
Genetic Landscape of a Pleural Mesothelioma in a Child Affected by NF2-Related Schwannomatosis
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
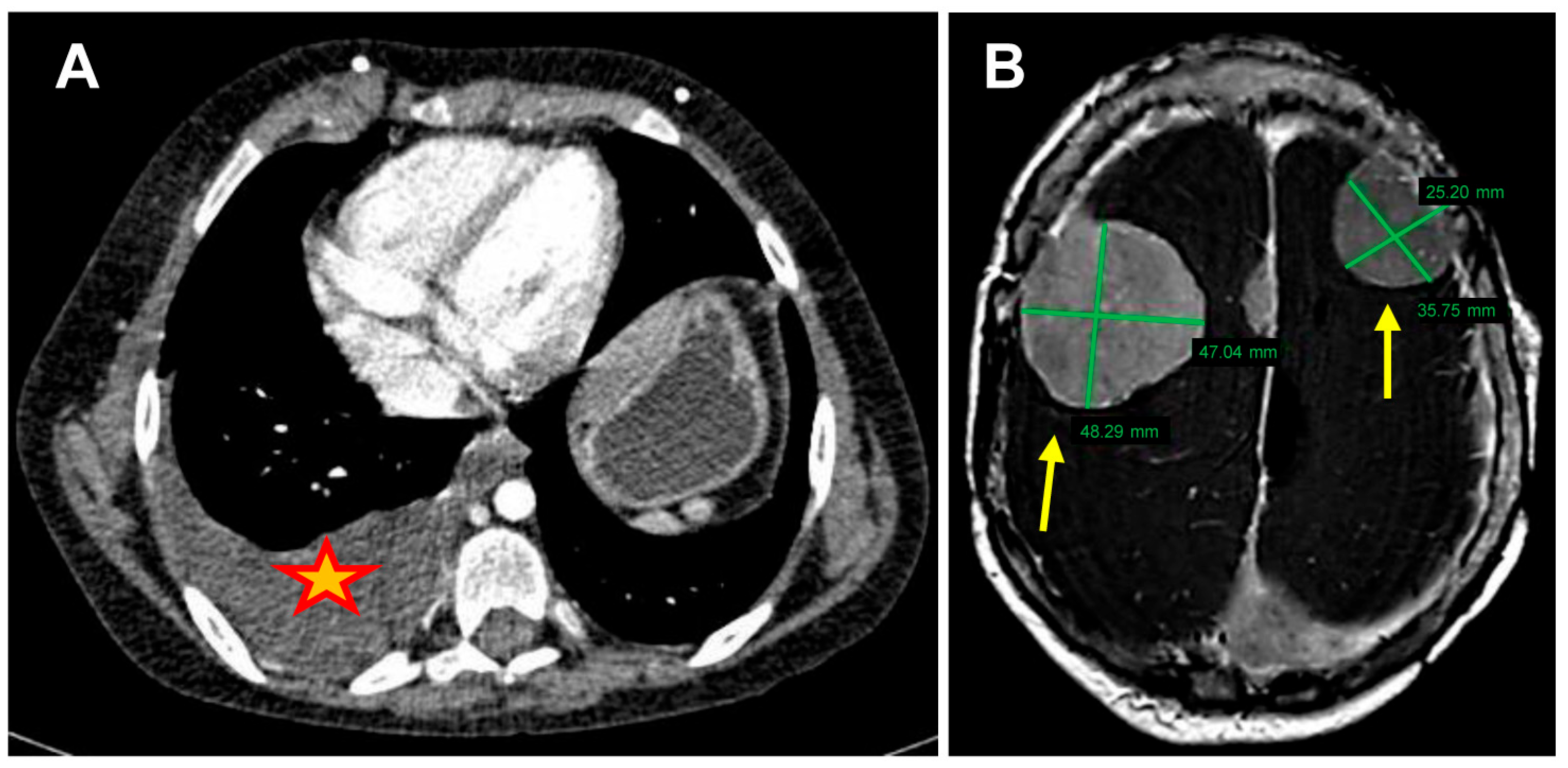
2.1. Patient Information
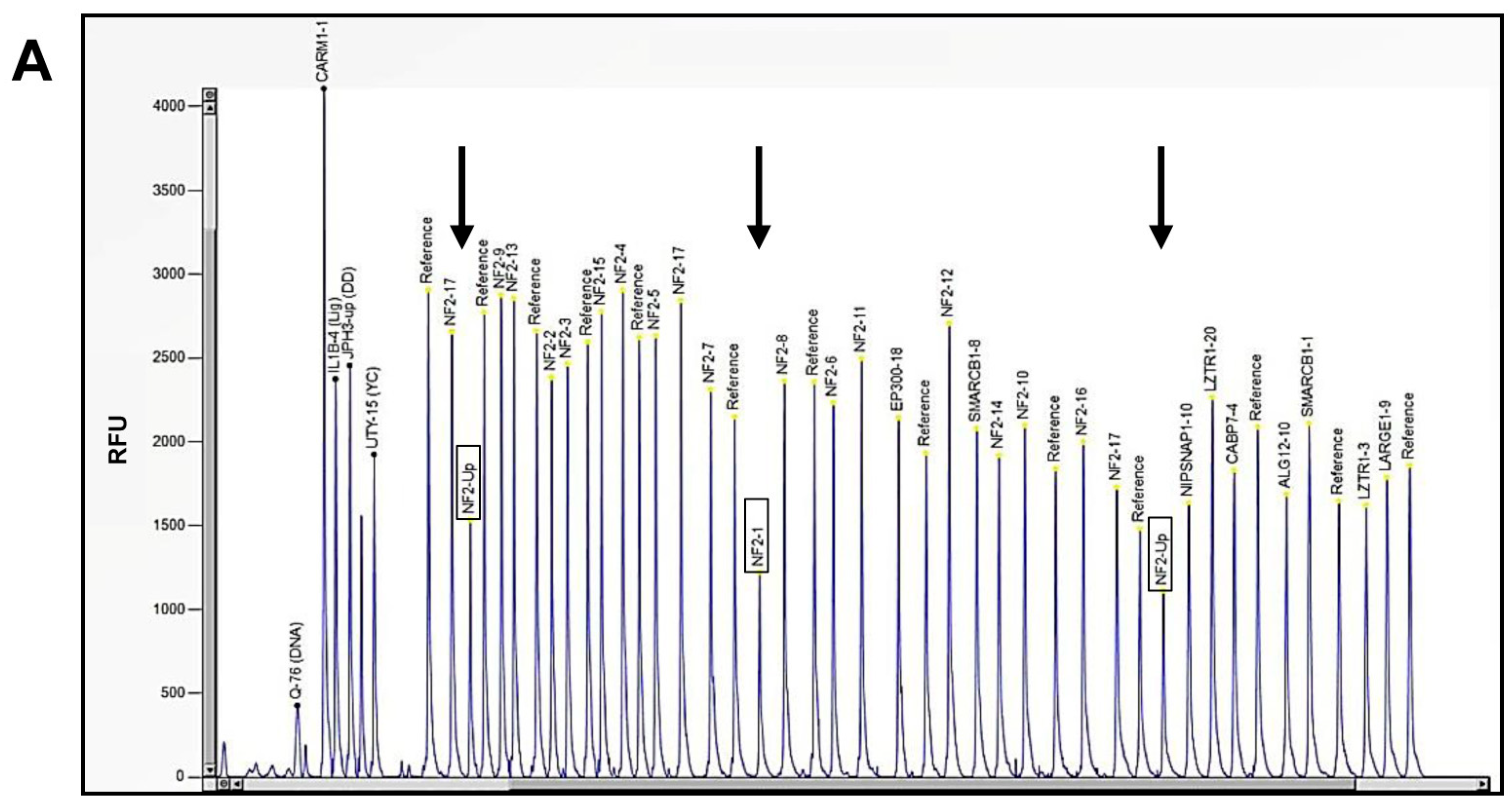
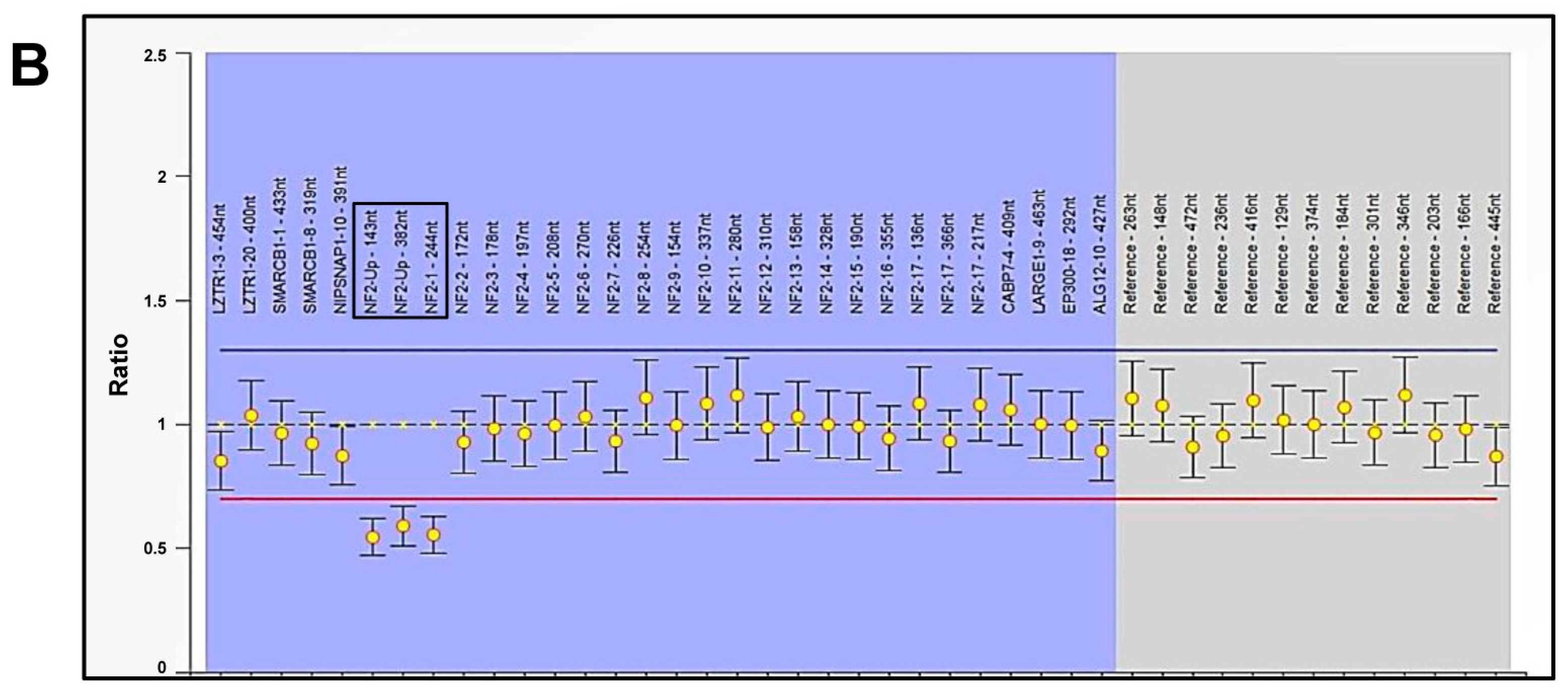
2.2. Molecular Diagnosis of NF2-Related Schwannomatosis
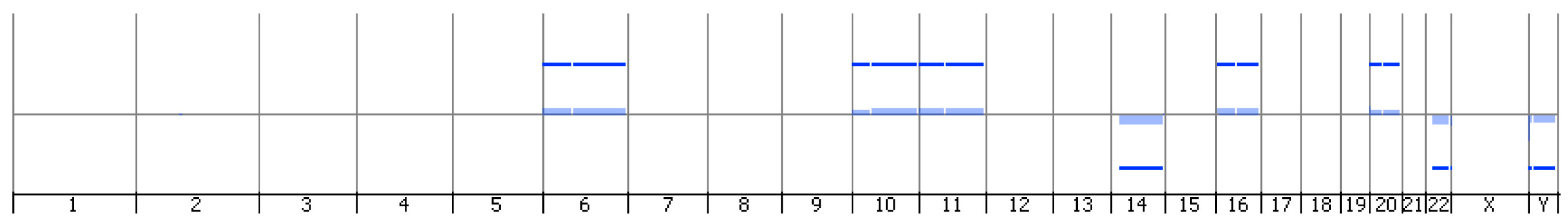
2.3. Genomic Profile Analysis of Mesothelioma Cells by Array-CGH
2.4. Genomic Profiling of Mesothelioma Cells by NGS Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Targeted Next-Generation Sequencing (NGS) of NF2-SWN-Related Genes
4.3. Multiplex Ligation-Dependent Probe Amplification Analysis (MLPA)
4.4. Genomic Profile Analysis by Array-Comparative Genomic Hybridization (a-CGH)
4.5. Targeted NGS of Cancer-Related Genes
4.6. Bioinformatics Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Orbach, D.; André, N.; Brecht, I.B.; López Almaraz, R.; Ben-Ami, T.; Vermersch, S.; Carton, M.; Virgone, C.; Bisogno, G.; Schneider, D.T.; et al. Mesothelioma in children and adolescents: The European Cooperative Study Group for Pediatric Rare Tumors (EXPeRT) contribution. Eur. J. Cancer 2020, 140, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.M.; Bortolotto, C.; Benvenuti, S.; Lancia, A.; Filippi, A.R.; Stella, G.M. Malignant Pleural Mesothelioma: Genetic and Microenviromental Heterogeneity as an Unexpected Reading Frame and Therapeutic Challenge. Cancers 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Aspesi, A.; Sculco, M.; Matullo, G.; Magnani, C.; Dianzani, I. Genetic predisposition for malignant mesothelioma: A concise review. Mutat. Res. Rev. Mutat. Res. 2019, 781, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Farioli, A.; Ottone, M.; Morganti, A.G.; Compagnone, G.; Romani, F.; Cammelli, S.; Mattioli, S.; Violante, F.S. Radiation-induced