

Circulating Extracellular Vesicles in Cardiovascular Disease



Abstract
1. Introduction
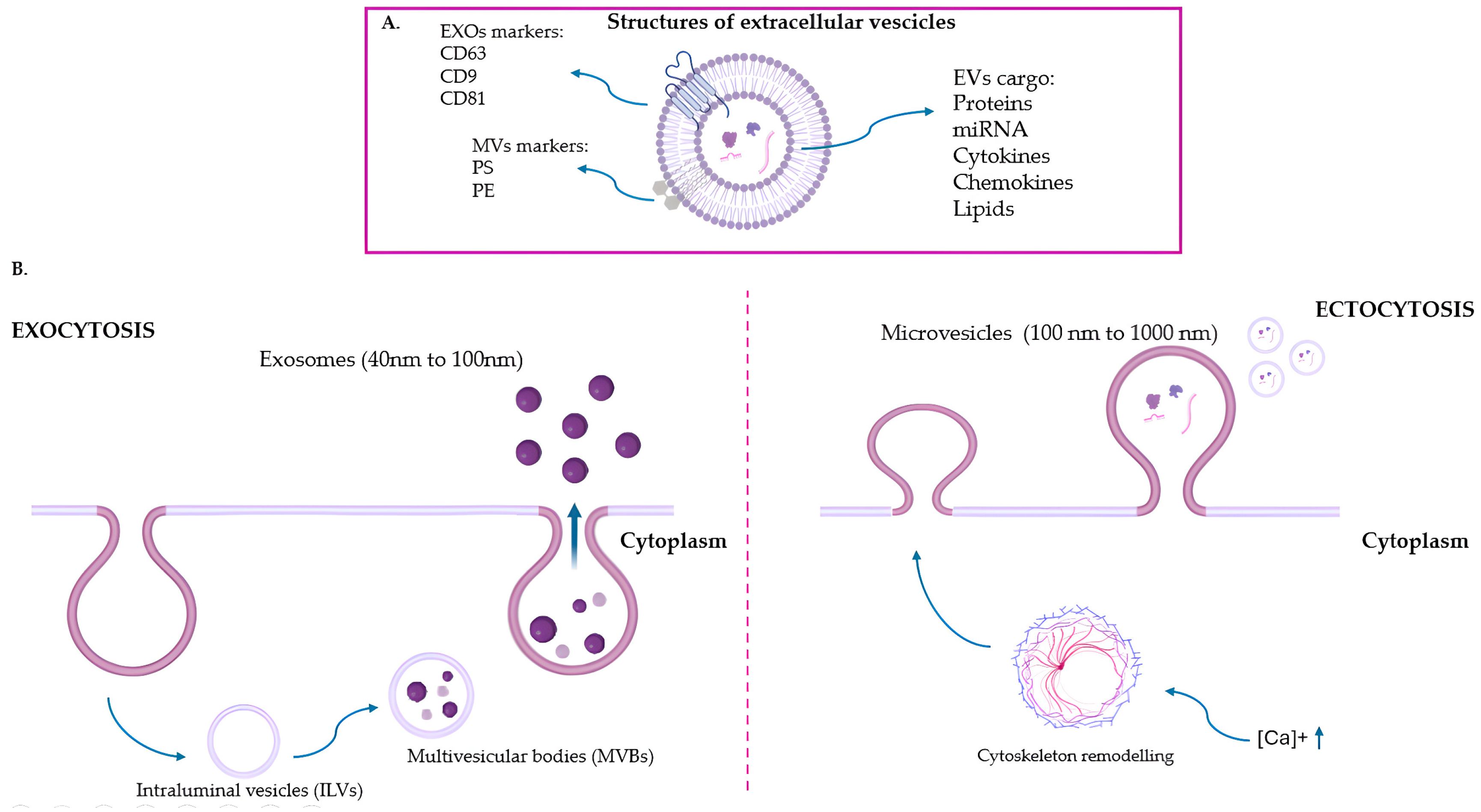
2. Extracellular Vesicles
3. Extracellular Vesicles in Bloodstream
4. EVs as Possible Novel Biomarkers for CVDs
4.1. EVs and Atherosclerosis
4.2. EVs and Myocardial Infarction
4.3. EVs and Heart Failure
4.4. EVs and Hypertension
5. Extracellular Vesicles in CVD Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palena, L.M.; Isernia, G.; Parlani, G.; Veroux, P.; Ficarelli, I.; Frascheri, A.; Pischedda, A.; Patrone, L.; Dionisi, C.P.; Cianni, R.; et al. A multicenter prospective observational study appraising the effectiveness of the Supera stent after subintimal recanalization of femoro-popliteal artery occlusion: The SUPERSUB II study. Catheter. Cardiovasc. Interv. 2024, 103, 963–971. [Google Scholar] [CrossRef]
- Ekanem, E.; Neuzil, P.; Reichlin, T.; Kautzner, J.; van der Voort, P.; Jais, P.; Chierchia, G.B.; Bulava, A.; Blaauw, Y.; Skala, T.; et al. Safety of pulsed field ablation in more than 17,000 patients with atrial fibrillation in the MANIFEST-17K study. Nat. Med. 2024, 30, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, L.; Mikus, E.; Tenti, E.; Sangiorgi, D.; Zannoni, S.; Cavallucci, A.; Ferroni, L.; Cimaglia, P.; Tolio, V.; Tremoli, E.; et al. Post-Operative Delirium and Cognitive Dysfunction in Aged Patients Undergoing Cardiac Surgery: A Randomized Comparison between Two Blood Oxygenators. Bioengineering 2023, 10, 1429. [Google Scholar] [CrossRef]
- Joseph, J.J.; Deedwania, P.; Acharya, T.; Aguilar, D.; Bhatt, D.L.; Chyun, D.A.; Di Palo, K.E.; Golden, S.H.; Sperling, L.S.; American Heart Association Diabetes Committee of the Council on Lifestyle and Cardiometabolic Health; et al. Comprehensive Management of Cardiovascular Risk Factors for Adults With Type 2 Diabetes: A Scientific Statement From the American Heart Association. Circulation 2022, 145, e722–e759. [Google Scholar] [CrossRef] [PubMed]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef]
- Markina, Y.V.; Kirichenko, T.V.; Tolstik, T.V.; Bogatyreva, A.I.; Zotova, U.S.; Cherednichenko, V.R.; Postnov, A.Y.; Markin, A.M. Target and Cell Therapy for Atherosclerosis and CVD. Int. J. Mol. Sci. 2023, 24, 10308. [Google Scholar] [CrossRef]
- Cullen, L.; Aldous, S.; Than, M.; Greenslade, J.H.; Tate, J.R.; George, P.M.; Hammett, C.J.; Richards, A.M.; Ungerer, J.P.; Troughton, R.W.; et al. Comparison of high sensitivity troponin T and I assays in the diagnosis of non-ST elevation acute myocardial infarction in emergency patients with chest pain. Clin. Biochem. 2014, 47, 321–326. [Google Scholar] [CrossRef]
- Huang, X.; Bai, S.; Luo, Y. Advances in research on biomarkers associated with acute myocardial infarction: A review. Medicine 2024, 103, e37793. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef]
- Corradi, D.; Saffitz, J.E.; Novelli, D.; Asimaki, A.; Simon, C.; Oldoni, E.; Masson, S.; Meessen, J.M.T.A.; Monaco, R.; Manuguerra, R.; et al. Prospective Evaluation of Clinico-Pathological Predictors of Postoperative Atrial Fibrillation: An Ancillary Study From the OPERA Trial. Circ. Arrhythm. Electrophysiol. 2020, 13, e008382. [Google Scholar] [CrossRef]
- Giannitsis, E.; Mair, J.; Christersson, C.; Siegbahn, A.; Huber, K.; Jaffe, A.S.; Peacock, W.F.; Plebani, M.; Thygesen, K.; Möckel, M.; et al. How to use D-dimer in acute cardiovascular care. Eur. Heart J. Acute Cardiovasc. Care 2017, 6, 69–80. [Google Scholar] [CrossRef]
