


Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions


Abstract
1. Introduction: Paradigm Shift in Tumor Glucose Metabolism Research
2. Multidimensional Characteristics of Spatiotemporal Heterogeneity in Tumor Glucose Metabolic Reprogramming
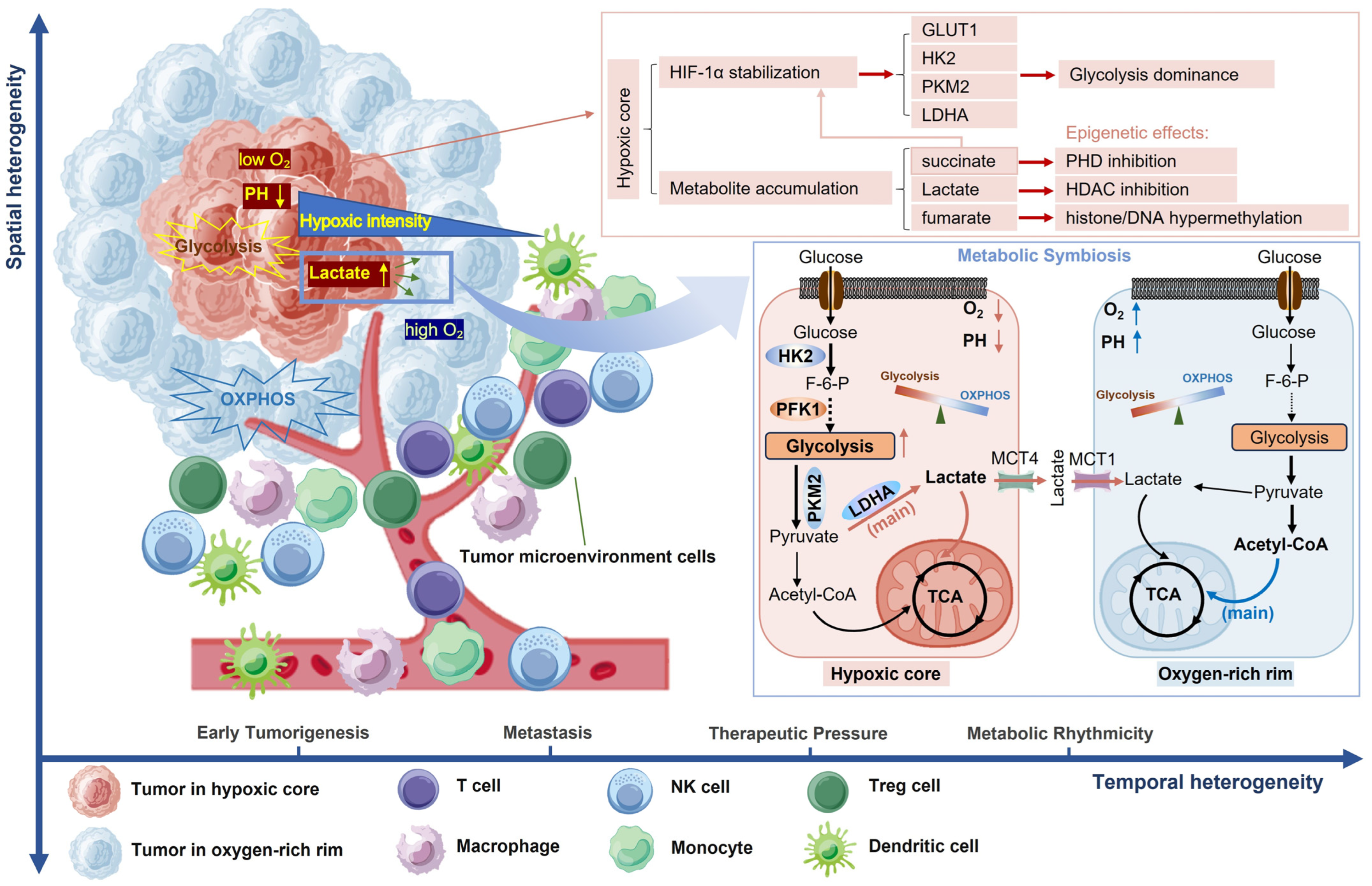
2.1. Spatial Heterogeneity
2.1.1. Intratumoral Metabolic Zonation
2.1.2. Metabolic Symbiosis Networks

{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Metabolic Characteristics of the Core Region | Metabolic Characteristics of the Marginal Zone | Key Molecules of Metabolic Interactions | Clinical Significance |
|---|---|---|---|---|
| Glioblastoma [12] | Enhanced glycolysis and hypoxia-induced HIF-1α | OXPHOS is active and aggressive. | LDHA, MCT4 | Hypoxic regions are resistant to radiotherapy. |
| Glioblastoma [13] | Significant glycolytic phenotype, high expression of hypoxia-related genes, significant chromosomal copy number variations (CNAs). | The metabolic state is more similar to that of normal tissues and more dependent on oxidative phosphorylation. Infiltration of T cells and myeloid cells was observed, but the degree of immunosuppression was low. | HIF-1α, VEGFA | Multimodal treatment: core zone (glycolysis inhibitor + radiotherapy) combined with marginal zone (OXPHOS inhibitor + immunotherapy). |
| OSCC [14] | Energy is obtained mainly through aerobic glycolysis, which produces large amounts of lactic acid. | Immune cells and stromal cells in the TME take up lactate to produce energy. | HIF-1α, CXCL12, TGF-β | Combined targeting of lactic acid metabolism (such as MCT inhibitors) and immune checkpoint inhibitors may enhance the therapeutic effect. |
| OSCC [15] | Significant glycolytic activity was enhanced, with localized enrichment of glycolytic metabolites. | The retention of higher glucose levels may serve as a “reservoir” of glucose to support tumor metabolic requirements. | Hexokinase, pyruvate kinase | Targeting key glycolytic enzymes (such as hexokinase and lactate dehydrogenase) or the inhibition of glycolytic pathways (such as the use of 2-deoxyglucose) may inhibit the tumor energy supply, especially for OSCC subtypes that are resistant to conventional chemoradiotherapy. |
| Cervical squamous cell carcinoma [16] | OXPHOS activity was low and hypoxia-related metabolic pathways dominated. | The OXPHOS pathway was significantly activated. | PRKCE, ITGA2, PKM | A combination strategy is appropriate. |
| Gastric cancer [17] | Glycolytic activity was highest and lactate accumulation was accompanied by the upregulation of tricarboxylic acid cycle intermediates, such as succinate. | Some regions may retain OXPHOS capacity, but are affected by tumor–stroma interactions and have complex metabolic phenotypes (e.g., immune cell-infiltrating regions may inhibit glycolysis). | PKM2, PFKL, ENO1 | Early screening and staging based on glycolysis imaging; combined strategies targeting glycolysis (e.g., PFKFB3 inhibitors) with immunometabolic regulation (e.g., GLS inhibitors). |
| Breast cancer [18] | The glucose content is high and more prone to glycolytic metabolism. | With a preference for mitochondrial metabolism. | PI3K, GLUT1 | Combined inhibition of PI3K and the bromodomain can overcome drug resistance and reduce metabolic heterogeneity. |
| Osteosarcoma [6,19] | Nucleotide/amino acid pathway amplification (alanine, aspartate, proline); glycolytic dominance; mitochondrial dysfunction. | FAO-enhanced invasion; ACLY-mediated acetyl-CoA production. | HIF-2α, PRODH, CPT1A | PRODH inhibitors sensitize to hypoxia-targeted therapy. |
| Leiomyosarcoma [20,21] | Highest glycolytic activity; lactate accumulation. | Retained OXPHOS capacity; modulated by tumor–stroma interactions. | RAS/PI3K pathway, FBP2 | High glycolysis correlates with poor prognosis. |
| PanNETs [7] | Homogeneous glycolysis (mTOR-VEGF axis dominance); under hypoxic conditions, invasive PanNET achieved metabolic adaptation by upregulating glycolysis and downregulating oxidative phosphorylation. | Lactate shuttling to stromal fibroblasts. | mTOR, VEGF, MCT4, HDAC1/2 | mTOR inhibitors reduce glycolytic flux but increase metastasis risk. |
| PDAC [22] | Enhanced glycolysis, hypoxia-induced HIF-1α stabilization, lactate accumulation, upregulation of glucose transporters (e.g., GLUT1), and key glycolytic enzymes (e.g., HK2, PFK1, LDHA). | OXPHOS activity is present but often compromised due to tumor–stroma interactions, lactate uptake via MCT1. | HIF-1α, GLUT1, HK2, PFK1, LDHA, MCT1, MCT4, CD147 | Hypoxic regions are resistant to radiotherapy; metabolic heterogeneity contributes to treatment resistance and relapse; targeting metabolic vulnerabilities (e.g., glycolysis inhibitors, MCT inhibitors) may enhance therapeutic efficacy. |
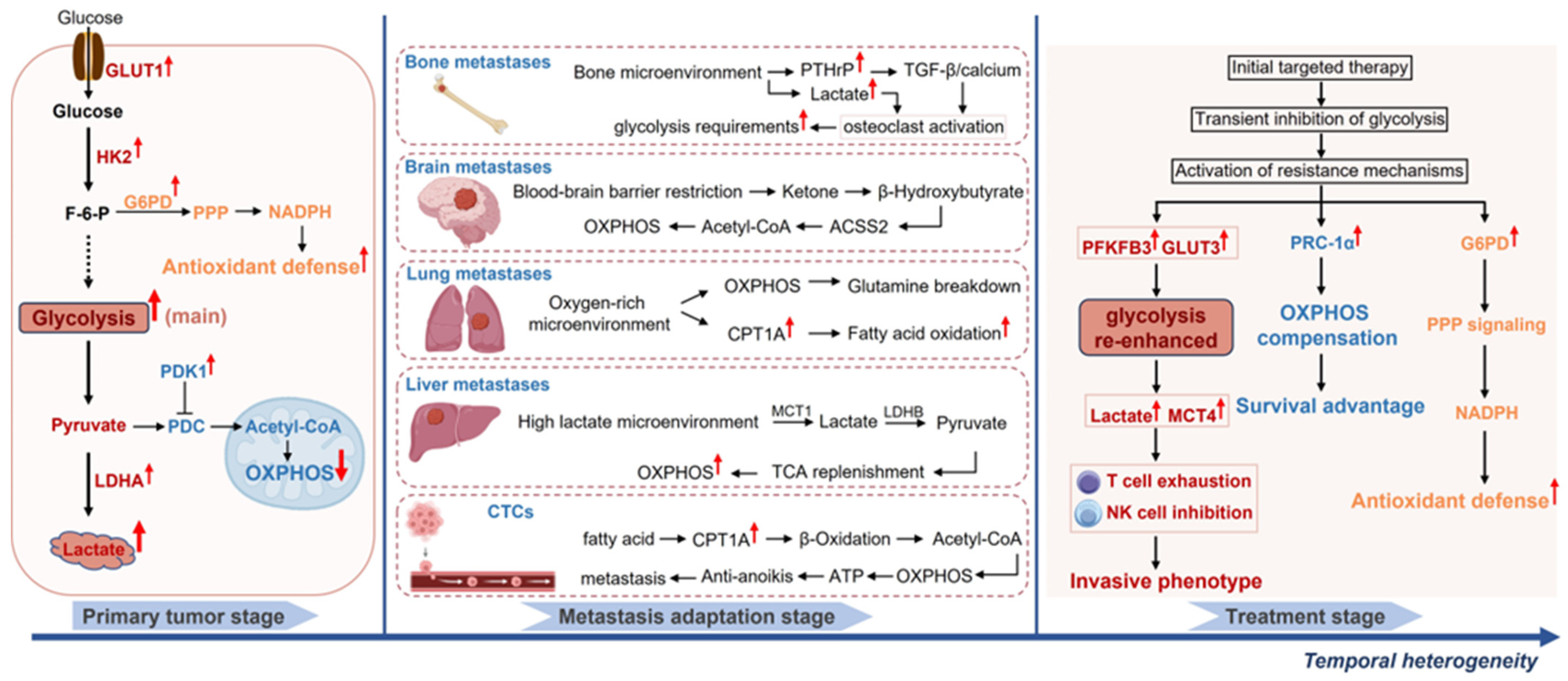
2.1.3. Organ-Specific Metabolic Adaptation of Metastatic Lesions
2.2. Temporal Heterogeneity: Metabolic Dynamic Evolution and Therapeutic Adaptation
2.2.1. Metabolic Phase Transitions During Tumor Progression
2.2.2. Metabolic Adaptive Remodeling Under Therapeutic Pressure
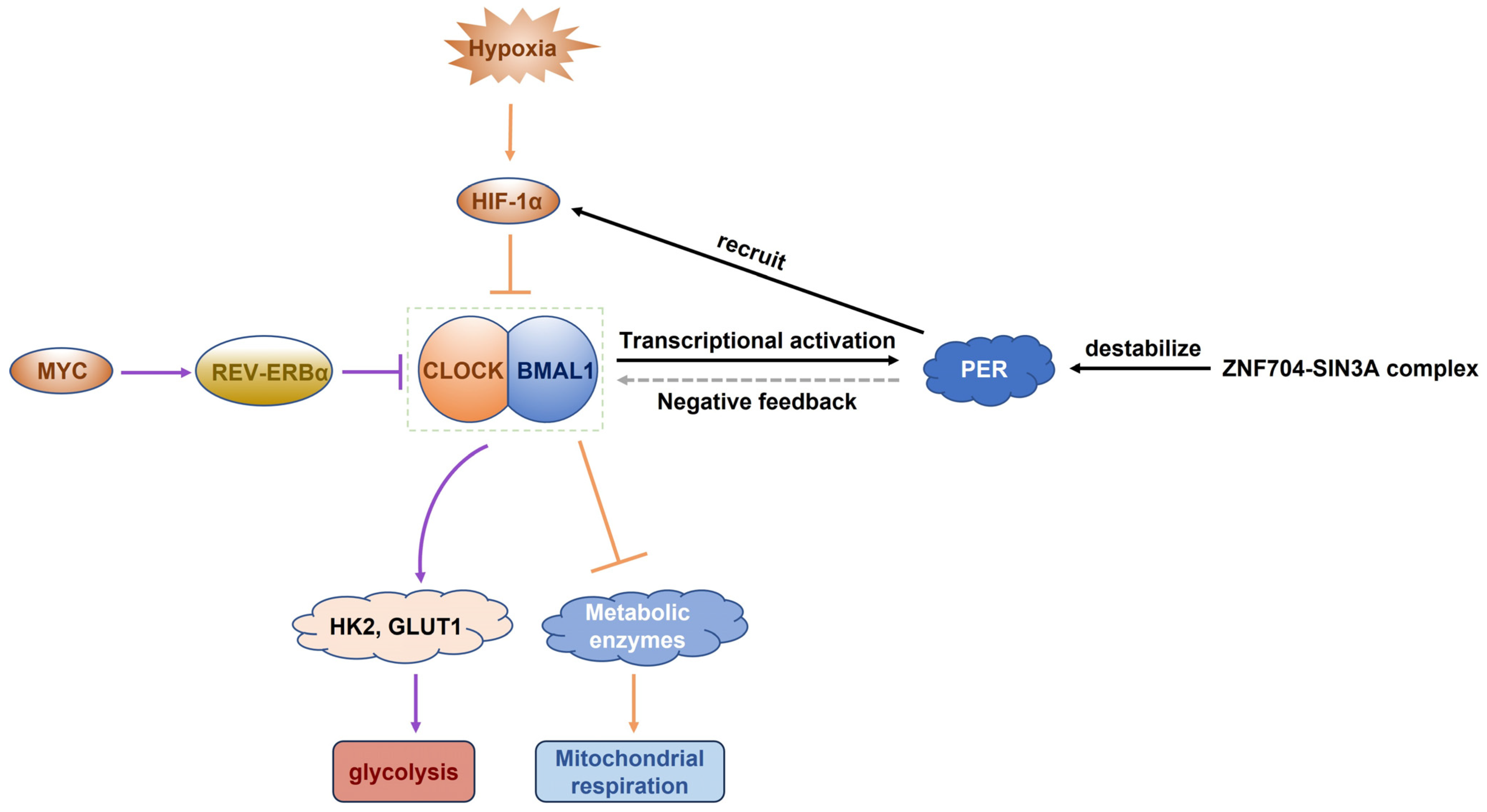
2.2.3. Circadian Regulation of Tumor Metabolism
3. Regulatory Networks of Spatiotemporal Heterogeneity in Tumor Glucose Metabolism Reprogramming
3.1. Spatial Heterogeneity: Topographic Metabolic Landscapes and Microenvironmental Crosstalk
3.1.1. Oxygen Gradient-Driven Metabolic Zonation Mechanism and Epigenetic Interplay
3.1.2. Metabolic Competition in the Immunometabolic Microenvironment
3.1.3. Metabolic Plasticity Drives Tumor Metastasis
- Metabolic Adaptations During Invasion and Dissemination
- Survival Mechanisms of Circulating Tumor Cells (CTCs)
- Organ-Specific Metabolic Strategies for Colonization
3.2. Temporal Dynamic Regulation: From Genetic Mutations to Epigenetic Memory
3.2.1. Metabolic Clonal Selection During Tumor Evolution
3.2.2. Treatment-Induced Metabolic and Epigenetic Remodeling Mechanisms
3.2.3. Molecular Basis of Metabolic Circadian Rhythm
4. Targeted Intervention Strategies for Spatiotemporal Heterogeneity
4.1. Compartment-Specific Metabolic Targeting
4.2. Temporal Modulation of Metabolic Evolution
| Category | Target | Mechanism of Action | Representative Drugs | Development Status |
|---|---|---|---|---|
| Glycolytic Key Enzyme Inhibitors | ||||
| GLUT Inhibitors | GLUT1, GLUT3 | Block glucose uptake; suppress glycolysis initiation. | 2-Deoxyglucose (2-DG), WZB117 [154] | Preclinical |
| Hexokinase (HK) Inhibitors | HK2, HK3 | Inhibit glycolysis initiation; reduce ATP/lactate production. | HK2 siRNA [180], 3-Bromopyruvate [181] | Preclinical |
