
Systemic Delivery Strategies for Oncolytic Viruses: Advancing Targeted and Efficient Tumor Therapy
Abstract
1. Introduction
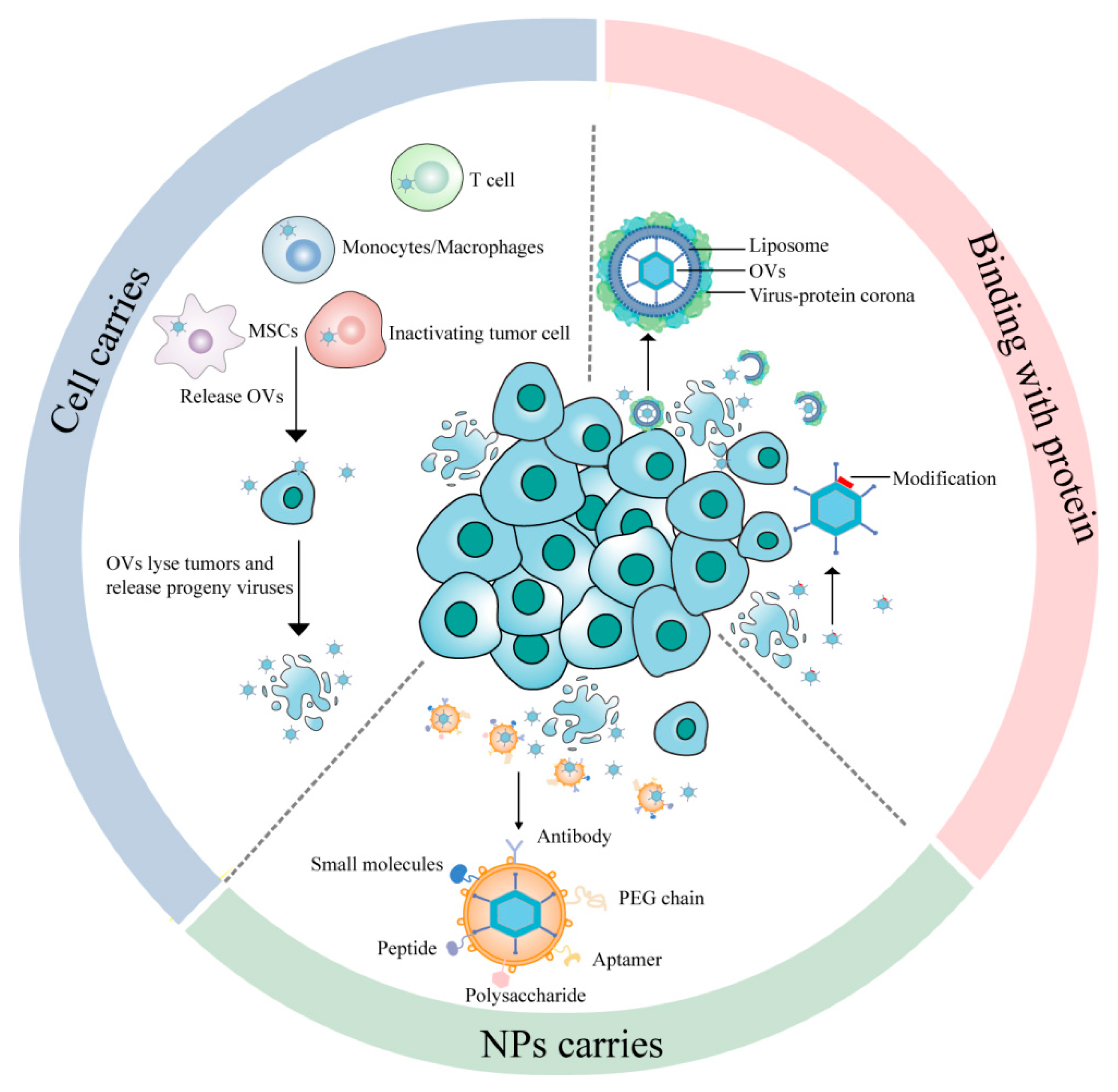
2. Cell-Mediated OV Systemic Delivery
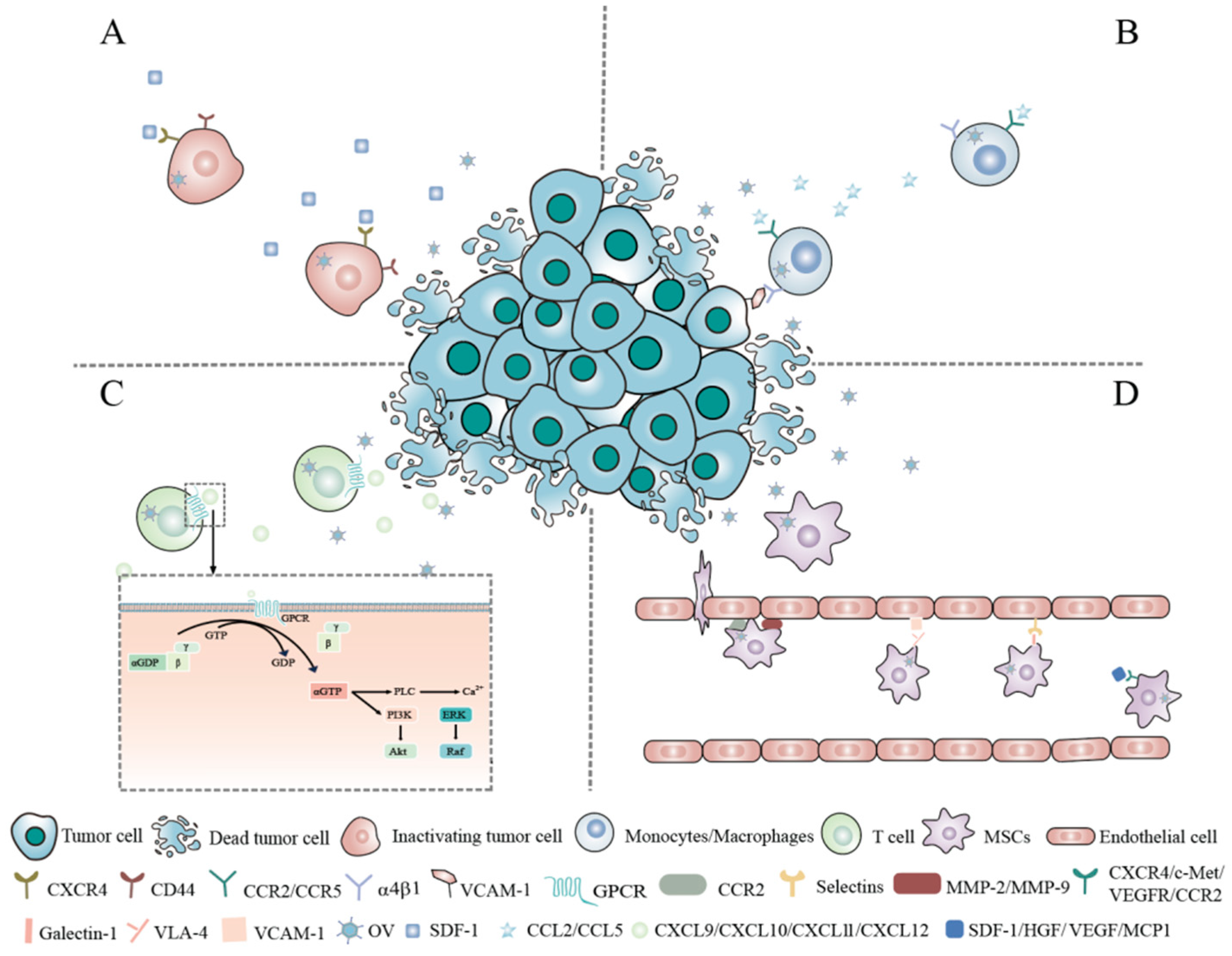
2.1. Tumor Cells
2.2. Monocytes/Macrophages
2.3. T Lymphocytes
2.3.1. Background
2.3.2. Application of T Lymphocytes as OV Carriers
Tumor-Infiltrating Lymphocytes (TILs)
Cytokine-Induced Killer (CIK) Cells
Chimeric Antigen Receptor T (CAR-T) Cells
2.3.3. Improvements
2.4. Mesenchymal Stem Cells (MSCs)
2.4.1. Background
2.4.2. Applications of MSCs as OV Carriers
2.4.3. Improvements
3. Binding with Proteins
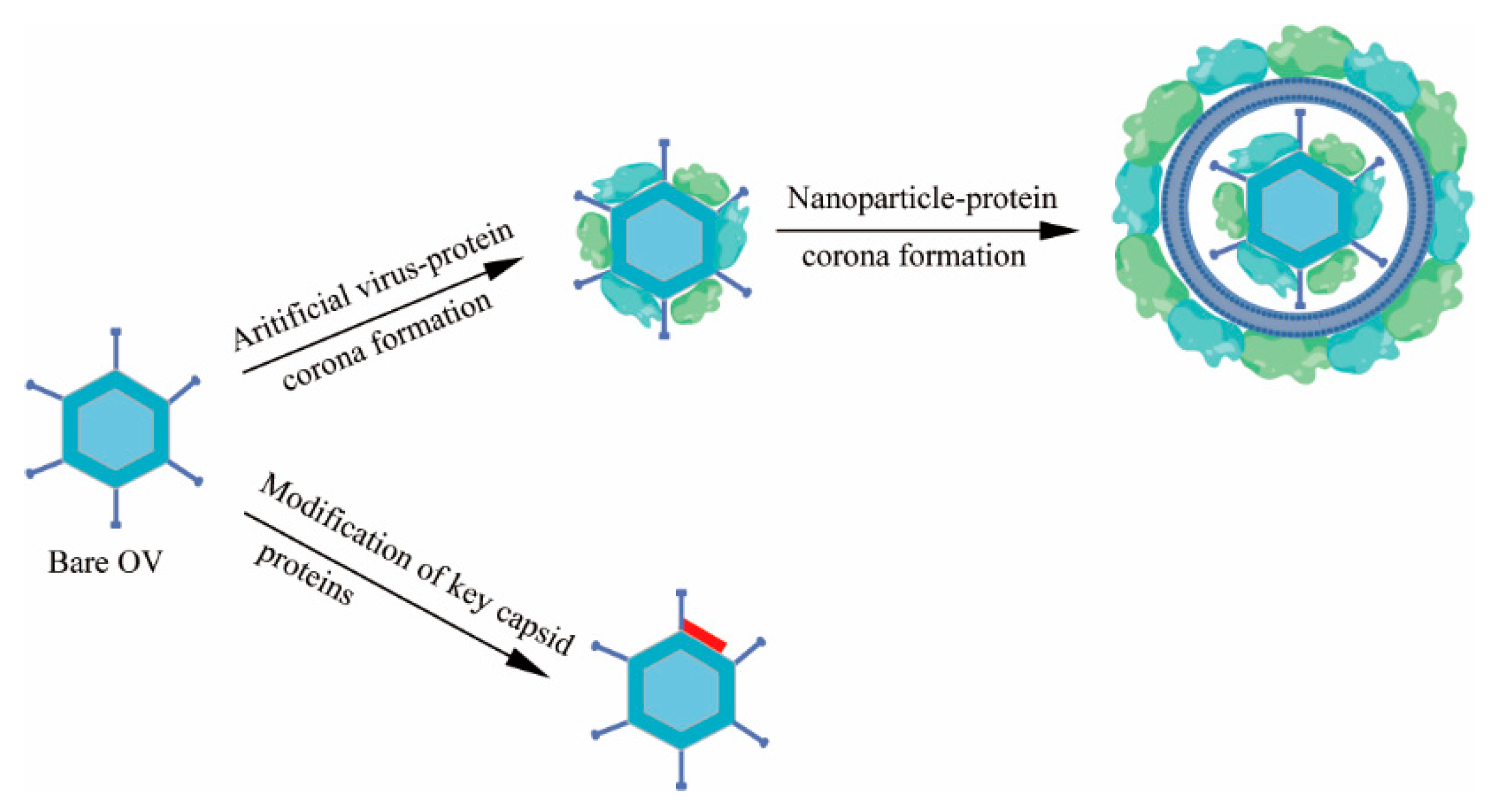
3.1. Virus-Protein Corona Replacement Strategy
3.2. Modification of Key Capsid Proteins
4. Nanoparticle (NP)-Based Delivery Systems
4.1. Microbial Nanocomposites
4.2. Biomineralization
4.3. Cell Membrane Nanovesicles
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OV | oncolytic virus |
| ICAM-1 | intercellular adhesion molecule-1 |
| IFN | interferon |
| RAS | rat sarcoma |
| RAF | rapidly accelerated fibrosarcoma |
| MEK | mitogen-activated protein kinase kinase |
| ERK | extracellular signal-regulated kinase |
| PKR | protein kinase R |
| TAAs | tumor-associated antigens |
| TNAs | tumor neoantigens |
| ICD | immunogenic cell death |
| DAMPs | damage-associated molecular patterns |
| PAMPs | pathogen-associated molecular patterns |
| TME | tumor microenvironment |
| APCs | antigen-presenting cells |
| FIX | factor IX |
| C4BP | C4b-binding protein |
| HSV-1 | herpes simplex virus |
| Adv | adenoviruses |
| IgM | immunoglobulin M |
| NPs | nanoparticles |
| CXCR4 | C-X-C chemokine receptor type 4 |
| CD44 | cluster of differentiation 44 |
| CTCs | circulating tumor cells |
| SDF-1 | stromal cell-derived factor-1 |
| MPI | magnetic particle imaging |
| Ad11 | adenovirus type 11 |
| LNT | liquid nitrogen treatment |
| VCAM-1 | vascular cell adhesion molecule-1 |
| MM6 | human acute monocytic leukemia |
| TAMs | tumor-associated macrophages |
| VEGF | vascular endothelial growth factor |
| TARP | tumor-associated receptor protein |
| PSA | prostate-specific antigen |
| PSMA | prostate-specific membrane antigen |
| GPCRs | G-protein-coupled receptors |
| AKT | protein kinase B |
| TILs | tumor-infiltrating lymphocytes |
| CIK | cytokine-induced killer |
| CAR T | chimeric antigen receptor T cell |
| TGF-β | transforming growth factor-beta |
| PD-L1 | programmed cell death protein 1 |
| ONCOTECH | oncolytic virus–T-cell chimera |
| NKG2D | natural killer group 2 member D |
| VV | vaccinia virus |
| TK | thymine kinase |
| VGF | viral growth factor |
| MAPK | mitogen-activated protein kinase |
| VSV | vesicular stomatitis virus |
| PDX | patient-derived xenograft |
| MSCs | mesenchymal stem cells |
| RSV | respiratory syncytial virus |
| COVID-19 | coronavirus disease 2019 |
| HVR1 | hyper-variable region 1 |
| EPR | enhanced permeability and retention |
| EVs | extracellular vesicles |
References
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Russo, S.; Ylösmäki, E.; Cerullo, V. Oncolytic ImmunoViroTherapy: A long history of crosstalk between viruses and immune system for cancer treatment. Pharmacol. Ther. 2022, 236, 108103. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, D.; Yang, L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 117. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers 2020, 12, 1699. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wojton, J.; Kaur, B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010, 21, 127–134. [Google Scholar] [CrossRef]