- Loffi, M.; Regazzoni, V.; Toselli, M.; Cereda, A.; Palmisano, A.; Vignale, D.; Moroni, F.; Pontone, G.; Andreini, D.; Mancini, E.M.; et al. Incidence and characterization of acute pulmonary embolism in patients with SARS-CoV-2 pneumonia: A multicenter Italian experience. PLoS ONE 2021, 16, e0245565. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, B.A.; Cabarkapa, V.S.; Ilic, T.A.; Salatic, I.R.; Lozanov-Crvenkovic, Z.S.; Mitic, G.P. Clinical significance of determining plasma homocysteine: Case-control study on arterial and venous thrombotic patients. Croat. Med. J. 2013, 54, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Bayes-Genis, A.; Mebazaa, A.; Bauersachs, J.; Cleland, J.G.F.; Coats, A.J.S.; Januzzi, J.L.; Maisel, A.S.; McDonald, K.; Mueller, T.; et al. Circulating heart failure biomarkers beyond natriuretic peptides: Review from the Biomarker Study Group of the Heart Failure Association (HFA), European Society of Cardiology (ESC). Eur. J. Heart Fail. 2021, 23, 1610–1632. [Google Scholar] [CrossRef] [PubMed]
- Loutati, R.; Bruoha, S.; Taha, L.; Karmi, M.; Perel, N.; Maller, T.; Sabouret, P.; Galli, M.; Zoccai, G.B.; De Rosa, S.; et al. Association between peak troponin level and prognosis among patients admitted to intensive cardiovascular care unit. Int. J. Cardiol. 2024, 417, 132556. [Google Scholar] [CrossRef]
- Katrukha, I.A. Human cardiac troponin complex. Structure and functions. Biochemistry 2013, 78, 1447–1465. [Google Scholar] [CrossRef]
- Chauin, A. The Main Causes and Mechanisms of Increase in Cardiac Troponin Concentrations Other Than Acute Myocardial Infarction (Part 1): Physical Exertion, Inflammatory Heart Disease, Pulmonary Embolism, Renal Failure, Sepsis. Vasc. Health Risk Manag. 2021, 17, 601–617. [Google Scholar] [CrossRef]
- Pavasini, R.; d’Ascenzo, F.; Campo, G.; Biscaglia, S.; Ferri, A.; Contoli, M.; Papi, A.; Ceconi, C.; Ferrari, R. Cardiac troponin elevation predicts all-cause mortality in patients with acute exacerbation of chronic obstructive pulmonary disease: Systematic review and meta-analysis. Int. J. Cardiol. 2015, 191, 187–193. [Google Scholar] [CrossRef]
- Samad, M.; Malempati, S.; Restini, C.B.A. Natriuretic Peptides as Biomarkers: Narrative Review and Considerations in Cardiovascular and Respiratory Dysfunctions. Yale J. Biol. Med. 2023, 96, 137–149. [Google Scholar] [CrossRef]
- Rahbar Kouibaran, F.; Sabatino, M.; Barozzi, C.; Diemberger, I. Atrial Natriuretic Peptides as a Bridge between Atrial Fibrillation, Heart Failure, and Amyloidosis of the Atria. Int. J. Mol. Sci. 2023, 24, 6470. [Google Scholar] [CrossRef]
- Castiglione, V.; Aimo, A.; Vergaro, G.; Saccaro, L.; Passino, C.; Emdin, M. Biomarkers for the diagnosis and management of heart failure. Heart Fail. Rev. 2022, 27, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Bytyçi, I.; Shenouda, R.; Wester, P.; Henein, M.Y. Carotid Atherosclerosis in Predicting Coronary Artery Disease: A Systematic Review and Meta-Analysis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e224–e237. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [PubMed]
- Gardin, C.; Ferroni, L.; Leo, S.; Tremoli, E.; Zavan, B. Platelet-Derived Exosomes in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 12546. [Google Scholar] [CrossRef]
