| LPS | Lipopolysaccharide |
References
- Jobe, A.H.; Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 163, 1723–1729. [Google Scholar] [CrossRef]
- Ehrenkranz, R.A.; Walsh, M.C.; Vohr, B.R.; Jobe, A.H.; Wright, L.L.; Fanaroff, A.A.; Wrage, L.A.; Poole, K.; National Institutes of Child, H.; Human Development Neonatal Research, N. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics 2005, 116, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Pinal, C. The developmental origins of adult disease. Matern. Child Nutr. 2005, 1, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Wenstrom, K.D.; Andrews, W.W.; Hauth, J.C.; Goldenberg, R.L.; DuBard, M.B.; Cliver, S.P. Elevated second-trimester amniotic fluid interleukin-6 levels predict preterm delivery. Am. J. Obstet. Gynecol. 1998, 178, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Zhao, J.; Zhang, Q.; Wang, T.; Ni, M.; Qi, S.; Shen, Q.; Li, W.; Li, B.; Ding, X.; et al. Aberrant methylation of Serpine1 mediates lung injury in neonatal mice prenatally exposed to intrauterine inflammation. Cell Biosci. 2022, 12, 164. [Google Scholar] [CrossRef]
- Huang, H.Y.; Chang, H.F.; Tsai, M.J.; Chen, J.S.; Wang, M.J. 6-Mercaptopurine attenuates tumor necrosis factor-alpha production in microglia through Nur77-mediated transrepression and PI3K/Akt/mTOR signaling-mediated translational regulation. J. Neuronflamm. 2016, 13, 78. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, G.X.; Yi, B.; Guo, Z.F.; Jang, S.; Yin, Y.; Yang, F.; Summer, R.; Sun, J. Nur77 limits endothelial barrier disruption to LPS in the mouse lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L615–L624. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, J.; Du, W.; Ren, J.; Zhang, Y.; Xie, F.; Xu, G. NR4A1 Promotes LPS-Induced Acute Lung Injury through Inhibition of Opa1-Mediated Mitochondrial Fusion and Activation of PGAM5-Related Necroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 6638244. [Google Scholar] [CrossRef]
- Tessem, J.S.; Moss, L.G.; Chao, L.C.; Arlotto, M.; Lu, D.; Jensen, M.V.; Stephens, S.B.; Tontonoz, P.; Hohmeier, H.E.; Newgard, C.B. Nkx6.1 regulates islet beta-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 5242–5247. [Google Scholar] [CrossRef]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.C.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular pathways: The role of NR4A orphan nuclear receptors in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef]
- Kurakula, K.; Vos, M.; Logiantara, A.; Roelofs, J.J.; Nieuwenhuis, M.A.; Koppelman, G.H.; Postma, D.S.; van Rijt, L.S.; de Vries, C.J. Nuclear Receptor Nur77 Attenuates Airway Inflammation in Mice by Suppressing NF-kappaB Activity in Lung Epithelial Cells. J. Immunol. 2015, 195, 1388–1398. [Google Scholar] [CrossRef]
- Principi, N.; Di Pietro, G.M.; Esposito, S. Bronchopulmonary dysplasia: Clinical aspects and preventive and therapeutic strategies. J. Transl. Med. 2018, 16, 36. [Google Scholar] [CrossRef]
- Pierro, M.; Van Mechelen, K.; van Westering-Kroon, E.; Villamor-Martínez, E.; Villamor, E. Endotypes of Prematurity and Phenotypes of Bronchopulmonary Dysplasia: Toward Personalized Neonatology. J. Pers. Med. 2022, 12, 687. [Google Scholar] [CrossRef]