- Yaacov, B.; Lazar, I.; Tayeb, S.; Frank, S.; Izhar, U.; Lotem, M.; Perlman, R.; Ben-Yehuda, D.; Zakay-Rones, Z.; Panet, A. Extracellular matrix constituents interfere with Newcastle disease virus spread in solid tissue and diminish its potential oncolytic activity. J. Gen. Virol. 2012, 93 Pt 8, 1664–1672. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Zhang, Y.N.; Yang, X.; Wang, S.B.; Hu, P.Y. Delivery systems for enhancing oncolytic adenoviruses efficacy. Int. J. Pharm. 2020, 591, 119971. [Google Scholar] [CrossRef]
- Kepp, O.; Marabelle, A.; Zitvogel, L.; Kroemer, G. Oncolysis without viruses-inducing systemic anticancer immune responses with local therapies. Nat. Rev. Clin. Oncol. 2020, 17, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Blechacz, B.; Liu, C.; Schmeckpeper, J.D.; Tarara, J.E.; Federspiel, M.J.; Caplice, N.; Russell, S.J. Infected cell carriers: A new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 114–122. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Liu, S.; Kon, T.; Krol, A.; Li, C.Y.; Yuan, F. Characterisation of systemic dissemination of nonreplicating adenoviral vectors from tumours in local gene delivery. Br. J. Cancer 2005, 92, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Shayakhmetov, D.M. Oncolytic Viruses for Systemic Administration: Engineering a Whole Different Animal. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 904–907. [Google Scholar] [CrossRef]
- Kulu, Y.; Dorfman, J.D.; Kuruppu, D.; Fuchs, B.C.; Goodwin, J.M.; Fujii, T.; Kuroda, T.; Lanuti, M.; Tanabe, K.K. Comparison of intravenous versus intraperitoneal administration of oncolytic herpes simplex virus 1 for peritoneal carcinomatosis in mice. Cancer Gene Ther. 2009, 16, 291–297. [Google Scholar] [CrossRef]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000, 81 Pt 11, 2605–2609. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef]
- Keller, B.A.; Bell, J.C. Oncolytic viruses-immunotherapeutics on the rise. J. Mol. Med. 2016, 94, 979–991. [Google Scholar] [CrossRef]
- Howard, F.; Conner, J.; Danson, S.; Muthana, M. Inconsistencies in Modeling the Efficacy of the Oncolytic Virus HSV1716 Reveal Potential Predictive Biomarkers for Tolerability. Front. Mol. Biosci. 2022, 9, 889395. [Google Scholar] [CrossRef]
- Mao, L.J.; Kan, Y.; Li, B.H.; Ma, S.; Liu, Y.; Yang, D.L.; Yang, C. Combination Therapy of Prostate Cancer by Oncolytic Adenovirus Harboring Interleukin 24 and Ionizing Radiation. Front. Oncol. 2020, 10, 421. [Google Scholar] [CrossRef]
- Collet, G.; Grillon, C.; Nadim, M.; Kieda, C. Trojan horse at cellular level for tumor gene therapies. Gene 2013, 525, 208–216. [Google Scholar] [CrossRef] [PubMed]
- De Lombaerde, E.; De Wever, O.; De Geest, B.G. Delivery routes matter: Safety and efficacy of intratumoral immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188526. [Google Scholar] [CrossRef]
- Na, T.Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Ci, T.; Li, H.; Chen, G.; Wang, Z.; Wang, J.; Abdou, P.; Tu, Y.; Dotti, G.; Gu, Z. Cryo-shocked cancer cells for targeted drug delivery and vaccination. Sci. Adv. 2020, 6, eabc3013. [Google Scholar] [CrossRef]
- Mokbel, K. Unlocking the Power of the Homing Phenomenon: Why Breast Conserving Surgery Outshines Mastectomy in Overall Survival. Clin. Breast Cancer 2024, 24, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, H.; Sun, M.; Zhang, R.; Wang, J.; Zheng, H.; Zhu, C.; Yang, S.; Shen, X.; Shi, J.; et al. Inhibition of Tumor Metastasis by Liquid-Nitrogen-Shocked Tumor Cells with Oncolytic Viruses Infection. Adv. Mater. 2023, 35, e2212210. [Google Scholar] [CrossRef]