| Pyruvate Kinase M2 (PKM2) Inhibitors | PKM2 | Block nuclear PKM2; inhibit HIF-1α/TGF-β-driven EMT. | Compound 3K [182] | Preclinical |
| Lactate Dehydrogenase A (LDHA) Inhibitors | LDHA | Suppress lactate production; reverse acidic TME and apoptosis resistance. | FX11 [183], GNE-140 [184] | FX11: Preclinical; GNE-140: Phase I (Breast/Pancreatic Cancer) |
| Pyruvate Dehydrogenase Kinase (PDK) Inhibitors | PDK1, PDK4 | Restore OXPHOS; inhibit glycolysis-dependent EMT. | DCA [185], KIS37 (Cryptotanshinone) [186] | DCA: Phase II (NCT01111097); KIS37: Preclinical |
| Other Glycolysis-Related Inhibitors | ||||
| Vitamin C (High-dose IV) | Indirect (GAPDH) | Depletes NAD+; disrupts microtubule dynamics; inhibits migration. | Ascorbate + Gemcitabine [187] | Phase I/II (NCT02905578, Pancreatic Cancer) |
| Aldolase A (ALDOA) Inhibitors | ALDOA | Block glycolysis intermediates; suppress cytoskeletal remodeling. | Raltegravir [188] | Preclinical |
| Enolase 1 (ENO1) Inhibitors | ENO1 | Inhibit glycolysis; regulate PI3K/AKT signaling. | ENO1 DNA vaccine [189,190] | Preclinical |
| Latest Developments | ||||
| 2-DG + Metformin | Glycolysis/AMPK | Synergistically inhibits EMT and stemness in glioblastoma. | 2-DG + Metformin [191] | Preclinical |
| DCA (Dichloroacetate) | PDK1–EGFR axis | Reverses cisplatin resistance in ovarian cancer. | DCA + Cisplatin [192] | Phase II (NCT01111097) |
| Repurposed Drugs | ||||
| Metformin | AMPK/mTOR | Indirectly inhibits glycolysis and EMT. | Metformin + Chemotherapy [193] | Phase II (NCT01864096, Breast Cancer/Glioblastoma) |
| Simvastatin | HMG-CoA reductase | Downregulates CXCR4, vimentin; inhibits TGF-β-induced EMT. | Simvastatin [194] | Phase II (NCT00944463, Pancreatic Cancer; NCT02161822, Advanced Rectal Cancer) |
| Immune-Metabolic Synergistic Approaches | ||||
| MCT4 Inhibitors | Lactate export | Block MCT4-mediated lactate efflux to restore T-cell function. | AZD3965 [195,196], Diclofenac [197,198], Syrosingopine [157,195] | Clinical (AZD3965: Phase I/II) |
| GLUT1 Inhibitors | Glucose uptake | Inhibit GLUT1-mediated glucose transport to counteract TME starvation. | BAY-876 [152], Phloretin [199], Glutor [200] | Preclinical |
| LDHA Inhibitors | Lactate production | Suppress LDHA activity to reduce immunosuppressive lactate accumulation. | Oxamate [174], Compound 7 [201,202] | Preclinical (Compound 7: Phase I) |
| GPR81 Antagonists | Lactate signaling | Block GPR81-mediated immunosuppressive signaling in dendritic cells. | 3-OBA [203] | Preclinical |
| SREBP2 Inhibitors | Lipid metabolism | Inhibit SREBP2 to disrupt lactate signaling in antigen-presenting cells. | Fatostatin [63], Botulin [204] | Preclinical |
| PI3K Inhibitors | Metabolic–immune crosstalk | Dual inhibition of PI3K-AKT-mTOR pathway and lactate signaling. | Copanlisib [205], Duvelisib [205] | Clinical (Copanlisib: FDA-approved for lymphoma) |
| mTOR Blockers | Immune metabolism | Suppress mTOR-driven metabolic reprogramming in Tregs. | Everolimus [206], CC-223 [207] | Clinical (Everolimus: Approved for multiple cancers) |
4.3. Immune–Metabolic Synergistic Approaches
4.4. Innovative Approaches for Targeting Metabolic–Immune Crosstalk
5. Challenges and Future Perspectives
5.1. Current Challenges and Technical Limitations
5.2. Clinical Translation Barriers
5.3. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, S.; Li, H.; Zhang, L.; Mu, W.; Zhang, Y.; Chen, T.; Wu, J.; Tang, H.; Zheng, S.; Liu, Y.; et al. Generic Diagramming Platform (GDP): A comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2025, 53, D1670–D1676. [Google Scholar] [CrossRef] [PubMed]
- Flint, L.E.; Hamm, G.; Ready, J.D.; Ling, S.; Duckett, C.J.; Cross, N.A.; Cole, L.M.; Smith, D.P.; Goodwin, R.J.A.; Clench, M.R. Characterization of an Aggregated Three-Dimensional Cell Culture Model by Multimodal Mass Spectrometry Imaging. Anal. Chem. 2020, 92, 12538–12547. [Google Scholar] [CrossRef] [PubMed]
- Demicco, M.; Liu, X.Z.; Leithner, K.; Fendt, S.M. Metabolic heterogeneity in cancer. Nat. Metab. 2024, 6, 18–38. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.E9. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Miallot, R.; Galland, F.; Millet, V.; Blay, J.Y.; Naquet, P. Metabolic landscapes in sarcomas. J. Hematol. Oncol. 2021, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Ogawa, M.; Zhou, Y.; Otani, Y.; Hendrickson, R.C.; Miele, M.M.; Li, Z.; Klimstra, D.S.; Wang, J.Y.; Roehrl, M.H. Proteogenomic characterization of pancreatic neuroendocrine tumors uncovers hypoxia and immune signatures in clinically aggressive subtypes. iScience 2024, 27, 110544. [Google Scholar] [CrossRef] [PubMed]
- Kueckelhaus, J.; Frerich, S.; Kada-Benotmane, J.; Koupourtidou, C.; Ninkovic, J.; Dichgans, M.; Beck, J.; Schnell, O.; Heiland, D.H. Inferring histology-associated gene expression gradients in spatial transcriptomic studies. Nat. Commun. 2024, 15, 7280. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.; Chen, Y.; Tian, H.; Chai, P.; Shen, Y.; Yao, Y.; Xu, S.; Ge, S.; Jia, R. Lactate and lactylation in cancer. Signal Transduct. Target. Ther. 2025, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Nair, L.; Kumar, D.; Arora, M.K.; Bajaj, S.; Gadewar, M.; Mishra, S.S.; Rath, S.K.; Dubey, A.K.; Kaithwas, G.; et al. Hypoxia induced lactate acidosis modulates tumor microenvironment and lipid reprogramming to sustain the cancer cell survival. Front. Oncol. 2023, 13, 1034205. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, J.L.; Jalasutram, A.; Asuthkar, S.; Sahagun, J.; Park, S.; Ravindra, A.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Beyond glucose: Alternative sources of energy in glioblastoma. Theranostics 2021, 11, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salie, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655e613. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Z.; Zhang, Y.; Zhou, W.; Zhang, X.; Peng, C.; Ji, T.; Zou, X.; Zhang, Z.; Ren, Z. Spatial transcriptomics reveals that metabolic characteristics define the tumor immunosuppression microenvironment via iCAF transformation in oral squamous cell carcinoma. Int. J. Oral. Sci. 2024, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Wang, Q.; Zi, M.; Zhang, S.; Ge, J.; Liu, K.; Lu, L.; Fan, C.; Yan, Q.; Shi, L.; et al. Spatial Transcriptomic and Metabolomic Landscapes of Oral Submucous Fibrosis-Derived Oral Squamous Cell Carcinoma and its Tumor Microenvironment. Adv. Sci. 2024, 11, e2306515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, J.; Yao, P.; Liu, X.; Chen, F.; Chen, Y.; Zhou, L.; Shen, C.; Zhou, Y.; Du, X.; et al. Spatial transcriptomics reveals unique metabolic profile and key oncogenic regulators of cervical squamous cell carcinoma. J. Transl. Med. 2024, 22, 1163. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, A.; Zhou, Y.; Chen, P.; Wang, X.; Huang, J.; Gao, J.; Wang, X.; Shu, L.; Lu, J.; et al. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 2023, 14, 2692. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Ratcliffe, C.D.H.; Hooper, S.; Ellis, J.; MacRae, J.I.; Hennequart, M.; Dunsby, C.W.; Anderson, K.I.; Sahai, E. Single-cell resolved imaging reveals intra-tumor heterogeneity in glycolysis, transitions between metabolic states, and their regulatory mechanisms. Cell Rep. 2021, 34, 108750. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Serada, N.; Sheehan, M.; Srinivasan, S.; Mason, N.; Guha, M.; Avadhani, N. Mitochondrial genome and functional defects in osteosarcoma are associated with their aggressive phenotype. PLoS ONE 2018, 13, e0209489. [Google Scholar] [CrossRef] [PubMed]