- Hansmann, G.; Sallmon, H.; Roehr, C.C.; Kourembanas, S.; Austin, E.D.; Koestenberger, M. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr. Res. 2021, 89, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Schreiber, M.D.; Msall, M.E.; Kuban, K.C.K.; Joseph, R.M.; TM, O.S.; Allred, E.N.; Leviton, A. Cognitive Development and Quality of Life Associated With BPD in 10-Year-Olds Born Preterm. Pediatrics 2018, 141, 2017–2719. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Siddiqui, N.H.; Stocker, J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum. Pathol. 1998, 29, 710–717. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Thannickal, V.J. The lung mesenchyme in development, regeneration, and fibrosis. J. Clin. Investig. 2023, 133, e170498. [Google Scholar] [CrossRef]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef]
- Leng, R.; Meng, Y.; Sun, X.; Zhao, Y. NUF2 overexpression contributes to epithelial ovarian cancer progression via ERBB3-mediated PI3K-AKT and MAPK signaling axes. Front. Oncol. 2022, 12, 1057198. [Google Scholar] [CrossRef]
- Osaki, L.H.; Gama, P. MAPKs and signal transduction in the control of gastrointestinal epithelial cell proliferation and differentiation. Int. J. Mol. Sci. 2013, 14, 10143–10161. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Cheng, W.H.; Kao, S.Y.; Chen, C.L.; Yuliani, F.S.; Lin, L.Y.; Lin, C.H.; Chen, B.C. Amphiregulin induces CCN2 and fibronectin expression by TGF-beta through EGFR-dependent pathway in lung epithelial cells. Respir. Res. 2022, 23, 381. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Marsh, L.M.; Cakarova, L.; Kwapiszewska, G.; von Wulffen, W.; Herold, S.; Seeger, W.; Lohmeyer, J. Surface expression of CD74 by type II alveolar epithelial cells: A potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L442–L452. [Google Scholar] [CrossRef]
- Bao, T.; Zhu, H.; Zheng, Y.; Hu, J.; Wang, H.; Cheng, H.; Zhang, Y.; Tian, Z. Expression of long noncoding RNA uc.375 in bronchopulmonary dysplasia and its function in the proliferation and apoptosis of mouse alveolar epithelial cell line MLE 12. Front. Physiol. 2022, 13, 971732. [Google Scholar] [CrossRef]
- Stancil, I.T.; Michalski, J.E.; Hennessy, C.E.; Hatakka, K.L.; Yang, I.V.; Kurche, J.S.; Rincon, M.; Schwartz, D.A. Interleukin-6-dependent epithelial fluidization initiates fibrotic lung remodeling. Sci. Transl. Med. 2022, 14, eabo5254. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Hirani, D.; Alvira, C.M.; Danopoulos, S.; Milla, C.; Donato, M.; Tian, L.; Mohr, J.; Dinger, K.; Vohlen, C.; Selle, J.; et al. Macrophage-derived IL-6 trans-signalling as a novel target in the pathogenesis of bronchopulmonary dysplasia. Eur. Respir. J. 2022, 59, 2002248. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Steach, H.; Gauld, S.B.; Reinke-Breen, L.; Karman, J.; Carr, T.L.; Wetter, J.B.; Phillips, L.; Hinchcliff, M.; Flavell, R.A. Epiregulin is a dendritic cell-derived EGFR ligand that maintains skin and lung fibrosis. Sci. Immunol. 2022, 7, eabq6691. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Han, Y.; Xia, W.; Zhang, L.; Xu, S.; Ju, H.; Zhang, X.; Ren, G.; Liu, L.; et al. EREG-driven oncogenesis of Head and Neck Squamous Cell Carcinoma exhibits higher sensitivity to Erlotinib therapy. Theranostics 2020, 10, 10589–10605. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, R.; Chen, X.; Lee, S.B.; Pan, J.; Xiong, D.; Hu, J.; Miller, M.S.; Szabo, E.; Lubet, R.A.; et al. Effect of weekly or daily dosing regimen of Gefitinib in mouse models of lung cancer. Oncotarget 2017, 8, 72447–72456. [Google Scholar] [CrossRef]