- Choi, M.R.; Stanton-Maxey, K.J.; Stanley, J.K.; Levin, C.S.; Bardhan, R.; Akin, D.; Badve, S.; Sturgis, J.; Robinson, J.P.; Bashir, R.; et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007, 7, 3759–3765. [Google Scholar] [CrossRef]
- Xia, Y.; Rao, L.; Yao, H.; Wang, Z.; Ning, P.; Chen, X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Adv. Mater. 2020, 32, e2002054. [Google Scholar] [CrossRef]
- Bunuales, M.; Garcia-Aragoncillo, E.; Casado, R.; Quetglas, J.I.; Hervas-Stubbs, S.; Bortolanza, S.; Benavides-Vallve, C.; Ortiz-de-Solorzano, C.; Prieto, J.; Hernandez-Alcoceba, R. Evaluation of monocytes as carriers for armed oncolytic adenoviruses in murine and Syrian hamster models of cancer. Hum. Gene Ther. 2012, 23, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Winder, N.; Atkinson, E.; Al-Janabi, H.; Allen, R.J.; Hughes, R.; Moamin, M.; Louie, R.; Evans, D.; Hutchinson, M.; et al. Macrophages Mediate the Antitumor Effects of the Oncolytic Virus HSV1716 in Mammary Tumors. Mol. Cancer Ther. 2021, 20, 589–601. [Google Scholar] [CrossRef]
- Berkeley, R.A.; Steele, L.P.; Mulder, A.A.; van den Wollenberg, D.J.M.; Kottke, T.J.; Thompson, J.; Coffey, M.; Hoeben, R.C.; Vile, R.G.; Melcher, A.; et al. Antibody-Neutralized Reovirus Is Effective in Oncolytic Virotherapy. Cancer Immunol. Res. 2018, 6, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.W.; Dogan, A.; Vrana, J.; Liu, C.; Ong, H.T.; Kumar, S.; Dispenzieri, A.; Dietz, A.B.; Russell, S.J. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am. J. Hematol. 2009, 84, 401–407. [Google Scholar] [CrossRef]
- Muthana, M.; Giannoudis, A.; Scott, S.D.; Fang, H.Y.; Coffelt, S.B.; Morrow, F.J.; Murdoch, C.; Burton, J.; Cross, N.; Burke, B.; et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011, 71, 1805–1815. [Google Scholar] [CrossRef]
- Reale, A.; Calistri, A.; Altomonte, J. Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics 2021, 13, 2192. [Google Scholar] [CrossRef]
- Huang, D.; Chen, X.; Zeng, X.; Lao, L.; Li, J.; Xing, Y.; Lu, Y.; Ouyang, Q.; Chen, J.; Yang, L.; et al. Targeting regulator of G protein signaling 1 in tumor-specific T cells enhances their trafficking to breast cancer. Nat. Immunol. 2021, 22, 865–879. [Google Scholar] [CrossRef]
- Santos, J.; Heiniö, C.; Quixabeira, D.; Zafar, S.; Clubb, J.; Pakola, S.; Cervera-Carrascon, V.; Havunen, R.; Kanerva, A.; Hemminki, A. Systemic Delivery of Oncolytic Adenovirus to Tumors Using Tumor-Infiltrating Lymphocytes as Carriers. Cells 2021, 10, 978. [Google Scholar] [CrossRef] [PubMed]
- Yotnda, P.; Savoldo, B.; Charlet-Berguerand, N.; Rooney, C.; Brenner, M. Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood 2004, 104, 2272–2280. [Google Scholar] [CrossRef]
- Lasner, T.M.; Tal-Singer, R.; Kesari, S.; Lee, V.M.; Trojanowski, J.Q.; Fraser, N.W. Toxicity and neuronal infection of a HSV-1 ICP34.5 mutant in nude mice. J. Neurovirology 1998, 4, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef]
- de Melo Gagliato, D.; Cortes, J.; Curigliano, G.; Loi, S.; Denkert, C.; Perez-Garcia, J.; Holgado, E. Tumor-infiltrating lymphocytes in Breast Cancer and implications for clinical practice. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Heiniö, C.; Havunen, R.; Santos, J.; de Lint, K.; Cervera-Carrascon, V.; Kanerva, A.; Hemminki, A. TNFa and IL2 Encoding Oncolytic Adenovirus Activates Pathogen and Danger-Associated Immunological Signaling. Cells 2020, 9, 798. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, X.; Bao, W.; Liu, G.; Wei, W.; Ping, Y. An oncolytic virus-T cell chimera for cancer immunotherapy. Nat. Biotechnol. 2024, 42, 1876–1887. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Vigolo, E.; D’Accardio, G.; Astori, G.; Rosato, A.; Sommaggio, R. How can Cytokine-induced killer cells overcome CAR-T cell limits. Front. Immunol. 2023, 14, 1229540. [Google Scholar] [CrossRef]