- Chachques, J.C.; Gardin, C.; Lila, N.; Ferroni, L.; Migonney, V.; Falentin-Daudre, C.; Zanotti, F.; Trentini, M.; Brunello, G.; Rocca, T.; et al. Elastomeric Cardiowrap Scaffolds Functionalized with Mesenchymal Stem Cells-Derived Exosomes Induce a Positive Modulation in the Inflammatory and Wound Healing Response of Mesenchymal Stem Cell and Macrophage. Biomedicines 2021, 9, 824. [Google Scholar] [CrossRef]
- Gardin, C.; Ferroni, L.; Erdoğan, Y.K.; Zanotti, F.; De Francesco, F.; Trentini, M.; Brunello, G.; Ercan, B.; Zavan, B. Nanostructured Modifications of Titanium Surfaces Improve Vascular Regenerative Properties of Exosomes Derived from Mesenchymal Stem Cells: Preliminary In Vitro Results. Nanomaterials 2021, 11, 3452. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Mobarrez, F.; Sjövik, C.; Soop, A.; Hållström, L.; Frostell, C.; Pisetsky, D.S.; Wallén, H. CD40L expression in plasma of volunteers following LPS administration: A comparison between assay of CD40L on platelet microvesicles and soluble CD40L. Platelets 2015, 26, 486–490. [Google Scholar] [CrossRef]
- Xu, M.; Ji, J.; Jin, D.; Wu, Y.; Wu, T.; Lin, R.; Zhu, S.; Jiang, F.; Ji, Y.; Bao, B.; et al. The biogenesis and secretion of exosomes and multivesicular bodies (MVBs): Intercellular shuttles and implications in human diseases. Genes. Dis. 2023, 10, 1894–1907. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef]
- Leo, S.; Tremoli, E.; Ferroni, L.; Zavan, B. Role of Epicardial Adipose Tissue Secretome on Cardiovascular Diseases. Biomedicines 2023, 11, 1653. [Google Scholar] [CrossRef]
- Xiong, M.; Chen, Z.; Tian, J.; Peng, Y.; Song, D.; Zhang, L.; Jin, Y. Exosomes derived from programmed cell death: Mechanism and biological significance. Cell Commun. Signal 2024, 22, 156. [Google Scholar] [CrossRef]
- Bellin, G.; Gardin, C.; Ferroni, L.; Chachques, J.C.; Rogante, M.; Mitrečić, D.; Ferrari, R.; Zavan, B. Exosome in Cardiovascular Diseases: A Complex World Full of Hope. Cells 2019, 8, 166. [Google Scholar] [CrossRef]
- Waldenström, A.; Gennebäck, N.; Hellman, U.; Ronquist, G. Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS ONE 2012, 7, e34653. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, B. Insight into endothelial cell-derived extracellular vesicles in cardiovascular disease: Molecular mechanisms and clinical implications. Pharmacol. Res. 2024, 207, 107309. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J.; Lasonder, E.; Groenen-Döpp, Y.A.; Willekens, F.L.; Werre, J.M. The proteome of erythrocyte-derived microparticles from plasma: New clues for erythrocyte aging and vesiculation. J. Proteom. 2012, 76, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Pugholm, L.H.; Bæk, R.; Søndergaard, E.K.; Revenfeld, A.L.; Jørgensen, M.M.; Varming, K. Phenotyping of Leukocytes and Leukocyte-Derived Extracellular Vesicles. J. Immunol. Res. 2016, 2016, 6391264. [Google Scholar] [CrossRef]
- Oba, R.; Isomura, M.; Igarashi, A.; Nagata, K. Circulating CD3+HLA-DR+ Extracellular Vesicles as a Marker for Th1/Tc1-Type Immune Responses. J. Immunol. Res. 2019, 2019, 6720819. [Google Scholar] [CrossRef]
- Amjadi, M.F.; Avner, B.S.; Greenlee-Wacker, M.C.; Horswill, A.R.; Nauseef, W.M. Neutrophil-derived extracellular vesicles modulate the phenotype of naïve human neutrophils. J. Leukoc. Biol. 2021, 110, 917–925. [Google Scholar] [CrossRef]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, W.; Xia, Y.; Wang, X.; Liang, J. Platelet-derived extracellular vesicles promote endothelial dysfunction in sepsis by enhancing neutrophil extracellular traps. BMC Immunol. 2023, 24, 22. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef]