- Babichev, Y.; Kabaroff, L.; Datti, A.; Uehling, D.; Isaac, M.; Al-Awar, R.; Prakesch, M.; Sun, R.X.; Boutros, P.C.; Venier, R.; et al. PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma. J. Transl. Med. 2016, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Dauchy, R.T.; Blask, D.E.; Dauchy, E.M.; Slakey, L.M.; Brimer, S.; Yuan, L.; Xiang, S.; Hauch, A.; Smith, K.; et al. Melatonin suppression of aerobic glycolysis (Warburg effect), survival signalling and metastasis in human leiomyosarcoma. J. Pineal Res. 2016, 60, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.; Scheckel, B.; Habsaoui, A.; Haque, M.; Kuhr, K.; Monsef, I.; Bohlius, J.; Skoetz, N. Intravenous iron versus oral iron versus no iron with or without erythropoiesis-stimulating agents (ESA) for cancer patients with anaemia: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2022, 6, CD012633. [Google Scholar] [CrossRef] [PubMed]
- Prayoonhong, W.; Sonsingh, W.; Permsuwan, U. Clinical outcomes and economic evaluation of patient-centered care system versus routine-service system for patients with type 2 diabetes in Thailand. Heliyon 2024, 10, e25093. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q. Changes in mitochondrial function during EMT induced by TGFbeta-1 in pancreatic cancer. Oncol. Lett. 2017, 13, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Shasha, T.; Gruijs, M.; van Egmond, M. Mechanisms of colorectal liver metastasis development. Cell Mol. Life Sci. 2022, 79, 607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Zeng, Z.L.; Lu, J.; Wang, Y.; Liu, Z.X.; He, M.M.; Zhao, Q.; Wang, Z.X.; Li, T.; Lu, Y.X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.D.; Weilandt, D.R.; Liang, L.; MacArthur, M.R.; Jaiswal, N.; Ong, O.; Mann, C.G.; Chu, Q.; Hunter, C.J.; Ryseck, R.P.; et al. Lactate homeostasis is maintained through regulation of glycolysis and lipolysis. Cell Metab. 2025, 37, 758–771.E8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Bartman, C.R.; Jankowski, C.S.R.; Zhang, Z.; Xu, X.; Xing, X.; Wang, L.; Lu, W.; Hui, S.; Rabinowitz, J.D. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab. 2021, 33, 367–378.E5. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, F.; Lang, L.; Yang, F.; Fu, Z.; Martinez, J.; Cho, A.; Saba, N.F.; Teng, Y. Therapeutic Targeting of the GLS1-c-Myc Positive Feedback Loop Suppresses Glutaminolysis and Inhibits Progression of Head and Neck Cancer. Cancer Res. 2024, 84, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Salhi, A.; Jordan, A.C.; Bochaca, I.I.; Izsak, A.; Darvishian, F.; Houvras, Y.; Giles, K.M.; Osman, I. Oxidative Phosphorylation Promotes Primary Melanoma Invasion. Am. J. Pathol. 2020, 190, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Huang, Y.; Li, P.; Yang, M.; Zeng, S.; Chen, D.; Wang, Q.; Liu, H.; Luo, K.; et al. Targeting nicotinamide N-methyltransferase overcomes resistance to EGFR-TKI in non-small cell lung cancer cells. Cell Death Discov. 2022, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Adhikari, S.; Bhattacharya, A.; Chakraborty, S.; Mondal, P.; Yadav, S.S.; Adhikary, S.; Hunt, C.R.; Yadav, K.K.; Pandita, S.; et al. Epigenetic-Metabolic Interplay in the DNA Damage Response and Therapeutic Resistance of Breast Cancer. Cancer Res. 2023, 83, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2021, 124, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chamoto, K. Immune metabolism in PD-1 blockade-based cancer immunotherapy. Int. Immunol. 2021, 33, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, K.; Sassone-Corsi, P. Metabolic rivalry: Circadian homeostasis and tumorigenesis. Nat. Rev. Cancer 2020, 20, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, L.; Meng, Y.; Fang, J.; Xu, D.; Lu, Z. The interplay of the circadian clock and metabolic tumorigenesis. Trends Cell Biol. 2024, 34, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, J.; Liu, X.; Wang, Y.; Liu, B.; Chen, X.; Wu, X.; Yan, D.; Han, L.; Liu, S.; et al. Circadian Rhythm Is Disrupted by ZNF704 in Breast Carcinogenesis. Cancer Res. 2020, 80, 4114–4128. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Zeng, D.; Zhang, W.; Peng, F.; Cui, B.; Liu, Q. Integrating cancer medicine into metabolic rhythms. Trends Endocrinol. Metab. 2025. [Google Scholar] [CrossRef] [PubMed]
- Yfantis, A.; Mylonis, I.; Chachami, G.; Nikolaidis, M.; Amoutzias, G.D.; Paraskeva, E.; Simos, G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells 2023, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gilkes, D.M. HIF-1 and HIF-2 in cancer: Structure, regulation, and therapeutic prospects. Cell Mol. Life Sci. 2025, 82, 44. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhao, L.; Gui, Z.; Liu, S.; Liu, C.; Yu, T.; Zhang, L. PI3K/AKT signaling activates HIF1alpha to modulate the biological effects of invasive breast cancer with microcalcification. NPJ Breast Cancer 2023, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Kilic-Eren, M.; Boylu, T.; Tabor, V. Targeting PI3K/Akt represses Hypoxia inducible factor-1alpha activation and sensitizes Rhabdomyosarcoma and Ewing’s sarcoma cells for apoptosis. Cancer Cell Int. 2013, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [PubMed]
- Huimin, W.; Xin, W.; Shan, Y.; Junwang, Z.; Jing, W.; Yuan, W.; Qingtong, L.; Xiaohui, L.; Jia, Y.; Lili, Y. Lactate promotes the epithelial-mesenchymal transition of liver cancer cells via TWIST1 lactylation. Exp. Cell Res. 2025, 447, 114474. [Google Scholar] [CrossRef] [PubMed]