- Guo, Z.S.; Thorne, S.H.; Bartlett, D.L. Oncolytic virotherapy: Molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim. Biophys. Acta 2008, 1785, 217–231. [Google Scholar] [CrossRef]
- Edinger, M.; Cao, Y.A.; Verneris, M.R.; Bachmann, M.H.; Contag, C.H.; Negrin, R.S. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 2003, 101, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Fang, J.; Xue, G.; Wang, Z.; Li, X.; Zhou, M.; Jin, G.; Rahman, M.M.; McFadden, G.; Lu, Y. Induction of tumor cell autosis by myxoma virus-infected CAR-T and TCR-T cells to overcome primary and acquired resistance. Cancer Cell 2022, 40, 973–985.e977. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Tian, W.; Zhang, C.; Wang, X.; Li, W.; Zhang, Q.; Zhang, Y.; Zheng, J. Oncolytic adenovirus-mediated expression of CCL5 and IL12 facilitates CA9-targeting CAR-T therapy against renal cell carcinoma. Pharmacol. Res. 2023, 189, 106701. [Google Scholar] [CrossRef] [PubMed]
- Mardi, A.; Shirokova, A.V.; Mohammed, R.N.; Keshavarz, A.; Zekiy, A.O.; Thangavelu, L.; Mohamad, T.A.M.; Marofi, F.; Shomali, N.; Zamani, A.; et al. Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction. Cancer Cell Int. 2022, 22, 168. [Google Scholar] [CrossRef]
- Evgin, L.; Kottke, T.; Tonne, J.; Thompson, J.; Huff, A.L.; van Vloten, J.; Moore, M.; Michael, J.; Driscoll, C.; Pulido, J.; et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef]
- VanSeggelen, H.; Tantalo, D.G.; Afsahi, A.; Hammill, J.A.; Bramson, J.L. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics 2015, 2, 15014. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, J.; Qiu, Y.; Cheng, H.; Tao, M.; Xie, E.; Pei, X.; Li, W.; Zhang, L.; Wu, A.; et al. Suppression of the METTL3-m(6)A-integrin β1 axis by extracellular acidification impairs T cell infiltration and antitumor activity. Cell Rep. 2024, 43, 113796. [Google Scholar] [CrossRef]
- Ponte, A.L.; Marais, E.; Gallay, N.; Langonné, A.; Delorme, B.; Hérault, O.; Charbord, P.; Domenech, J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells 2007, 25, 1737–1745. [Google Scholar] [CrossRef]
- Mohr, A.; Zwacka, R. The future of mesenchymal stem cell-based therapeutic approaches for cancer-From cells to ghosts. Cancer Lett. 2018, 414, 239–249. [Google Scholar] [CrossRef]
- Rüster, B.; Göttig, S.; Ludwig, R.J.; Bistrian, R.; Müller, S.; Seifried, E.; Gille, J.; Henschler, R. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 2006, 108, 3938–3944. [Google Scholar] [CrossRef]
- Hernanda, P.Y.; Pedroza-Gonzalez, A.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q. Multipotent mesenchymal stromal cells in liver cancer: Implications for tumor biology and therapy. Biochim. Biophys. Acta 2014, 1846, 439–445. [Google Scholar] [CrossRef]
- Naji, A.; Eitoku, M.; Favier, B.; Deschaseaux, F.; Rouas-Freiss, N.; Suganuma, N. Biological functions of mesenchymal stem cells and clinical implications. Cell. Mol. Life Sci. CMLS 2019, 76, 3323–3348. [Google Scholar] [CrossRef]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Thanuja, M.Y.; Anupama, C.; Ranganath, S.H. Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv. Drug Deliv. Rev. 2018, 132, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Bolontrade, M.F.; Sganga, L.; Piaggio, E.; Viale, D.L.; Sorrentino, M.A.; Robinson, A.; Sevlever, G.; García, M.G.; Mazzolini, G.; Podhajcer, O.L. A specific subpopulation of mesenchymal stromal cell carriers overrides melanoma resistance to an oncolytic adenovirus. Stem Cells Dev. 2012, 21, 2689–2702. [Google Scholar] [CrossRef]