- Terrisse, A.D.; Puech, N.; Allart, S.; Gourdy, P.; Xuereb, J.M.; Payrastre, B.; Sié, P. Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow. J. Thromb. Haemost. 2010, 8, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Sadallah, S.; Eken, C.; Martin, P.J.; Schifferli, J.A. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J. Immunol. 2011, 186, 6543–6552. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Dashevsky, O.; Rivo, J.; Gozal, Y.; Varon, D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc. Res. 2005, 67, 30–38. [Google Scholar] [CrossRef]
- Gao, Y.; Jin, H.; Tan, H.; Cai, X.; Sun, Y. Erythrocyte-derived extracellular vesicles aggravate inflammation by promoting the proinflammatory macrophage phenotype through TLR4-MyD88-NF-κB-MAPK pathway. J. Leukoc. Biol. 2022, 112, 693–706. [Google Scholar] [CrossRef]
- Yadid, M.; Lind, J.U.; Ardoña, H.A.M.; Sheehy, S.P.; Dickinson, L.E.; Eweje, F.; Bastings, M.M.C.; Pope, B.; O’Connor, B.B.; Straubhaar, J.R.; et al. Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chi. Sci. Transl. Med. 2020, 12, eaax8005. [Google Scholar] [CrossRef]
- van Balkom, B.W.; de Jong, O.G.; Smits, M.; Brummelman, J.; den Ouden, K.; de Bree, P.M.; van Eijndhoven, M.A.; Pegtel, D.M.; Stoorvogel, W.; Würdinger, T.; et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013, 121, 3997–4006, S1-15. [Google Scholar] [CrossRef]
- Todor, S.B.; Ichim, C.; Boicean, A.; Mihaila, R.G. Cardiovascular Risk in Philadelphia-Negative Myeloproliferative Neoplasms: Mechanisms and Implications-A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 8407–8423. [Google Scholar] [CrossRef]
- Waldenström, A.; Ronquist, G. Role of exosomes in myocardial remodeling. Circ. Res. 2014, 114, 315–324. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Zhu, H.; Kranias, E.G.; Tang, Y.; Peng, T.; Chang, J.; Fan, G.C. Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS ONE 2012, 7, e32765. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Guo, X.; Liu, X.M.; Liu, L.; Weng, Q.F.; Dong, S.J.; Knowlton, A.A.; Yuan, W.J.; Lin, L. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc. Res. 2013, 98, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Zhan, R.; Leng, X.; Liu, X.; Wang, X.; Gong, J.; Yan, L.; Wang, L.; Wang, Y.; Wang, X.; Qian, L.J. Heat shock protein 70 is secreted from endothelial cells by a non-classical pathway involving exosomes. Biochem. Biophys. Res. Commun. 2009, 387, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.A.; Moncayo-Arlandi, J.; Sepulveda, P.; Diez-Juan, A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc. Res. 2016, 109, 397–408. [Google Scholar] [CrossRef]
- Felekkis, K.; Papaneophytou, C. Challenges in Using Circulating Micro-RNAs as Biomarkers for Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 561. [Google Scholar] [CrossRef]
- Liu, P.; Wang, S.; Wang, G.; Zhao, M.; Du, F.; Li, K.; Wang, L.; Wu, H.; Chen, J.; Yang, Y.; et al. Macrophage-derived exosomal miR-4532 promotes endothelial cells injury by targeting SP1 and NF-κB P65 signalling activation. J. Cell Mol. Med. 2022, 26, 5165–5180. [Google Scholar] [CrossRef]