- Atallah, R.; Olschewski, A.; Heinemann, A. Succinate at the Crossroad of Metabolism and Angiogenesis: Roles of SDH, HIF1alpha and SUCNR1. Biomedicines 2022, 10, 3089. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 16, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Fernie, G. Consent and the individual detained in custody. Med. Law. 2005, 24, 515–523. [Google Scholar] [PubMed]
- Fitzsimmons, C.M.; Mandler, M.D.; Lunger, J.C.; Chan, D.; Maligireddy, S.S.; Schmiechen, A.C.; Gamage, S.T.; Link, C.; Jenkins, L.M.; Chan, K.; et al. Rewiring of RNA methylation by the oncometabolite fumarate in renal cell carcinoma. NAR Cancer 2024, 6, zcae004. [Google Scholar] [CrossRef] [PubMed]
- Pianka, S.T.; Li, T.; Prins, T.J.; Eldred, B.S.C.; Kevan, B.M.; Liang, H.; Zapanta Rinonos, S.; Kornblum, H.I.; Nathanson, D.A.; Pellegrini, M.; et al. D-2-HG Inhibits IDH1mut Glioma Growth via FTO Inhibition and Resultant m6A Hypermethylation. Cancer Res. Commun. 2024, 4, 876–894. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; O’Sullivan, D.; Pearce, E.L. Glucose makes Treg lose their temper. Cancer Cell 2021, 39, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Yang, H.; Liang, T.; Bai, X. Hurdle or thruster: Glucose metabolism of T cells in anti-tumour immunity. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189022. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Llibre, A.; Kucuk, S.; Gope, A.; Certo, M.; Mauro, C. Lactate: A key regulator of the immune response. Immunity 2025, 58, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Plebanek, M.P.; Xue, Y.; Nguyen, Y.V.; DeVito, N.C.; Wang, X.; Holtzhausen, A.; Beasley, G.M.; Theivanthiran, B.; Hanks, B.A. A lactate-SREBP2 signaling axis drives tolerogenic dendritic cell maturation and promotes cancer progression. Sci. Immunol. 2024, 9, eadi4191. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Li, J.; Wei, J.; Lim, S.A. Regulatory T Cell Metabolism: A Promising Therapeutic Target for Cancer Treatment? Immune Netw. 2025, 25, e13. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Kumagai, S.; Nishikawa, H. Metabolic advantages of regulatory T cells dictated by cancer cells. Int. Immunol. 2024, 36, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.E9. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Hasko, G. Anti-CD73 in cancer immunotherapy: Awakening new opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Tang, M.; Su, Y.; Xie, J.; Shang, Q.; Guo, M.; An, X.; Lin, L.; Wang, R.; Huang, Q.; et al. Nanomedicine-driven tumor glucose metabolic reprogramming for enhanced cancer immunotherapy. Acta Pharm. Sin. B 2025, 15, 2845–2866. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Tong, Y.; Jiang, X.; Meng, Y.; Jiang, H.; Du, L.; Wu, Q.; Li, S.; Luo, S.; Li, M.; et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IkappaBalpha. Cell Metab. 2022, 34, 1312–1324.E6. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Fang, W.; Xiang, Z.; Wang, Q.; Cheng, H.; Chen, S.; Fang, J.; Liu, J.; Wang, Q.; Lu, Z.; et al. Glycolytic enzyme HK2 promotes PD-L1 expression and breast cancer cell immune evasion. Front. Immunol. 2023, 14, 1189953. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Liu, R.; Li, J.; Wang, Y.; Tan, L.; Li, X.J.; Qian, X.; Zhang, C.; Xia, Y.; Xu, D.; et al. EGFR-Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol. Cell 2018, 70, 197–210.E7. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zhang, X.; Wang, R.J.; Ma, Q.Y.; Xu, L.; Wang, Y.; Liao, H.P.; Wang, H.L.; Hu, L.D.; Kong, X.; et al. PI3Kalpha inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+T cells and promoting fatty acid metabolism. J. Immunother. Cancer 2021, 9, e003093. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Huang, X.; Lulu, T.B.; Jia, W.; Zhang, S.; Cohen, L.; Huang, S.; Fan, J.; Chen, X.; Liu, S.; et al. A novel pan-PI3K inhibitor KTC1101 synergizes with anti-PD-1 therapy by targeting tumor suppression and immune activation. Mol. Cancer 2024, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Sckell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar] [PubMed]
- Muri, J.; Kopf, M. The thioredoxin system: Balancing redox responses in immune cells and tumors. Eur. J. Immunol. 2023, 53, e2249948. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, S.; Liu, J.; Tian, Y.; Ma, B.; Xu, S.; Fu, Y.; Luo, Y. Secreted Pyruvate Kinase M2 Promotes Lung Cancer Metastasis through Activating the Integrin Beta1/FAK Signaling Pathway. Cell Rep. 2020, 30, 1780–1797. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987e974. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, H.J.; Wu, X.; Huo, L.; Kim, S.J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Stresing, V.; Baltziskueta, E.; Rubio, N.; Blanco, J.; Arriba, M.C.; Valls, J.; Janier, M.; Clezardin, P.; Sanz-Pamplona, R.; Nieva, C.; et al. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene 2013, 32, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Sellers, K.; Fox, M.P.; Bousamra, M., 2nd; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, F.; Tabaries, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Kakonen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Di Pompo, G.; Porporato, P.E.; Sboarina, M.; Russell, S.; Gillies, R.J.; Baldini, N.; Sonveaux, P.; Avnet, S. MDA-MB-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhang, W.; Wu, K.; Shi, L. The roles of KRAS in cancer metabolism, tumor microenvironment and clinical therapy. Mol. Cancer 2025, 24, 14. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Liaghat, M.; Ferdousmakan, S.; Mortazavi, S.H.; Yahyazadeh, S.; Irani, A.; Banihashemi, S.; Seyedi Asl, F.S.; Akbari, A.; Farzam, F.; Aziziyan, F.; et al. The impact of epithelial-mesenchymal transition (EMT) induced by metabolic processes and intracellular signaling pathways on chemo-resistance, metastasis, and recurrence in solid tumors. Cell Commun. Signal 2024, 22, 575. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Uslu, C.; Kapan, E.; Lyakhovich, A. Cancer resistance and metastasis are maintained through oxidative phosphorylation. Cancer Lett. 2024, 587, 216705. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Raghu, D.; Haupt, Y. Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Front. Oncol. 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Beck, B.H.; Vaidya, K.S.; Nash, K.T.; Feeley, K.P.; Ballinger, S.W.; Pounds, K.M.; Denning, W.L.; Diers, A.R.; Landar, A.; et al. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014, 74, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, R.A.; Fischer, G.M.; Kircher, D.A.; Joon, A.Y.; Cortez, J.R.; Grossman, A.H.; Hudgens, C.W.; Ledesma, D.A.; Lazcano, R.; Onana, C.Y.; et al. Oxidative Phosphorylation (OXPHOS) Promotes the Formation and Growth of Melanoma Lung and Brain Metastases. bioRxiv 2025. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Resistance to energy metabolism—Targeted therapy of AML cells residual in the bone marrow microenvironment. Cancer Drug Resist. 2023, 6, 138–150. [Google Scholar] [CrossRef] [PubMed]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia 2022, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xian, H.C.; Tang, Y.J.; Liang, X.H.; Tang, Y.L. Fatty acid oxidation: Driver of lymph node metastasis. Cancer Cell Int. 2021, 21, 339. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; Geldhof, V.; Carmeliet, P. How glucose, glutamine and fatty acid metabolism shape blood and lymph vessel development. Dev. Biol. 2019, 447, 90–102. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Stern, A.; Chiu, D.T. G6PD: A hub for metabolic reprogramming and redox signaling in cancer. Biomed. J. 2021, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Li, X.; Sun, W.; Sun, M.; Li, Z.; Sheng, H.; Xie, F.; Zhang, S.; Shan, C. Targeting G6PD reverses paclitaxel resistance in ovarian cancer by suppressing GSTP1. Biochem. Pharmacol. 2020, 178, 114092. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, Y.; Wang, G.; Feng, S.; Ge, X.; Ye, W.; Wang, Z.; Zhu, Y.; Cai, W.; Bai, J.; et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. 2022, 53, 102344. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Van Nyen, T.; Planque, M.; van Wagensveld, L.; Duarte, J.A.G.; Zaal, E.A.; Talebi, A.; Rossi, M.; Korner, P.R.; Rizzotto, L.; Moens, S.; et al. Serine metabolism remodeling after platinum-based chemotherapy identifies vulnerabilities in a subgroup of resistant ovarian cancers. Nat. Commun. 2022, 13, 4578. [Google Scholar] [CrossRef] [PubMed]
- Pranzini, E.; Pardella, E.; Muccillo, L.; Leo, A.; Nesi, I.; Santi, A.; Parri, M.; Zhang, T.; Uribe, A.H.; Lottini, T.; et al. SHMT2-mediated mitochondrial serine metabolism drives 5-FU resistance by fueling nucleotide biosynthesis. Cell Rep. 2022, 40, 111233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Ma, Y.; Zhang, Q.; Liu, R.; Luo, H.; Wang, Z. Review of possible mechanisms of radiotherapy resistance in cervical cancer. Front. Oncol. 2023, 13, 1164985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Mo, F.; Patel, G.; Butterworth, K.; Shao, C.; Prise, K.M. The Roles of HIF-1alpha in Radiosensitivity and Radiation-Induced Bystander Effects Under Hypoxia. Front. Cell Dev. Biol. 2021, 9, 637454. [Google Scholar] [CrossRef]
- Read, G.H.; Bailleul, J.; Vlashi, E.; Kesarwala, A.H. Metabolic response to radiation therapy in cancer. Mol. Carcinog. 2022, 61, 200–224. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Li, Z.; Xiao, L.; Hu, W.; Zhang, L.; Xie, B.; Zhou, Q.; He, J.; Qiu, Y.; Wen, M.; et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep. 2019, 28, 1136–1143.E4. [Google Scholar] [CrossRef] [PubMed]
- Falcone, M.; Uribe, A.H.; Papalazarou, V.; Newman, A.C.; Athineos, D.; Stevenson, K.; Sauve, C.G.; Gao, Y.; Kim, J.K.; Del Latto, M.; et al. Sensitisation of cancer cells to radiotherapy by serine and glycine starvation. Br. J. Cancer 2022, 127, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Solomou, G.; Finch, A.; Asghar, A.; Bardella, C. Mutant IDH in Gliomas: Role in Cancer and Treatment Options. Cancers 2023, 15, 2883. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Wu, Y.; Li, S.; Yu, X. Interplay Between Glucose Metabolism and Chromatin Modifications in Cancer. Front. Cell Dev. Biol. 2021, 9, 654337. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.M.; Chu, D.T.; Lin, S.C.; Lee, J.S.; Vu, T.D.; Vu, H.T.; Ramasamy, T.S.; Lin, S.P.; Wu, C.C. Enhanced mitochondrial function and delivery from adipose-derived stem cell spheres via the EZH2-H3K27me3-PPARgamma pathway for advanced therapy. Stem Cell Res. Ther. 2025, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jing, X.; Du, Q.; Sun, X.; Holgersson, K.; Gao, J.; He, X.; Hosaka, K.; Zhao, C.; Tao, W.; et al. Disruption of the Clock Component Bmal1 in Mice Promotes Cancer Metastasis through the PAI-1-TGF-beta-myoCAF-Dependent Mechanism. Adv. Sci. 2023, 10, e2301505. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Morinibu, A.; Koyasu, S.; Goto, Y.; Hiraoka, M.; Harada, H. A circadian clock gene, PER2, activates HIF-1 as an effector molecule for recruitment of HIF-1alpha to promoter regions of its downstream genes. FEBS J. 2017, 284, 3804–3816. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.M.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Cazarin, J.; DeRollo, R.E.; Shahidan, S.; Burchett, J.B.; Mwangi, D.; Krishnaiah, S.; Hsieh, A.L.; Walton, Z.E.; Brooks, R.; Mello, S.S.; et al. MYC disrupts transcriptional and metabolic circadian oscillations in cancer and promotes enhanced biosynthesis. PLoS Genet. 2023, 19, e1010904. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, Z.; Ye, L.; Duan, Y.; Jiang, H.; He, H.; Xiao, L.; Wu, Q.; Xia, Y.; Yang, M.; et al. Nucleus-exported CLOCK acetylates PRPS to promote de novo nucleotide synthesis and liver tumour growth. Nat. Cell Biol. 2023, 25, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Tang, H.; Yang, K. PER1 suppresses glycolysis and cell proliferation in oral squamous cell carcinoma via the PER1/RACK1/PI3K signaling complex. Cell Death Dis. 2021, 12, 276. [Google Scholar] [CrossRef] [PubMed]
- Gowda, P.; Lathoria, K.; Sharma, S.; Patrick, S.; Umdor, S.B.; Sen, E. Rewiring of Lactate-Interleukin-1beta Autoregulatory Loop with Clock-Bmal1: A Feed-Forward Circuit in Glioma. Mol. Cell Biol. 2021, 41, e0044920. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Yang, T.; Mu, J.; Zhao, J.; Yang, Y.; Yan, Z.; Hou, Y.; Chen, C.; Xing, J.; Zhang, H.; et al. Circadian clock gene NPAS2 promotes reprogramming of glucose metabolism in hepatocellular carcinoma cells. Cancer Lett. 2020, 469, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Enya, S.; Arai, M.; Suzuki, Y.; Nojiri, T.; Kangawa, K.; Koyama, S.; Kawaoka, S. Remote reprogramming of hepatic circadian transcriptome by breast cancer. Oncotarget 2017, 8, 34128–34140. [Google Scholar] [CrossRef] [PubMed]