mesothelioma among long-term solid cancer survivors: A longitudinal analysis of SEER database. Cancer Med. 2016, 5, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Attanoos, R.L.; Churg, A.; Galateau-Salle, F.; Gibbs, A.R.; Roggli, V.L. Malignant Mesothelioma and Its Non-Asbestos Causes. Arch. Pathol. Lab. Med. 2018, 142, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tomson, B.N.; Buys, T.P.H.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. Genomic Landscape of Malignant Mesotheliomas. Mol. Cancer Ther. 2016, 15, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Neragi-Miandoab, S.; Sugarbaker, D.J. Chromosomal deletion in patients with malignant pleural mesothelioma. Interact Cardiovasc. Thorac. Surg. 2009, 9, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Messiaen, L.; Legius, E.; Pancza, P.; Avery, R.A.; Blakeley, J.O.; Babovic-Vuksanovic, D.; Ferner, R.; Fisher, M.J.; Friedman, J.M.; et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet. Med. 2022, 24, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Baser, M.E.; De Rienzo, A.; Altomare, D.; Balsara, B.R.; Hedrick, N.M.; Gutmann, D.H.; Pitts, L.H.; Jackler, R.K.; Testa, J.R. Neurofibromatosis 2 and malignant mesothelioma. Neurology 2002, 59, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.; Sholl, L.M.; Landgraf, J.R.; Chirieac, L.; Roggli, V.R. Molecular Analysis of a Patient With Neurofibromatosis 2 (NF2) and Peritoneal Malignant Mesothelioma. Am. J. Surg. Pathol. 2020, 44, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Evaristo, G.; Fiset, P.O.; Camilleri-Broët, S.; Vanounou, T.; Kavan, P.; Spatz, A.; Wang, H. Molecular Analysis of a Patient With Neurofibromatosis 2 (NF2) and Peritoneal Malignant Mesothelioma. Am. J. Surg. Pathol. 