- Hosseinkhani, B.; Kuypers, S.; van den Akker, N.M.S.; Molin, D.G.M.; Michiels, L. Extracellular Vesicles Work as a Functional Inflammatory Mediator Between Vascular Endothelial Cells and Immune Cells. Front. Immunol. 2018, 9, 1789. [Google Scholar] [CrossRef]
- Hosseinkhani, B.; van den Akker, N.; D’Haen, J.; Gagliardi, M.; Struys, T.; Lambrichts, I.; Waltenberger, J.; Nelissen, I.; Hooyberghs, J.; Molin, D.G.M.; et al. Direct detection of nano-scale extracellular vesicles derived from inflammation-triggered endothelial cells using surface plasmon resonance. Nanomedicine 2017, 13, 1663–1671. [Google Scholar] [CrossRef]
- He, S.; Wu, C.; Xiao, J.; Li, D.; Sun, Z.; Li, M. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand. J. Immunol. 2018, 87, e12648. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Zhang, S.; Yan, S.; Wang, Z.; Wang, C.; Zhang, X. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol. Med. Rep. 2019, 19, 3298–3304. [Google Scholar] [CrossRef]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Chen, X.; Cheng, X.; Liao, Y.; Yu, X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J. Mol. Med. 2016, 94, 711–724. [Google Scholar] [CrossRef]
- Burrello, J.; Biemmi, V.; Dei Cas, M.; Amongero, M.; Bolis, S.; Lazzarini, E.; Bollini, S.; Vassalli, G.; Paroni, R.; Barile, L. Sphingolipid composition of circulating extracellular vesicles after myocardial ischemia. Sci. Rep. 2020, 10, 16182. [Google Scholar] [CrossRef]
- de Hoog, V.C.; Timmers, L.; Schoneveld, A.H.; Wang, J.W.; van de Weg, S.M.; Sze, S.K.; van Keulen, J.K.; Hoes, A.W.; den Ruijter, H.M.; de Kleijn, D.P.; et al. Serum extracellular vesicle protein levels are associated with acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2013, 2, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Nickenig, G.; Werner, N. Extracellular Vesicles in Cardiovascular Disease: Potential Applications in Diagnosis, Prognosis, and Epidemiology. Circ. Res. 2017, 120, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef]
- Deng, W.; Tang, T.; Hou, Y.; Zeng, Q.; Wang, Y.; Fan, W.; Qu, S. Extracellular vesicles in atherosclerosis. Clin. Chim. Acta 2019, 495, 109–117. [Google Scholar] [CrossRef]
- Srikanthan, S.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J. Thromb. Haemost. 2014, 12, 1906–1917. [Google Scholar] [CrossRef]
- Blaser, M.C.; Buffolo, F.; Halu, A.; Turner, M.E.; Schlotter, F.; Higashi, H.; Pantano, L.; Clift, C.L.; Saddic, L.A.; Atkins, S.K.; et al. Multiomics of Tissue Extracellular Vesicles Identifies Unique Modulators of Atherosclerosis and Calcific Aortic Valve Stenosis. Circulation 2023, 148, 661–678. [Google Scholar] [CrossRef]
- Charla, E.; Mercer, J.; Maffia, P.; Nicklin, S.A. Extracellular vesicle signalling in atherosclerosis. Cell Signal 2020, 75, 109751. [Google Scholar] [CrossRef]
- Młynarska, E.; Czarnik, W.; Fularski, P.; Hajdys, J.; Majchrowicz, G.; Stabrawa, M.; Rysz, J.; Franczyk, B. From Atherosclerotic Plaque to Myocardial Infarction-The Leading Cause of Coronary Artery Occlusion. Int. J. Mol. Sci. 2024, 25, 7295. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 2012, 96, 64–72. [Google Scholar] [CrossRef]