- Masri, S.; Papagiannakopoulos, T.; Kinouchi, K.; Liu, Y.; Cervantes, M.; Baldi, P.; Jacks, T.; Sassone-Corsi, P. Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell 2016, 165, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Samanta, P.; Sarkar, R.; Biswas, S.; Saha, P.; Hajra, S.; Bhowmik, A. Targeting HIF-1alpha by Natural and Synthetic Compounds: A Promising Approach for Anti-Cancer Therapeutics Development. Molecules 2022, 27, 5192. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Xue, Z.; Zhang, L.; Ruan, X.; Yang, J.; Fan, Z.; Zhao, H.; Cao, Y.; Chen, G.; et al. Adamantaniline Derivatives Target ATP5B to Inhibit Translation of Hypoxia Inducible Factor-1alpha. Adv. Sci. 2023, 10, e2301071. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Sivaraman, A.; Kim, M.; Min, K.H.; Song, M.E.; Choi, Y.; Choi, W.J.; Han, H.K.; Han, J.; Jang, J.P.; et al. HIF-1alpha inhibition by MO-2097, a novel chiral-free benzofuran targeting hnRNPA2B1. J. Adv. Res. 2024, 64, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.T.; Chau, C.H.; Strope, J.D.; Huitema, A.D.R.; Sissung, T.M.; Price, D.K.; Figg, W.D. Antitumor Activity of NLG207 (Formerly CRLX101) in Combination with Enzalutamide in Preclinical Prostate Cancer Models. Mol. Cancer Ther. 2021, 20, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Jung, Y.; Song, Y.S.; Park, S.; Park, Y.; Lee, H.J. Enhanced anti-angiogenic activity of novel melatonin-like agents. J. Pineal Res. 2021, 71, e12739. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Tao, F.; Lu, Y.; Fang, M.; Huang, H.; Zhou, Y. The Role of HK2 in Tumorigenesis and Development: Potential for Targeted Therapy with Natural Products. Int. J. Med. Sci. 2025, 22, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Lee, E.J.; Park, W.; Ha, K.T.; Chung, H.S. Natural compounds as lactate dehydrogenase inhibitors: Potential therapeutics for lactate dehydrogenase inhibitors-related diseases. Front. Pharmacol. 2023, 14, 1275000. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.A.; Muthuswami, S.; Mueller, A.; Ma, R.Z.; Sywanycz, S.M.; Naik, A.; Huang, L.; Brody, R.M.; Diab, A.; Carey, R.M.; et al. GLUT1 inhibitor BAY-876 induces apoptosis and enhances anti-cancer effects of bitter receptor agonists in head and neck squamous carcinoma cells. Cell Death Discov. 2024, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, M.Z.; Sun, H.W.; Chai, Y.T.; Li, X.; Jiang, Q.; Hou, J. A Novel Microcrystalline BAY-876 Formulation Achieves Long-Acting Antitumor Activity Against Aerobic Glycolysis and Proliferation of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 783194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Bryniarski, M.A.; Morris, M.E. In Vitro and In Vivo Efficacy of the Monocarboxylate Transporter 1 Inhibitor AR-C155858 in the Murine 4T1 Breast Cancer Tumor Model. AAPS J. 2018, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef] [PubMed]
- Esquea, E.M.; Ciraku, L.; Young, R.G.; Merzy, J.; Talarico, A.N.; Ahmed, N.N.; Karuppiah, M.; Ramesh, A.; Chatoff, A.; Crispim, C.V.; et al. Selective and brain-penetrant ACSS2 inhibitors target breast cancer brain metastatic cells. Front. Pharmacol. 2024, 15, 1394685. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lin, H.; Tavares, C.D.J.; Dominy, J.E.; Camporez, J.P.; Perry, R.J.; Schilling, R.; Rines, A.K.; Lee, J.; Hickey, M.; et al. Selective Chemical Inhibition of PGC-1alpha Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 2017, 169, 148–160.E15. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef] [PubMed]
- Szeliga, M.; Rola, R. Conoidin A, a Covalent Inhibitor of Peroxiredoxin 2, Reduces Growth of Glioblastoma Cells by Triggering ROS Production. Cells 2023, 12, 1934. [Google Scholar] [CrossRef] [PubMed]
- Udumula, M.P.; Rashid, F.; Singh, H.; Pardee, T.; Luther, S.; Bhardwaj, T.; Anjaly, K.; Piloni, S.; Hijaz, M.; Gogoi, R.; et al. Targeting mitochondrial metabolism with CPI-613 in chemoresistant ovarian tumors. J. Ovarian Res. 2024, 17, 226. [Google Scholar] [CrossRef] [PubMed]
- Arlt, B.; Mastrobuoni, G.; Wuenschel, J.; Astrahantseff, K.; Eggert, A.; Kempa, S.; Deubzer, H.E. Inhibiting PHGDH with NCT-503 reroutes glucose-derived carbons into the TCA cycle, independently of its on-target effect. J. Enzyme Inhib. Med. Chem. 2021, 36, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.; Pickarski, M.; Zhuo, Y.; Masarachia, P.J.; Duong, L.T. The effects of the cathepsin K inhibitor odanacatib on osteoclastic bone resorption and vesicular trafficking. Bone 2011, 49, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Boike, L.; Cioffi, A.G.; Majewski, F.C.; Co, J.; Henning, N.J.; Jones, M.D.; Liu, G.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; et al. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol. 2021, 28, 4–13e17. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.N.; Hursting, S.D.; Haines, D.C.; James, S.J.; Miller, B.J.; Phang, J.M. Chemoprevention of spontaneous tumorigenesis in nullizygous p53-deficient mice by dehydroepiandrosterone and its analog 16alpha-fluoro-5-androsten-17-one. Carcinogenesis 1997, 18, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.G. Dehydroepiandrosterone, Cancer, and Aging. Aging Dis. 2022, 13, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, F.; Zhang, Y.; Lin, Z.; Yang, J.; Han, X.; Feng, Y.; Pei, X.; Li, F.; Liu, Q.; et al. Targeting glucose-6-phosphate dehydrogenase by 6-AN induces ROS-mediated autophagic cell death in breast cancer. FEBS J. 2023, 290, 763–779. [Google Scholar] [CrossRef] [PubMed]
- Zemnou, C.T. In silico Comparative Study of the Anti-Cancer Potential of Inhibitors of Glucose-6-Phosphate Dehydrogenase Enzyme Using ADMET Analysis, Molecular Docking, and Molecular Dynamic Simulation. Adv. Theor. Simul. 2025, 8, 2400757. [Google Scholar] [CrossRef]
- Gupta, T.; Mondal, A.K.; Pani, I.; Chattopadhyay, K.; Pal, S.K. Elucidating liquid crystal-aqueous interface for the study of cholesterol-mediated action of a beta-barrel pore forming toxin. Soft Matter 2022, 18, 5293–5301. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Huang, C.H.; Lin, Y.Q.; Han, B.F.; Chen, Y.Z.; Li, C.J.; Li, J.W.; Ding, Y.Y.; Song, X.C.; Wang, W.; et al. Generation of inactivated IL2RG and RAG1 monkeys with severe combined immunodeficiency using base editing. Signal Transduct. Tar. 2023, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Huang, X.; Floberg, J.M.; Elhammali, A.E.; McCormick, M.L.; Patti, G.J.; Spitz, D.R.; Schwarz, J.K. Radioresistant Cervical Cancers Are Sensitive to Inhibition of Glycolysis and Redox Metabolism. Cancer Res. 2018, 78, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Sotgia, F.; Sisci, D.; Cappello, A.R.; Lisanti, M.P. Mitochondrial “power” drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget 2017, 8, 20309–20327. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Yu, H.; Liang, R.; Jia, R.; Wang, J.; Jiang, K.; Wang, Z. Rev-erbalpha inhibits proliferation by reducing glycolytic flux and pentose phosphate pathway in human gastric cancer cells. Oncogenesis 2019, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.; Zhou, X.; Hu, Y.; Shi, S.; Yang, G. Rev-erbalpha Inhibits Proliferation and Promotes Apoptosis of Preadipocytes through the Agonist GSK4112. Int. J. Mol. Sci. 2019, 20, 4524. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—Potential clinical relevance. Cell Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, R.; Xu, Z.; Ke, Y.; Sun, R.; Yang, H.; Zhang, X.; Zhen, X.; Zheng, L.T. Early glycolytic reprogramming controls microglial inflammatory activation. J. Neuroinflamm. 2021, 18, 129. [Google Scholar] [CrossRef] [PubMed]