- Liu, R.; Wei, S.; Chen, J.; Xu, S. Mesenchymal stem cells in lung cancer tumor microenvironment: Their biological properties, influence on tumor growth and therapeutic implications. Cancer Lett. 2014, 353, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Huang, B.; Wu, H.B.; Wu, J.H.; Li, L.M.; Li, Y.X.; Hu, Y.L.; Han, M.; Shen, Y.Q.; Tabata, Y.; et al. Synergistic effects of co-administration of suicide gene expressing mesenchymal stem cells and prodrug-encapsulated liposome on aggressive lung melanoma metastases in mice. J. Control. Release Off. J. Control. Release Soc. 2015, 209, 260–271. [Google Scholar] [CrossRef]
- Li, M.; Sun, S.; Dangelmajer, S.; Zhang, Q.; Wang, J.; Hu, F.; Dong, F.; Kahlert, U.D.; Zhu, M.; Lei, T. Exploiting tumor-intrinsic signals to induce mesenchymal stem cell-mediated suicide gene therapy to fight malignant glioma. Stem Cell Res. Ther. 2019, 10, 88. [Google Scholar] [CrossRef]
- Shimizu, Y.; Gumin, J.; Gao, F.; Hossain, A.; Shpall, E.J.; Kondo, A.; Parker Kerrigan, B.C.; Yang, J.; Ledbetter, D.; Fueyo, J.; et al. Characterization of patient-derived bone marrow human mesenchymal stem cells as oncolytic virus carriers for the treatment of glioblastoma. J. Neurosurg. 2022, 136, 757–767. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 134–153. [Google Scholar] [CrossRef]
- Josiah, D.T.; Zhu, D.; Dreher, F.; Olson, J.; McFadden, G.; Caldas, H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Jazowiecka-Rakus, J.; Sochanik, A.; Rusin, A.; Hadryś, A.; Fidyk, W.; Villa, N.; Rahman, M.M.; Chmielik, E.; Franco, L.S.; McFadden, G. Myxoma Virus-Loaded Mesenchymal Stem Cells in Experimental Oncolytic Therapy of Murine Pulmonary Melanoma. Mol. Ther. Oncolytics 2020, 18, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774–34787. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Seah, I.; Bougazzoul, O.; Choi, G.; Meeth, K.; Bosenberg, M.W.; Wakimoto, H.; Fisher, D.; Shah, K. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc. Natl. Acad. Sci. USA 2017, 114, E6157–E6165. [Google Scholar] [CrossRef]
- Ong, H.T.; Federspiel, M.J.; Guo, C.M.; Ooi, L.L.; Russell, S.J.; Peng, K.W.; Hui, K.M. Systemically delivered measles virus-infected mesenchymal stem cells can evade host immunity to inhibit liver cancer growth. J. Hepatol. 2013, 59, 999–1006. [Google Scholar] [CrossRef]
- Babaei, A.; Soleimanjahi, H.; Soleimani, M.; Arefian, E. Mesenchymal stem cells loaded with oncolytic reovirus enhances antitumor activity in mice models of colorectal cancer. Biochem. Pharmacol. 2021, 190, 114644. [Google Scholar] [CrossRef]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Reis, M.; Mavin, E.; Nicholson, L.; Green, K.; Dickinson, A.M.; Wang, X.N. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Dendritic Cell Maturation and Function. Front. Immunol. 2018, 9, 2538. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Fleury-Cappellesso, S.; Gadelorge, M.; Dietrich, G.; Bourin, P.; Fournié, J.J.; Poupot, R. A regulatory cross-talk between Vgamma9Vdelta2 T lymphocytes and mesenchymal stem cells. Eur. J. Immunol. 2009, 39, 752–762. [Google Scholar] [CrossRef]