- van der Zee, P.M.; Biró, E.; Trouw, L.A.; Ko, Y.; de Winter, R.J.; Hack, C.E.; Sturk, A.; Nieuwland, R. C-reactive protein in myocardial infarction binds to circulating microparticles but is not associated with complement activation. Clin. Immunol. 2010, 135, 490–495. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Zhao, R.; Qiu, Z.; Shen, C.; Wang, Z.; Liu, W.; Zhang, W.; Ge, J.; Shi, B. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics 2021, 11, 6315–6333. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Liu, L.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Shi, J.; Zhou, W.; Wang, L.; Fang, W.; Zhong, Y.; Chen, X.; Chen, Y.; Sabri, A.; et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic. Res. Cardiol. 2020, 115, 22. [Google Scholar] [CrossRef] [PubMed]
- Gabisonia, K.; Khan, M.; Recchia, F.A. Extracellular vesicle-mediated bidirectional communication between heart and other organs. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H769–H784. [Google Scholar] [CrossRef]
- Abassi, Z.; Khoury, E.E.; Karram, T.; Aronson, D. Edema formation in congestive heart failure and the underlying mechanisms. Front. Cardiovasc. Med. 2022, 9, 933215. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Tang, X.; Yang, Z.; Liu, C.; Zhang, X.; Jin, J.; Lyu, J. Plasma-derived exosomes contribute to inflammation via the TLR9-NF-κB pathway in chronic heart failure patients. Mol. Immunol. 2017, 87, 114–121. [Google Scholar] [CrossRef]
- Kou, Y.; Zou, L.; Liu, R.; Zhao, X.; Wang, Y.; Zhang, C.; Dong, Z.; Kou, J.; Bi, Y.; Fu, L.; et al. Intravascular cells and circulating microparticles induce procoagulant activity via phosphatidylserine exposure in heart failure. J. Thromb. Thrombolysis 2019, 48, 187–194. [Google Scholar] [CrossRef]
- Acelajado, M.C.; Hughes, Z.H.; Oparil, S.; Calhoun, D.A. Treatment of Resistant and Refractory Hypertension. Circ. Res. 2019, 124, 1061–1070. [Google Scholar] [CrossRef]
- Mayet, J.; Hughes, A. Cardiac and vascular pathophysiology in hypertension. Heart 2003, 89, 1104–1109. [Google Scholar] [CrossRef]
- Martinez-Quinones, P.; McCarthy, C.G.; Watts, S.W.; Klee, N.S.; Komic, A.; Calmasini, F.B.; Priviero, F.; Warner, A.; Chenghao, Y.; Wenceslau, C.F. Hypertension Induced Morphological and Physiological Changes in Cells of the Arterial Wall. Am. J. Hypertens. 2018, 31, 1067–1078. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Jose, P.A.; Yang, J.; Zeng, C. Importance of extracellular vesicles in hypertension. Exp. Biol. Med. 2021, 246, 342–353. [Google Scholar] [CrossRef]
- Mostefai, H.A.; Agouni, A.; Carusio, N.; Mastronardi, M.L.; Heymes, C.; Henrion, D.; Andriantsitohaina, R.; Martinez, M.C. Phosphatidylinositol 3-kinase and xanthine oxidase regulate nitric oxide and reactive oxygen species productions by apoptotic lymphocyte microparticles in endothelial cells. J. Immunol. 2008, 180, 5028–5035. [Google Scholar] [CrossRef]
- Burrello, J.; Gai, C.; Tetti, M.; Lopatina, T.; Deregibus, M.C.; Veglio, F.; Mulatero, P.; Camussi, G.; Monticone, S. Characterization and Gene Expression Analysis of Serum-Derived Extracellular Vesicles in Primary Aldosteronism. Hypertension 2019, 74, 359–367. [Google Scholar] [CrossRef]
- Nomura, S.; Tandon, N.N.; Nakamura, T.; Cone, J.; Fukuhara, S.; Kambayashi, J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 2001, 158, 277–287. [Google Scholar] [CrossRef]
- Xu, H.; Ni, Y.Q.; Liu, Y.S. Mechanisms of Action of MiRNAs and LncRNAs in Extracellular Vesicle in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 733985. [Google Scholar] [CrossRef]