- Heijink, I.H.; Postma, D.S.; Noordhoek, J.A.; Broekema, M.; Kapus, A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am. J. Respir. Cell Mol. Biol. 2010, 42, 69–79. [Google Scholar] [CrossRef]
- Huber, O.; Bierkamp, C.; Kemler, R. Cadherins and catenins in development. Curr. Opin. Cell Biol. 1996, 8, 685–691. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef]
- Yue, L.; Lu, X.; Dennery, P.A.; Yao, H. Metabolic dysregulation in bronchopulmonary dysplasia: Implications for identification of biomarkers and therapeutic approaches. Redox Biol. 2021, 48, 102104. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Furnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | Count | % | Genes | p Value |
|---|---|---|---|---|
| Thyroid cancer | 2 | 5.41 | Cdkn1a,Ncoa4 | 0.00098 |
| Mineral absorption | 2 | 4.26 | Slc39a4,Steap1 | 0.00194 |
| Hematopoietic cell lineage | 3 | 3.19 | Cd55,Cd24a,Kitl, | 0.00131 |
| Viral myocarditis | 2 | 2.41 | Cd55,Cd80 | 0.00957 |
| ErbB signaling pathway | 2 | 2.3 | Cdkn1a,Ereg | 0.01088 |
| Colorectal cancer | 2 | 2.22 | Cdkn1a,Ereg | 0.01193 |
| Protein digestion and absorption | 2 | 2.2 | Col11a1,Ace2 | 0.01230 |
| Small cell lung cancer | 2 | 2.08 | Cdkn1a,Lamb1 | 0.01422 |
| HIF-1 signaling pathway | 2 | 1.82 | Cdkn1a,Eno3 | 0.02048 |
| Cell adhesion molecules | 3 | 1.81 | Cd80,Cdh5,Selp | 0.00995 |
| PI3K-Akt signaling pathway | 6 | 1.66 | Cdkn1a,Gngt2,Nr4a1,Lamb1,Kitl,Ereg | 0.00191 |
| Relaxin signaling pathway | 2 | 1.52 | Gngt2,Acta2 | 0.03307 |
| Apelin signaling pathway | 2 | 1.43 | Gngt2,Acta2 | 0.03850 |
| MAPK signaling pathway | 4 | 1.32 | Nr4a1,Dusp4,Kitl,Ereg | 0.01704 |
| Cushing syndrome | 2 | 1.23 | Cdkn1a,Nr4a1 | 0.05578 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, X.; Li, R.; Yao, D.; Lei, Z.; Li, W.; Shen, Q.; Chen, Z.; Ni, M.; Li, B.; Liu, X.; et al. NR4A1 Mediates Bronchopulmonary Dysplasia-Like Lung Injury Induced by Intrauterine Inflammation in Mouse Offspring. Int. J. Mol. Sci. 2025, 26, 6931. https://doi.org/10.3390/ijms26146931
Ding X, Li R, Yao D, Lei Z, Li W, Shen Q, Chen Z, Ni M, Li B, Liu X, et al. NR4A1 Mediates Bronchopulmonary Dysplasia-Like Lung Injury Induced by Intrauterine Inflammation in Mouse Offspring. International Journal of Molecular Sciences. 2025; 26(14):6931. https://doi.org/10.3390/ijms26146931
Chicago/Turabian StyleDing, Xiya, Ruoxuan Li, Dongting Yao, Zhimin Lei, Wei Li, Qianwen Shen, Ze Chen, Meng Ni, Baihe Li, Xiaorui Liu, and et al. 2025. "NR4A1 Mediates Bronchopulmonary Dysplasia-Like Lung Injury Induced by Intrauterine Inflammation in Mouse Offspring" International Journal of Molecular Sciences 26, no. 14: 6931. https://doi.org/10.3390/ijms26146931
APA StyleDing, X., Li, R., Yao, D., Lei, Z., Li, W., Shen, Q., Chen, Z., Ni, M., Li, B., Liu, X., Zhao, J., Zhang, Q., & Liu, Z. (2025). NR4A1 Mediates Bronchopulmonary Dysplasia-Like Lung Injury Induced by Intrauterine Inflammation in Mouse Offspring. International Journal of Molecular Sciences, 26(14), 6931. https://doi.org/10.3390/ijms26146931