- Huang, H.; Liu, M.; Sun, M.; Duan, S.; Pan, S.; Liu, P.; Cheng, Z.; Ergonul, O.; Can, F.; Wang, Z.; et al. Virus-Protein Corona Replacement Strategy to Improve the Antitumor Efficacy of Intravenously Injected Oncolytic Adenovirus. ACS Nano 2023, 17, 14461–14474. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zeng, L.; Yao, L.; Wang, Z.; Yang, X.; Shi, J.; Hu, L.; Liu, Q.; Chen, C.; Xia, T.; et al. Inherited and acquired corona of coronavirus in the host: Inspiration from the biomolecular corona of nanoparticles. Nano Today 2021, 39, 101161. [Google Scholar] [CrossRef]
- Atasheva, S.; Emerson, C.C.; Yao, J.; Young, C.; Stewart, P.L.; Shayakhmetov, D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020, 12, eabc6659. [Google Scholar] [CrossRef]
- Bradley, R.R.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus serotype 5 neutralizing antibodies target both hexon and fiber following vaccination and natural infection. J. Virol. 2012, 86, 625–629. [Google Scholar] [CrossRef]
- Rojas, L.A.; Condezo, G.N.; Moreno, R.; Fajardo, C.A.; Arias-Badia, M.; San Martín, C.; Alemany, R. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J. Control. Release Off. J. Control. Release Soc. 2016, 237, 78–88. [Google Scholar] [CrossRef]
- Tesfay, M.Z.; Ammayappan, A.; Federspiel, M.J.; Barber, G.N.; Stojdl, D.; Peng, K.W.; Russell, S.J. Vesiculovirus neutralization by natural IgM and complement. J. Virol. 2014, 88, 6148–6157. [Google Scholar] [CrossRef]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geiβ, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef]
- Rangaswamy, U.S.; Cotter, C.R.; Cheng, X.; Jin, H.; Chen, Z. CD55 is a key complement regulatory protein that counteracts complement-mediated inactivation of Newcastle Disease Virus. J. Gen. Virol. 2016, 97, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Doroudian, M.; MacLoughlin, R.; Poynton, F.; Prina-Mello, A.; Donnelly, S.C. Nanotechnology based therapeutics for lung disease. Thorax 2019, 74, 965–976. [Google Scholar] [CrossRef]
- Feola, S.; Russo, S.; Martins, B.; Lopes, A.; Vandermeulen, G.; Fluhler, V.; De Giorgi, C.; Fusciello, M.; Pesonen, S.; Ylösmäki, E.; et al. Peptides-Coated Oncolytic Vaccines for Cancer Personalized Medicine. Front. Immunol. 2022, 13, 826164. [Google Scholar] [CrossRef]
- Zein, R.; Sharrouf, W.; Selting, K. Physical Properties of Nanoparticles That Result in Improved Cancer Targeting. J. Oncol. 2020, 2020, 5194780. [Google Scholar] [CrossRef] [PubMed]
- Iscaro, A.; Jones, C.; Forbes, N.; Mughal, A.; Howard, F.N.; Janabi, H.A.; Demiral, S.; Perrie, Y.; Essand, M.; Weglarz, A.; et al. Targeting circulating monocytes with CCL2-loaded liposomes armed with an oncolytic adenovirus. Nanomed. Nanotechnol. Biol. Med. 2022, 40, 102506. [Google Scholar] [CrossRef] [PubMed]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Ciernikova, S.; Sevcikova, A.; Stevurkova, V.; Mego, M. Tumor microbiome-an integral part of the tumor microenvironment. Front. Oncol. 2022, 12, 1063100. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Cummins, J.; Tangney, M. Bacteria and tumours: Causative agents or opportunistic inhabitants? Infect. Agents Cancer 2013, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.H.; Nguyen, V.H.; Jiang, S.N.; Park, S.H.; Tan, W.; Hong, S.H.; Shin, M.G.; Chung, I.J.; Hong, Y.; Bom, H.S.; et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 2017, 9, eaak9537. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.S.; Munn, L.L.; Fukumura, D.; Jain, R.K. Sparse initial entrapment of systemically injected Salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 2003, 63, 5188–5193. [Google Scholar]