- Cal, M.; Matyjaszczyk, I.; Litwin, I.; Augustyniak, D.; Ogorek, R.; Ko, Y.; Ulaszewski, S. The Anticancer Drug 3-Bromopyruvate Induces DNA Damage Potentially Through Reactive Oxygen Species in Yeast and in Human Cancer Cells. Cells 2020, 9, 1161. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kundu, A.; Lee, S.H.; Jiang, C.; Lee, S.H.; Kim, Y.S.; Kyung, S.Y.; Park, S.H.; Kim, H.S. Specific Pyruvate Kinase M2 Inhibitor, Compound 3K, Induces Autophagic Cell Death through Disruption of the Glycolysis Pathway in Ovarian Cancer Cells. Int. J. Biol. Sci. 2021, 17, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.S.; Liu, J.; Wu, Q.C.; Zhou, X.L. Lactate regulates pathological cardiac hypertrophy via histone lactylation modification. J. Cell Mol. Med. 2024, 28, e70022. [Google Scholar] [CrossRef] [PubMed]
- Forteza, M.J.; Berg, M.; Edsfeldt, A.; Sun, J.; Baumgartner, R.; Kareinen, I.; Casagrande, F.B.; Hedin, U.; Zhang, S.; Vuckovic, I.; et al. Pyruvate dehydrogenase kinase regulates vascular inflammation in atherosclerosis and increases cardiovascular risk. Cardiovasc. Res. 2023, 119, 1524–1536. [Google Scholar] [CrossRef] [PubMed]
- Tambe, Y.; Terado, T.; Kim, C.J.; Mukaisho, K.I.; Yoshida, S.; Sugihara, H.; Tanaka, H.; Chida, J.; Kido, H.; Yamaji, K.; et al. Antitumor activity of potent pyruvate dehydrogenase kinase 4 inhibitors from plants in pancreatic cancer. Mol. Carcinog. 2019, 58, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Bodeker, K.L.; Smith, B.J.; Berg, D.J.; Chandrasekharan, C.; Sharif, S.; Fei, N.; Vollstedt, S.; Brown, H.; Chandler, M.; Lorack, A.; et al. A randomized trial of pharmacological ascorbate, gemcitabine, and nab-paclitaxel for metastatic pancreatic cancer. Redox Biol. 2024, 77, 103375. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chiou, J.; Yang, Y.F.; Su, C.Y.; Lin, Y.F.; Yang, C.N.; Lu, P.J.; Huang, M.S.; Yang, C.J.; Hsiao, M. Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs Survival. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.; Mucciolo, G.; Roux, C.; Brugiapaglia, S.; Scagliotti, A.; Guadagnin, G.; Conti, L.; Longo, D.; Grosso, D.; Papotti, M.G.; et al. PI3Kgamma inhibition combined with DNA vaccination unleashes a B-cell-dependent antitumor immunity that hampers pancreatic cancer. J. Exp. Clin. Cancer Res. 2024, 43, 157. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology 2013, 144, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xu, J.; Ma, C.; Wang, Z.; Yang, X.; Song, X.; Zheng, H. Metformin and 2-Deoxy-d-glucose synergistically inhibit the viability of thyroid papillary carcinoma with BRAF mutation by ROS-dependent PI3K/AKT signaling pathway. Anticancer. Agents Med. Chem. 2023, 12, 357. [Google Scholar] [CrossRef]
- Qin, H.; Zheng, G.; Li, Q.; Shen, L. Metabolic reprogramming induced by DCA enhances cisplatin sensitivity through increasing mitochondrial oxidative stress in cholangiocarcinoma. Front. Pharmacol. 2023, 14, 1128312. [Google Scholar] [CrossRef] [PubMed]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, Z.; Qian, K.; Du, W.; Ju, L.; Shan, D.; Yu, M.; Fang, Y.; Zhang, Y.; Xiao, Y.; et al. Unveiling the association between HMG-CoA reductase inhibitors and bladder cancer: A comprehensive analysis using Mendelian randomization, animal models, and transcriptomics. Pharmacogenomics J. 2024, 24, 24. [Google Scholar] [CrossRef] [PubMed]
- Pouyssegur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Zdralevic, M.; Vucetic, M. ‘Warburg effect’ controls tumor growth, bacterial, viral infections and immunity—Genetic deconstruction and therapeutic perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Futagi, Y.; Ideno, M.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Effect of diclofenac on SLC16A3/MCT4 by the Caco-2 cell line. Drug Metab. Pharmacokinet. 2016, 31, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Vander Linden, C.; Corbet, C.; Bastien, E.; Martherus, R.; Guilbaud, C.; Petit, L.; Wauthier, L.; Loriot, A.; De Smet, C.; Feron, O. Therapy-induced DNA methylation inactivates MCT1 and renders tumor cells vulnerable to MCT4 inhibition. Cell Rep. 2021, 35, 109202. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Yadav, A.; Bhattacharya, S.; Dagar, A.; Kumar, V.; Rani, R. GLUT and HK: Two primary and essential key players in tumor glycolysis. Semin. Cancer Biol. 2024, 100, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Reckzeh, E.S.; Karageorgis, G.; Schwalfenberg, M.; Ceballos, J.; Nowacki, J.; Stroet, M.C.M.; Binici, A.; Knauer, L.; Brand, S.; Choidas, A.; et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem. Biol. 2019, 26, 1214–1228.E25. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colon, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Nezhady, M.A.; Chemtob, S. 3-OBA Is Not an Antagonist of GPR81. Front. Pharmacol. 2021, 12, 803907. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mu, G.; Xue, Z.; Wang, S.; Li, X.; Han, M. Targeting cholesterol homeostasis through inhibiting SREBP-2: An Achilles’ heel for glioblastoma. Neuro Oncol. 2023, 25, 2100–2102. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo, B.; Li, Q.; Nie, J. mTOR in metabolic homeostasis and disease. Exp. Cell Res. 2024, 441, 114173. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wang, J.; Liu, M.; Chen, D.; Qiu, C.; Sun, K. CC-223 blocks mTORC1/C2 activation and inhibits human hepatocellular carcinoma cells in vitro and in vivo. PLoS ONE 2017, 12, e0173252. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yi, J.; Han, G.; Qiao, L. MALDI-TOF Mass Spectrometry in Clinical Analysis and Research. ACS Meas. Sci. Au 2022, 2, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, S.V.; Kreuzaler, P.; Maclachlan, C.; McMahon, G.; Greenidge, G.; Collinson, L.; Bunch, J.; Yuneva, M. A multimodal imaging pipeline to decipher cell-specific metabolic functions and tissue microenvironment dynamics. Nat. Protoc. 2025, 20, 1678–1699. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Hoiem, T.S.; Claes, B.S.R.; Balluff, B.; Martin-Lorenzo, M.; Richardsen, E.; Krossa, S.; Bertilsson, H.; Heeren, R.M.A.; Rye, M.B.; et al. Spatial differentiation of metabolism in prostate cancer tissue by MALDI-TOF MSI. Cancer Metab. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Krestensen, K.K.; Hendriks, T.F.E.; Grgic, A.; Derweduwe, M.; De Smet, F.; Heeren, R.M.A.; Cuypers, E. Molecular Profiling of Glioblastoma Patient-Derived Single Cells Using Combined MALDI-MSI and MALDI-IHC. Anal. Chem. 2025, 97, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Gu, H.; Chen, C.; Huang, D.; Zhao, Y.; Xie, L.; Zou, Y.; Shu, H.S.; Zhang, Y.; He, X.; et al. Metabolic Imaging Reveals a Unique Preference of Symmetric Cell Division and Homing of Leukemia-Initiating Cells in an Endosteal Niche. Cell Metab. 2019, 29, 950–965.E6. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017, 26, 361–374e364. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Ahn, J.; Kim, S.; Lee, Y.; Lee, J.; Park, D.; Jeon, N.L. Tumor spheroid-on-a-chip: A standardized microfluidic culture platform for investigating tumor angiogenesis. Lab. Chip 2019, 19, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, J.M.; Rehman, S.; Virumbrales-Munoz, M.; McMinn, P.H.; Geiger, P.; Fitzgerald, C.; Heaster, T.; Skala, M.C.; Beebe, D.J. Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion. Sci. Adv. 2021, 7, eabc2331. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Loong, H.H.; Wong, A.M.; Chan, D.T.; Cheung, M.S.; Chow, C.; Ding, X.; Chan, A.K.; Johnston, P.A.; Lau, J.Y.; Poon, W.S.; et al. Patient-derived tumor organoid predicts drugs response in glioblastoma: A step forward in personalized cancer therapy? J. Clin. Neurosci. 2020, 78, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Zhang, Y.; Xu, H.; Shen, L.; Cheng, J.; Xu, X.; Tan, H.; Chen, X.; Li, J. Tumor Microenvironment-Adaptive Nanoplatform Synergistically Enhances Cascaded Chemodynamic Therapy. Bioact. Mater. 2023, 22, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Ye, H.; Li, Y.; Liang, Q.; Li, X.; Yin, L. Multivalency-assisted membrane-penetrating siRNA delivery sensitizes photothermal ablation via inhibition of tumor glycolysis metabolism. Biomaterials 2019, 223, 119463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Qiu, W.X.; Zhang, M.; Zhang, L.; Zhang, X.Z. MnO2 Motor: A Prospective Cancer-Starving Therapy Promoter. ACS Appl. Mater. Interfaces 2018, 10, 15030–15039. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Luo, G.F.; Lei, Q.; Hong, S.; Qiu, W.X.; Liu, L.H.; Cheng, S.X.; Zhang, X.Z. Overcoming the Heat Endurance of Tumor Cells by Interfering with the Anaerobic Glycolysis Metabolism for Improved Photothermal Therapy. ACS Nano 2017, 11, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Hao, Q.; Cai, M.; Wang, X.; An, W. Harnessing glucose metabolism with nanomedicine for cancer treatment. Theranostics 2024, 14, 6831–6882. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Moret, M.; Salvy, P.; Weilandt, D.; Hatzimanikatis, V.; Miskovic, L. Reconstructing Kinetic Models for Dynamical Studies of Metabolism using Generative Adversarial Networks. Nat. Mach. Intell. 2022, 4, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Stephanopoulos, G.; Kelleher, J.K. Evaluation of regression models in metabolic physiology: Predicting fluxes from isotopic data without knowledge of the pathway. Metabolomics 2006, 2, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Azad, R.K.; Shulaev, V. Metabolomics technology and bioinformatics for precision medicine. Brief. Bioinform. 2019, 20, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Rasmussen, M.H.; Piening, B.; Shen, X.; Chen, S.; Rost, H.; Snyder, J.K.; Tibshirani, R.; Skotte, L.; Lee, N.C.; et al. Metabolic Dynamics and Prediction of Gestational Age and Time to Delivery in Pregnant Women. Cell 2020, 181, 1680–1692.E15. [Google Scholar] [CrossRef] [PubMed]
| Therapeutic Context | Resistance Mechanism | Key Metabolic Alterations | Associated Biomarkers/Pathways |
|---|---|---|---|
| Gefitinib-Resistant EGFR-Mutant NSCLC [35] | Enhanced Glycolysis | Upregulation of GLUT1, HK2, LDHA | Increased lactate production |
| Vemurafenib-Resistant BRAF-Mutant Melanoma [36] | Enhanced Glycolysis | Upregulation of GLUT1/3, HK2 via MEK/ERK signaling | Elevated glucose uptake and glycolytic flux |
| Trastuzumab-Resistant HER2+ Breast Cancer [37] | Enhanced Glycolysis | PFKFB3-mediated fructose-2,6-bisphosphate synthesis | Increased glycolytic activity |
| Osimertinib-Resistant NSCLC [38] | Mitochondrial Reprogramming | Enhanced OXPHOS | Increased OXPHOS activity |
| BRAF Inhibitor-Resistant Melanoma [39] | Mitochondrial Biogenesis | PGC-1α-mediated increase in mitochondrial density and oxidative capacity | Elevated cristae density, increased OXPHOS |
| Immunotherapy Resistance (e.g., PD-1 Inhibition) [40] | Metabolic Reprogramming Linked to Immune Evasion | Selection for clones with enhanced OXPHOS, co-expression of TIM-3 | Link between metabolic reprogramming and immune evasion |
| Metabolite | Target Enzymes | Epigenetic Effects | Key Mechanisms |
|---|---|---|---|
| Lactate [51,52] | HDACs, GPR81 | Increased H3K18 lactylation | Lactate promotes the epithelial–mesenchymal transition of liver cancer cells via TWIST1 lactylation. |
| Succinate [53] | PHDs, KDMs | DNA hypermethylation | Succinic acid stabilizes HIF-1α activity by inhibiting its degradation. Succinate promotes angiogenesis by inducing the expression of VEGF. |
| Fumarate [56] | TETs, PHDs | Histone succinylation | The accumulation of fumaric acid results in pseudohypoxia by stabilizing the transcription factor HIF-1α and DNA hypermethylation, and inhibiting enzymes of the 2OGDD family. |
| 2-HG [57,58] | FTO, ATP synthase | Decreased m6A modification | Selective inhibition of the m6A epitranscriptomic regulator FTO attenuates growth in IDH1wt glioma. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, X.; Tao, Q.; Li, L. Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions. Int. J. Mol. Sci. 2025, 26, 6901. https://doi.org/10.3390/ijms26146901
Chai X, Tao Q, Li L. Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions. International Journal of Molecular Sciences. 2025; 26(14):6901. https://doi.org/10.3390/ijms26146901
Chicago/Turabian StyleChai, Xiaoxue, Qian Tao, and Lili Li. 2025. "Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions" International Journal of Molecular Sciences 26, no. 14: 6901. https://doi.org/10.3390/ijms26146901
APA StyleChai, X., Tao, Q., & Li, L. (2025). Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions. International Journal of Molecular Sciences, 26(14), 6901. https://doi.org/10.3390/ijms26146901