- Wang, Z.; Mo, H.; He, Z.; Chen, A.; Cheng, P. Extracellular vesicles as an emerging drug delivery system for cancer treatment: Current strategies and recent advances. Biomed. Pharmacother. 2022, 153, 113480. [Google Scholar] [CrossRef]
- Jurgielewicz, B.; Stice, S.; Yao, Y. Therapeutic Potential of Nucleic Acids when Combined with Extracellular Vesicles. Aging Dis. 2021, 12, 1476–1493. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, S.; Ren, X.; Wu, Z.; Liu, S.; Yao, X. Current Strategies for Exosome Cargo Loading and Targeting Delivery. Cells 2023, 12, 1416. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, Q.; Zhang, J.; Sun, L.; Hong, X.; Du, W.; Duan, R.; Jiang, J.; Ji, Y.; Wang, H.; et al. Exosomes derived from mir-214-3p overexpressing mesenchymal stem cells promote myocardial repair. Biomater. Res. 2023, 27, 77. [Google Scholar] [CrossRef]
- Wei, Z.; Chen, Z.; Zhao, Y.; Fan, F.; Xiong, W.; Song, S.; Yin, Y.; Hu, J.; Yang, K.; Yang, L.; et al. Mononuclear phagocyte system blockade using extracellular vesicles modified with CD47 on membrane surface for myocardial infarction reperfusion injury treatment. Biomaterials 2021, 275, 121000. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, M.; Deng, S.; Lu, J.; Huang, H.; Zhang, Y.; Gong, P.; Shen, X.; Ruan, H.; Jin, M.; et al. miR-93-5p-Containing Exosomes Treatment Attenuates Acute Myocardial Infarction-Induced Myocardial Damage. Mol. Ther. Nucleic Acids 2018, 11, 103–115. [Google Scholar] [CrossRef]
- Vrijsen, K.R.; Sluijter, J.P.; Schuchardt, M.W.; van Balkom, B.W.; Noort, W.A.; Chamuleau, S.A.; Doevendans, P.A. Cardiomyocyte progenitor cell-derived exosomes stimulate migration of endothelial cells. J. Cell Mol. Med. 2010, 14, 1064–1070. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, J.; Zhao, Q.; Zhuang, W.; Ding, J.; Zhang, C.; Gao, H.; Pang, D.W.; Pu, K.; Xie, H.Y. Molecularly Engineered Macrophage-Derived Exosomes with Inflammation Tropism and Intrinsic Heme Biosynthesis for Atherosclerosis Treatment. Angew. Chem. Int. Ed. Engl. 2020, 59, 4068–4074. [Google Scholar] [CrossRef] [PubMed]
- Sutaria, D.S.; Jiang, J.; Elgamal, O.A.; Pomeroy, S.M.; Badawi, M.; Zhu, X.; Pavlovicz, R.; Azevedo-Pouly, A.C.P.; Chalmers, J.; Li, C.; et al. Low active loading of cargo into engineered extracellular vesicles results in inefficient miRNA mimic delivery. J. Extracell. Vesicles 2017, 6, 1333882. [Google Scholar] [CrossRef] [PubMed]
- Komuro, H.; Aminova, S.; Lauro, K.; Harada, M. Advances of engineered extracellular vesicles-based therapeutics strategy. Sci. Technol. Adv. Mater. 2022, 23, 655–681. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Circulating Biomarkers | Release Time | Disease | Half Time | Normal Range | Refs. |
|---|---|---|---|---|---|
| cTn-I and cTn-T 1 | 3–4 h | Endocarditis, myocardial infarction, myocarditis, pericarditis | 7–10 days | ≤0.04 ng/mL | [7] |
| BNP 2 and NT-proBNP 3 | 1.5–2 h | Atrial fibrillation, heart failure, stroke, transient ischemic attack | 5–7 days | ≤125 pg/mL | [8] |
| CK-MB 4 | 3–4 h | Cardiac trauma, heart transplantation, myocardial infarction, myocarditis, pulmonary embolism | 24–72 h | 10 to 25 IU/L | [8] |
| CRP 5 | 12 h | Coronary heart disease, ischemic stroke | 24 h | ≤0.3 mg/dL | [9,10] |
| Myoglobin | 3 h | Acute myocardial infarction, acute myocarditis | 5 min | 50–85 Ug/L | [8] |
| D-dimer | 2 h | Deep venous thrombosis, pulmonary embolism | 8 h | ≤0.50 mg/L | [11,12] |