- Ban, W.; Sun, M.; Huang, H.; Huang, W.; Pan, S.; Liu, P.; Li, B.; Cheng, Z.; He, Z.; Liu, F.; et al. Engineered bacterial outer membrane vesicles encapsulating oncolytic adenoviruses enhance the efficacy of cancer virotherapy by augmenting tumor cell autophagy. Nat. Commun. 2023, 14, 2933. [Google Scholar] [CrossRef]
- Sun, M.; Yang, S.; Huang, H.; Gao, P.; Pan, S.; Cheng, Z.; He, Z.; Wang, Z.; Sun, J.; Liu, F. Boarding Oncolytic Viruses onto Tumor-Homing Bacterium-Vessels for Augmented Cancer Immunotherapy. Nano Lett. 2022, 22, 5055–5064. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef]
- Shen, W.; Pei, P.; Zhang, C.; Li, J.; Han, X.; Liu, T.; Shi, X.; Su, Z.; Han, G.; Hu, L.; et al. A Polymeric Hydrogel to Eliminate Programmed Death-Ligand 1 for Enhanced Tumor Radio-Immunotherapy. ACS Nano 2023, 17, 23998–24011. [Google Scholar] [CrossRef]
- Li, Y.S.; Ye, L.Y.; Luo, Y.X.; Zheng, W.J.; Si, J.X.; Yang, X.; Zhang, Y.N.; Wang, S.B.; Zou, H.; Jin, K.T.; et al. Tumor-targeted delivery of copper-manganese biomineralized oncolytic adenovirus for colorectal cancer immunotherapy. Acta Biomater. 2024, 179, 243–255. [Google Scholar] [CrossRef]
- Zhang, X.; Bai, X.C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Li, X.; Zhang, J.; Zhao, Q.R.; Zhang, M.J.; Liu, A.A.; Pang, D.W.; Xie, H.Y. MnCaCs-Biomineralized Oncolytic Virus for Bimodal Imaging-Guided and Synergistically Enhanced Anticancer Therapy. Nano Lett. 2019, 19, 8002–8009. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; El Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release Off. J. Control. Release Soc. 2019, 294, 165–175. [Google Scholar] [CrossRef]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release Off. J. Control. Release Soc. 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Lv, P.; Chen, X.; Fu, S.; Ren, E.; Liu, C.; Liu, X.; Jiang, L.; Zeng, Y.; Wang, X.; Liu, G. Surface engineering of oncolytic adenovirus for a combination of immune checkpoint blockade and virotherapy. Biomater. Sci. 2021, 9, 7392–7401. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Wang, S.; Qiao, L.; Yu, X.; Wang, N.; Chen, L.; Zhang, X.; Zhao, X.; Liu, H.; Wang, T.; et al. Oncolytic Virus-Driven Biotherapies from Bench to Bedside. Small 2023, 19, e2206948. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, R.; Huang, H.; Liu, P.; Zhao, X.; Wu, H.; He, Y.; Xu, R.; Qin, X.; Cheng, Z.; et al. Erythrocyte-Leveraged Oncolytic Virotherapy (ELeOVt): Oncolytic Virus Assembly on Erythrocyte Surface to Combat Pulmonary Metastasis and Alleviate Side Effects. Adv. Sci. 2024, 11, e2303907. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kholodenko, R.V.; Majouga, A.G.; Yarygin, K.N. Apoptotic MSCs and MSC-Derived Apoptotic Bodies as New Therapeutic Tools. Curr. Issues Mol. Biol. 2022, 44, 5153–5172. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery System | Tumor Targeting | Delivery Efficiency | Immune Escape | Antitumor Effect In Vivo | References | ||
|---|---|---|---|---|---|---|---|
| Cell-based OV delivery | Tumor cells | CD44 and E-cadherin on TC-1 mouse lung cancer cells. | More than 110-fold enrichment of LNT-Ad11. | 83% of OVs were protected from antibody neutralization. | The number of M2 cells and Treg cells was reduced; CD4+ and CD8+ and T cells priming is enhanced. | [28] | |
| Monocytes/Macrophages | Binding to CCL2 in TME via CCR2 on the cell surface. | 500 out of the 106 intravenously injected cells accumulated in tumors. | \ | Tumor-bearing mice exhibited a 1.5-fold increase in overall survival. | [35] | ||