2020, 44, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Baser, M.E.; Rai, H.; Wallace, A.J.; Evans, D.G. Neurofibromatosis 2 (NF2) and malignant mesothelioma in a man with a constitutional NF2 missense mutation. Fam. Cancer 2005, 4, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Miller, J.; Lucito, R.; Tang, C.; Ivanova, A.V.; Pei, J.; Carbone, M.; Cruz, C.; Beck, A.; Webb, C.; et al. Genomic events associated with progression of pleural malignant mesotheliom. Int. J. Cancer 2009, 124, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Fleischmann, Z.; Sokol, E.S.; Zoche, M.; Felley-Bosco, E.; Curioni-Fontecedro, A. Genomic landscape of pleural and peritoneal mesotelioma tumours. Br. J. Cancer 2022, 127, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Mariño-Enríquez, A.; Hammer, M.M.; Padera, R.F.; Sholl, L.M. Recurrent Tumor Suppressor Alterations in Primary Pericardial Mesothelioma. Mod. Pathol. 2023, 36, 100237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Luo, J.L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef] [PubMed]
- Hylebos, M.; Van Camp, G.; van Meerbeeck, J.P.; Op de Beeck, K. The Genetic Landscape of Malignant Pleural Mesothelioma: Results from Massively Parallel Sequencing. J. Thorac. Oncol. 2016, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Hylebos, M.; Van Camp, G.; Vandeweyer, G.; Fransen, E.; Beyens, M.; Cornelissen, R.; Suls, A.; Pauwels, P.; van Meerbeeck, J.P.; Op de Beeck, K. Large-scale copy number analysis reveals variations in genes not previously associated with malignant pleural mesothelioma. Oncotarget 2017, 8, 113673–113686. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, O.D.; Gilliam, K.; Del Gaudio, D.; McNeely, K.E.; Smith, S.; Acevedo, M.; Gaduraju, M.; Hodge, R.; Ramsland, A.S.S.; Segal, J.; et al. Germline Variants Incidentally Detected via Tumor-Only Genomic Profiling of Patients With Mesothelioma. JAMA Netw. Open 2023, 6, e2327351. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Gemignani, F.; Landi, S. The genetic susceptibility in the development of malignant pleural mesothelioma. J. Thorac. Dis. 2018, 10 (Suppl. S2), S246–S252. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Ogata, S.; Nakashima, H.; Urabe, S.; Murakami, I.; Hiroshima, K. Clinicopathologic study of deciduoid mesothelioma using SMARCB1/INI1 immunohistochemistry and fluorescence in situ hybridization. Hum. Pathol. 2019, 93, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, L.A.; Liang, C.C.; Belan, O.; Kunzelmann, S.; Maslen, S.; Rodrigo-Brenni, M.C.; Anand, R.; Skehel, M.; Boulton, S.J.; West, S.C. Structure and function of the RAD51B-RAD51C-RAD51D-XRCC2 tumour suppressor. Nature 2023, 619, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Ozaslan, M. Exonic sequencing and MLH3 gene expression analysis of breast cancer patients. Cell Mol. Biol. 2021, 67, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Scharf, J.B.; Lees, G.M.; Sergi, C.M. Malignant pleural mesothelioma in a child. J. Pediatr. Surg. Case Rep. 2015, 3, 440–443. [Google Scholar] [CrossRef]
- Patel, R.A.; Lin, M.; Harper, M.M.; Beck, S.J.; Dietrich, C.S.; Kolesar, J.M.; Arnold, S.M.; Hahn, J.; Pandalai, P.K.; Qasem, S.A.; et al. Pediatric patient with peritoneal mesothelioma harboring ALK rearrangement. Curr. Probl. Cancer Case Rep. 2021, 4, 100074. [Google Scholar] [CrossRef]
- Redzepagic, J.; Zvizdic, Z.; Bilalovic, N.; Milisic, E.; Bukvic, M.; Vranic, S. Comprehensive genomic profiling of pediatric peritoneal mesothelioma: Case report with a literature review. J. Surg. Case Rep. 2024, 5, rjae324. [Google Scholar] [CrossRef] [PubMed]