| Homocysteine | 6–8 h | Thrombosis | 3–4 h | ≤10 µmol/L | [13] |
| EV Origin | Marker | Location | Refs. |
|---|---|---|---|
| Cardiomyocyte | Caveolin-3, flotillin-1 | Membrane-embedded | [34] |
| Endothelial cell | CD31 (PECAM-1), CD144 (VE-cadherin), CD146 (MCAM), CD62E (E-selectin), | Surface | [35] |
| von Willebrand Factor (vWF) | Intravesicular | ||
| Erythrocyte | CD55, CD59, CD235a (Glycophorin A) | Surface | [36] |
| Hemoglobin (α and β chains), | Intravesicular | ||
| Band 3 (Anion Exchanger 1) | Membrane-embedded | ||
| T helper cells | CD4, CD45, CD28, CD16 | Surface | [37] |
| T cytotoxic cells | CD8, CD45, CD28, CD16 | Surface | [37] |
| B cells and Natural killer cells | CD49d | Surface | [37] |
| Monocyte | CD11b, CD14, CD64, CD142, HLA-DR | Surface | [37,38] |
| Neutrophil | CD35, CD66b | Surface | [39] |
| Platelet | CD41a (Integrin αIIb), CD42b (Glycoprotein Ib; GPIb), CD61 (Integrin β3), CD62P (P-selectin) | Surface | [40,41] |
| Disease | Cell Origin | EV Cargo | Clinical Outcomes | Species | Ref. |
|---|---|---|---|---|---|
| AS 1 | Macrophages | miR-4532 | Promote endothelial cell dysfunction | Human | [56] |
| AS | Macrophages | TNF-α | Propagation of inflammatory signals | Human | [43] |
| AS | Endothelial cells | CXCL-10 | Promote endothelial cell dysfunction | Human | [57] |
| AS | Endothelial cells | ICAM-1, VCAM | Recruitment of leukocytes | Human | [58] |
| AS, MI 2 | Macrophages | miR-155 | Suppression fibroblast proliferation and promotion of inflammation | Human | [59] |
| Coronary AS | N/A | miR-30, miR-92a | Regulation of cellular cholesterol and phospholipid homeostasis | Human | [60] |
| HF 3 | Myoblasts | miR 192, miR 194, and miR-34a | Induction of ventricular remodeling | Human | [61] |
| HT 4, AS | Leukocytes and platelets | IL-1, IL-8 | Promotion of inflammatory responses in endothelial cells | Human | [57] |
| MI | N/A | miR-30a | Regulate autophagy | Human | [62] |
| MI | N/A | Ceramides | Induction of cardiomyocyte apoptosis and inflammation | Human | [63] |
| MI | N/A | Sphingolipids | Modulation of endothelial function and inflammation | Human | [63] |
| MI | N/A | PIgR 5, Cystatin C, and Complement factor C5a | Promote inflammatory responses | Human | [64] |
| MI, AS | Cardiomyocytes | CCL2, CCL7 | Promotion of inflammatory responses | Human | [57] |
| MI, HT | Leukocytes, platelet, and cardiomyocytes | IL-6 | Promotion of inflammatory responses | Human | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappucci, I.P.; Tremoli, E.; Zavan, B.; Ferroni, L. Circulating Extracellular Vesicles in Cardiovascular Disease. Int. J. Mol. Sci. 2025, 26, 6817. https://doi.org/10.3390/ijms26146817
Cappucci IP, Tremoli E, Zavan B, Ferroni L. Circulating Extracellular Vesicles in Cardiovascular Disease. International Journal of Molecular Sciences. 2025; 26(14):6817. https://doi.org/10.3390/ijms26146817
Chicago/Turabian StyleCappucci, Ilenia Pia, Elena Tremoli, Barbara Zavan, and Letizia Ferroni. 2025. "Circulating Extracellular Vesicles in Cardiovascular Disease" International Journal of Molecular Sciences 26, no. 14: 6817. https://doi.org/10.3390/ijms26146817
APA StyleCappucci, I. P., Tremoli, E., Zavan, B., & Ferroni, L. (2025). Circulating Extracellular Vesicles in Cardiovascular Disease. International Journal of Molecular Sciences, 26(14), 6817. https://doi.org/10.3390/ijms26146817