| T lymphocytes | Chemokines, such as CXCL9, CXCL19 ect, secreted by the tumor promoted T-cell migration into tumors. | The delivery of the OVs–T cell chimera (ONCOTECH) achieved a 3.8-fold increase in viral accumulation. | B16OVA cell membrane with MHC-I–OVA could protect eOA from neutralization by anti-Ad5 antibody. | A single administration of ONCOTECH resulted in an 80% survival rate over 70 days. | [48] | ||
| MSCs | Chemokines, such as SDF-1, HGF ect, secreted by the tumor recruited MSCs into tumors. | Shielded MYXV accumulated in lung tumors at 2 h after post-IV injection. | \ | The number of pulmonary foci was reduced twofold. | [72] | ||
| Binding with proteins | Virus-Protein Corona Replacement Strategy | DSPE-Poly(2-ethyl-2-oxazoline) (DSPE-PEOZ), a pH-sensitive phospholipid, enabled viral release in the acidic TME. | Increased OVs loading in tumors by more than 10-fold. | Prolonged the circulation time of OVs by more than 30-fold. | Tumor growth was suppressed by more than sevenfold. | [83] | |
| Modification of key capsid proteins | The HAdv penton base RGD loop interacted with cellular integrins of αvβ3 andαvβ5, promoting efficient virus infection. | Ad5-3M was detected in tumors 12 days after post-IV injection and persisted for 80 days. | Modified Ad5-3M avoided sequestration in liver tissue and poorly activated inflammatory cytokines. | Tumor-bearing mice exhibited a 6.6-fold increase in overall survival. | [86] | ||
| Nanoparticle (NP)-based delivery systems | Microbial Nanocomposites | Hypoxia, aberrant tumor vasculature, and the immunosuppressive TME promoted bacterial self-colonization in tumors. | Self-propelled E. coli-lipo-OAs demonstrated over 170-fold enrichment in tumors. | Liposomes possessed both bioprotective and biocompatible properties. | E. coli-lipo-OAs significantly promoted dendritic cell (DC) activation and induced long-term immune memory in mice. | [106] | |
| Biomineralization | Mn2+ achieved tumor-targeted accumulation via pH-gating effects in acidic TME. | At 48 h after post-IV injection, OA@CuMnCs showed over 50-fold higher viral titers in tumors. | Metal cations formed a biomineralized Adv coating to prevent immune clearance. | OA@CuMnCs enhanced T-cell infiltration and converted “cold” tumors to “hot” tumors. | [110] | ||
| Cell Membrane Nanovesicles | PD-1-engineered biomimetic nanovesicles targeted tumor tissues by binding to PD-L1 on tumor cell surfaces. | PD1-BCMN@OA showed approximately twofold higher accumulation in the tumor than naked virus. | PD1-BCMN (bioengineered cell membrane nanovesicles with PD-1) masked viral epitopes recognized by neutralizing antibodies. | PD1-BCMNs could bind to PD-L1 to activate TILs and elicit a strong antitumor immune response. | [116] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, Y.; Li, D.; Yang, K.; Ou, X. Systemic Delivery Strategies for Oncolytic Viruses: Advancing Targeted and Efficient Tumor Therapy. Int. J. Mol. Sci. 2025, 26, 6900. https://doi.org/10.3390/ijms26146900
Xia Y, Li D, Yang K, Ou X. Systemic Delivery Strategies for Oncolytic Viruses: Advancing Targeted and Efficient Tumor Therapy. International Journal of Molecular Sciences. 2025; 26(14):6900. https://doi.org/10.3390/ijms26146900
Chicago/Turabian StyleXia, Yunxin, Dan Li, Kai Yang, and Xia Ou. 2025. "Systemic Delivery Strategies for Oncolytic Viruses: Advancing Targeted and Efficient Tumor Therapy" International Journal of Molecular Sciences 26, no. 14: 6900. https://doi.org/10.3390/ijms26146900
APA StyleXia, Y., Li, D., Yang, K., & Ou, X. (2025). Systemic Delivery Strategies for Oncolytic Viruses: Advancing Targeted and Efficient Tumor Therapy. International Journal of Molecular Sciences, 26(14), 6900. https://doi.org/10.3390/ijms26146900