- Cedres, S.; Valdivia, A.; Priano, I.; Rocha, P.; Iranzo, P.; Pardo, N.; Martinez-Marti, A.; Felip, E. BAP1 Mutations and Pleural Mesothelioma: Genetic Insights, Clinical Implications, and Therapeutic Perspectives. Cancers 2025, 17, 1581. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; De Marco, P.; Amoroso, L.; Fragola, M.; Zara, F.; Parodi, S.; Pezzolo, A. Neuroblastoma Patients’ Outcome and Chromosomal Instability. Int. J. Mol. Sci. 2023, 24, 15514. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus * | Filter | Variant Class | Variant ID | Cytoband and Copy Number ** |
|---|---|---|---|---|
| chr6:350767 | GAIN | CNV | ARM 6p | 6p25.3p12.1 (350,767–55,739,590) × 3 |
| chr6:62442567 | GAIN | CNV | ARM 6q | 6q11.1q27 (62,442,567–170,627,672) × 3 |
| chr10:323466 | GAIN | CNV | ARM 10p | 10p15.3p11.22 (323,466–32,856,813) × 3 |
| chr10:43292608 | GAIN | CNV | ARM 10q | 10q11.21q26.2 (43,292,608–129,902,370) × 3 |
| chr11:403974 | GAIN | CNV | ARM 11p | 11p15.5p11.12 (403,974–50,003,230) × 3 |
| chr11:55032426 | GAIN | CNV | ARM 11q | 11q11q24.3 (55,032,426–130,776,619) × 3 |
| chr14:20248397 | LOSS | CNV | ARM 14q | 14q11.2q32.33 (20,248,397–105,246,558) × 1 |
| chr14:20811781 | LOSS | CNV | PARP2 | 14q11.2 (20,811,781–20,825,977) × 0.942029 |
| chr14:45605157 | LOSS | CNV | FANCM | 14q21.2 (45,605,157–45,669,234) × 1.05797 |
| chr14:68290164 | LOSS | CNV | RAD51B | 14q24.1 (68,290,164–69,061,406) × 1 |
| chr14:68290164 | LOSS | LOH | RAD51B | 14q24.1 (68,290,164–69,061,406) × 1 |
| chr14:75483761 | LOSS | CNV | MLH3 | 14q24.3 (75,483,761–75,516,400) × 0.971014 |
| chr14:95556791 | LOSS | CNV | DICER1 | 14q32.13 (95,556,791–95,599,859) × 1.07246 |
| chr14:104165043 | LOSS | CNV | XRCC3 | 14q32.33 (104,165,043–104,177,450) × 1.05797 |
| chr16:336630 | GAIN | CNV | ARM 16p | 16p13.3p11.2 (336,630–31,092,113) × 3 |
| chr16:47697595 | GAIN | CNV | ARM 16q | 16q12.1q24.3 (47,697,595–89,883,082) × 3 |
| chr20:391069 | GAIN | CNV | ARM 20p | 20p13p11.21 (391,069–25,187,259) × 3 |
| chr20:30954155 | GAIN | CNV | ARM 20q | 20q11.21q13.33 (30,954,155–62,595,178) × 3 |
| chr22:22123473 | LOSS | CNV | MAPK1 | 22q11.21 (22,123,473–22,162,093) × 1.07246 |
| chr22:24129273 | LOSS | CNV | SMARCB1 | 22q11.23 (24,129,273–24,176,467) × 0.913043 |
| chr22:29083868 | LOSS | CNV | CHEK2 | 22q12.1 (29,083,868–29,130,729) × 1 |
| chr22:29083868 | LOSS | LOH | CHEK2 | 22q12.1 (29,083,868–29,130,729) × 1 |
| chr22:29999923 | LOSS | CNV | NF2 | 22q12.2 (29,999,923–30,090,863) × 0.855072 |
| chr22:41489001 | LOSS | CNV | EP300 | 22q13.2 (41,489,001–41,574,996) × 0.971014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ognibene, M.; Piccolo, G.; Crocco, M.; Di Duca, M.; Verrico, A.; Molteni, M.; Romano, F.; Capra, V.; Rossi, A.; Zara, F.; et al. Genetic Landscape of a Pleural Mesothelioma in a Child Affected by NF2-Related Schwannomatosis. Int. J. Mol. Sci. 2025, 26, 6848. https://doi.org/10.3390/ijms26146848
Ognibene M, Piccolo G, Crocco M, Di Duca M, Verrico A, Molteni M, Romano F, Capra V, Rossi A, Zara F, et al. Genetic Landscape of a Pleural Mesothelioma in a Child Affected by NF2-Related Schwannomatosis. International Journal of Molecular Sciences. 2025; 26(14):6848. https://doi.org/10.3390/ijms26146848
Chicago/Turabian StyleOgnibene, Marzia, Gianluca Piccolo, Marco Crocco, Marco Di Duca, Antonio Verrico, Marta Molteni, Ferruccio Romano, Valeria Capra, Andrea Rossi, Federico Zara, and et al. 2025. "Genetic Landscape of a Pleural Mesothelioma in a Child Affected by NF2-Related Schwannomatosis" International Journal of Molecular Sciences 26, no. 14: 6848. https://doi.org/10.3390/ijms26146848
APA StyleOgnibene, M., Piccolo, G., Crocco, M., Di Duca, M., Verrico, A., Molteni, M., Romano, F., Capra, V., Rossi, A., Zara, F., De Marco, P., & Milanaccio, C. (2025). Genetic Landscape of a Pleural Mesothelioma in a Child Affected by NF2-Related Schwannomatosis. International Journal of Molecular Sciences, 26(14), 6848. https://doi.org/10.3390/ijms26146848